

Abstract
The nicotinamide adenine dinucleotide (NAD+) is an essential redox cofactor, involved in various physiological and molecular processes, including energy metabolism, epigenetics, aging and metabolic diseases. NAD+ repletion ameliorates muscular dystrophy and improves the mitochondrial and muscle stem cell function, and thereby increase lifespan in mice. Accordingly, NAD+ is considered as an antioxidant and anti-aging molecule. NAD+ plays a central role in energy metabolism and the energy produced is used for movements, thermoregulation, and defense against foreign bodies. The dietary precursors of NAD+ synthesis is targeted to improve NAD+ biosynthesis, however, studies have revealed conflicting results regarding skeletal muscle-specific effects. Recent advances in the activation of nicotinamide phosphoribosyltransferase (NAMPT) in the NAD+ salvage pathway and supplementation of NAD+ precursors have led to beneficial effects in skeletal muscle pathophysiology and function during aging and associated metabolic diseases. NAD+ is also involved in the epigenetic regulation and post-translational modifications of proteins that are involved in various cellular processes to maintain tissue homeostasis. This review provides detailed insights into the roles of NAD+ along with molecular mechanisms during aging and disease conditions such as the impacts of age-related NAD+ deficiencies on NAD+-dependent enzymes, including poly (ADP-ribose) polymerase (PARPs), CD38, and sirtuins within skeletal muscle, and the most recent studies on the potential of nutritional supplementation and distinct modes of exercise to replenish the NAD+ pool.
Keywords: NAD+, Epigenetics, Muscle Diseases, Aging, Redox, Diabetes
Introduction
The aging human population is increasing worldwide along with an increase in healthcare spending [1, 2]. Logically, questions about treating the aging population ensure the demand to understand the aging process for healthy living and to restore the functional independence during old age [3]. Knowing the specific processes that occur within the body during aging could shape geriatric treatment, improving the prospect of individualized therapy based on the genetic makeup and environmental factors. Consequently, the anti-oxidant and anti-aging effects of NAD+ has gained interest in reducing the signs of aging [4].
The nicotinic adenine dinucleotide (NAD+) is a cofactor that plays a central role in energy metabolism for adenosine triphosphate (ATP) production in all cells and regulates tissue homeostasis. The availability of NAD+ is critical for the optimal functioning of cellular processes and normal physiology. The NAD+ fluxes vary widely in different tissues and is highly abundant in small intestines and spleen, and low in the skeletal muscle. The levels of NAD+ is reduced during aging and in metabolic diseases [5-8]. Studies have implicated lower levels of NAD+ to deleterious effects in skeletal muscle health, and the substitution of NAD+ and its precursors augment muscle health [4, 9]. NAD+ is largely known for its role as an essential redox molecule in metabolic pathways [10, 11]. However, NAD+ also plays an important role as a substrate for enzymes such as poly-adenosine diphosphate (ADP)-ribose polymerases (PARPs), cluster of differentiation (CD)38, CD73, CD157 and SARM1 (sterile alpha and toll interleukin receptor motif-containing protein 1), and sirtuins [11-14]. PARPs are implicated in DNA repair and the regulation of DNA metabolism [12]. However, recent evidences also suggest a role for PARPs in RNA biogenesis and metabolism [15]. CD38 is a cyclic (ADP)-ribose that plays a role in calcium homeostasis and signaling through the mobilization of Ca2+ stores as a second messenger [13]. The nicotinamide mononucleotide (NMN/β-NMN) acts a substrate for both CD38 and CD73, with the formation of nicotinamide (NAM) and nicotinamide riboside (NR), respectively [16]. Likewise, NAD+ also serves as a substrate for CD38 and CD157 [17], and the ectoenzymes are involved in immune response and aging [18, 19]. Increased SARM1 activity causes axon degeneration during injury with rapid breakdown of NAD+, which is protected by increasing the NAD+ synthesis [20]. Sirtuins are histone deacetylases (HDACs) with enzymatic activities that targets and affect cellular processes, such as mitochondrial biogenesis, glucose and lipid metabolism, inflammation, insulin sensitivity, and apoptosis [21]. In mammals, 7 sirtuin (SIRT) isoforms are reported, viz. SIRT1-7 that are classified as class III histone deacetylases [22]. Sirtuins play crucial roles in metabolism and stress response and are dependent on their localization within the cell. Isoforms of sirtuins 3, 4, and 5 are detected within the mitochondria, whereas sirtuins 1, 6, and 7 are located within the nucleus, and sirtuin 2 shuttles between both the nucleus and the cytoplasm. Overall, upon use by PARPs, CD38 and sirtuins, NAD+ is converted to NAM [23] (Fig. 1A), lowering cellular amounts of the available NAD+. Recent evidence suggests that it also acts as a nucleotide analog in DNA ligation and RNA capping [24, 25]. The present review provides an overview of the key aspects of NAD+ and its relation to epigenetics, inflammation, aging and metabolism as it relates to pathological states or injury. In addition, the strategies for increasing the NAD+ content in the tissues is also discussed for pharmacological targeting and therapeutics. While previous reviews provide focus on NAD+ levels in heart, or during aging related processes, the present review elaborates on the skeletal muscle specific roles of NAD+ and the molecular determinants during aging.
Figure 1. The NAD+ de novo and salvage pathways.

(A) NAD+ is utilized by CD38 by hydrolyzing NAD+ to NAM. SIRTs and PARPs use NAD+ as a co-substrate for deacetylation and PARYlation, respectively. This process generates NAM as a byproduct and inhibits the activity of NAD-dependent enzymes; (B) NAM is salvaged to regenerate NAD+. NAMPT catalyzes NAM into NMN, which allows to regenerate NAD+ by NMNAT1-3. NR is salvaged into NMN by NKR1/2 and regenerate NAD+ [23, 26, 27]. (C) Chemical structures of the NAD+ precursors; tryptophan, nicotinamide, nicotinic acid, nicotinamide riboside, nicotinamide mononucleotide, and the Nampt enzymatic activator P7C3 compound.
The depletion of the NAD+ pool by NADases can be replenished by NAD+ salvage enzymes [23, 26] (Fig. 1B). Nicotinamide riboside kinase 1 and 2 (NRK1, 2) are NAD+ salvage enzymes that regenerate NAD+ from NR [27]. Nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenyltransferases (NMNAT) 1-3 are key enzymes that are involved in the NAD+ salvage pathway. NAMPT enzymatically converts NAM to NMN, which is then converted to NAD+ by NMNATs (Fig. 1B). Alternatively, using the dietary nicotinic acid (NA) through Preiss-Handler pathway NAD+ can be replenished [28]. However, the Preiss-Handler pathway has limited use in skeletal muscle compared with the salvage pathway [29]. Figure 1C shows the chemical structure of the NAD+ precursors involved in the de novo (Tryptophan), Preiss-Handler (NA), and salvage (NAM, NR, and NMN) pathways, and the key rate limiting enzyme Nampt activator P7C3 involved in the salvage pathway.
In skeletal muscle, NAD+ is mainly produced through NAD+ salvage pathway and protects the muscle from metabolic stress and structural defects through increased mitochondrial biogenesis, gene expression and organization of the extracellular matrix [9]. We recently reported that activation of Nampt by P7C3, a small molecule ameliorates type 2 diabetes and skeletal muscle function in db/db mice [30]. Skeletal muscle serves as the major site of glycogen storage, insulin-mediated glucose uptake, lipid metabolism, and fatty acid oxidation within the body [31-33]. Skeletal muscle is composed of different types of myofibers (Table 1) such as fast twitch (glycolytic) and slow twitch (oxidative) fiber types [31, 33]. Slow twitch fibers are found in muscles that contract slowly and are mainly fueled by aerobic energy mechanisms such as oxidative phosphorylation, and consequently have high mitochondrial contents [34]. Whereas, fast twitch fibers are used for quick bursts of energy, relying on glycolytic mechanisms, and have low mitochondrial content [31, 35]. During aging and in metabolic diseases such as obesity and diabetes the fiber type distribution gets altered to meet the energy demand [34]. Aging causes a decline in the levels of NAMPT and correspondingly the NAD+ levels fluctuate [36]. This impairs the replenishment of NAD+ and has consequent effects on NAD-dependent co-substrates [37]. The overall effect of this age-related change is seen in a multitude of ways, including mitochondrial dysfunctions, insulin resistance, inflammation, and impairments in repair mechanisms within the skeletal muscle [38].
Table 1.
Skeletal muscle fiber types and their properties.
| Myofiber type/properties | Type 1 | Type 2a | Type 2b | Type 2x/d |
|---|---|---|---|---|
| Myosin heavy chain isoform | MyHC1 | MyHC2a | MyHC2b | MyHC2x/d |
| myofiber cross-sectional area | Small | Intermediate | Large | Large |
| Myosin ATPase activity | Slow | Fast | Fast | Fast |
| Energy metabolism | Oxidative | Predominantly oxidative | Glycolytic | Glycolytic |
| Twitch | Slow | Moderate | Very fast | Fast |
| Fatigue | Resistant | Moderately resistant | Fast fatigue | Fast fatigue |
| Force production | Weak | Intermediate | Very high | Strong |
| Endurance capacity | High | Intermediate | Low | Low |
| Myoglobin content | High | High | Low | Low |
| Appearance | Red | Red | White | White |
| Mitochondria numbers | High | High | Low | Low |
Skeletal muscle aging also known as sarcopenia results in loss of muscle mass and function that is exacerbated by myopathies, inflammation, metabolic diseases, and muscle damage. In this review, we provide insights into the roles of NAD+ within skeletal muscle and its epigenetic implications on skeletal muscle aging and disease. The review also provides insights into various physical, nutritional, and pharmacological interventions to reduce mitochondrial dysfunctions, inflammation, and increased insulin resistance within aging skeletal muscle by alleviating NAD+ deficiencies.
NAD+ functioning and metabolism in healthy skeletal muscle
In healthy skeletal muscle, NAD+ levels are maintained in a range from 100 μM to 1000μM within the mitochondria and cytosol that permit and regulate various NAD-dependent processes involved in cellular metabolism [39]. Further, the role of NAD+ is implicated in distinct subcellular compartments such as the nucleus, cytosol, and mitochondria [9, 40]. Previous reports show the important role of NAD+ in the process of muscle development and gene expression [9, 41, 42].
NAD+-dependent Sirtuins, PARPs and CD38 function
The NAD-dependent deacetylase, sirtuins play an important role in maintaining cellular processes in skeletal muscle health. The skeletal muscle stem cells change the substrate use from oxidative to glycolytic state during myogenesis, and in the muscle stem cells that undergo differentiation the NAD+ levels were decreased with reduced SIRT1 activity and increased H4K16 acetylation, leading to an increase in myogenic gene expression [43]. Similarly, the muscle-specific inactivation of SIRT1 in mice increased H4K16 acetylation and activation of muscle genes during myogenesis. These muscle-specific SIRT1-null mice also displayed a decrease in myofiber size, and impaired muscle regeneration and gene expression [43]. While the NAD+/NADH ratio is decreased during myogenic differentiation, an increase in the NAD+/NADH ratio has been shown to inhibit muscle gene expression through Sir2 [44].
The NAD+ supply also controls sirtuin’s ability to regulate various processes such as mitochondrial function, metabolism and cell cycle [45]. SIRT1 deacetylates the transcriptional regulators such as FOXO (Forkhead Box O) and PGC-1α (Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha), which increases mitochondrial biogenesis and reduces the oxidative stress [46]. SIRT1 is also important in responding to muscle injury through the regulation of NF-κB (Nuclear Factor-κB), FOXOs, MYOD1 (Myogenic Determination Protein1), and MEF2 (Myocyte Enhancer Factor2) [47]. Further, SIRT1 has been demonstrated to deacetylate the circadian core clock genes, CLOCK and BMAL1, promoting gene expression in mouse embryonic fibroblasts [47].
SIRT1 and SIRT6 both downregulate IGF-1 (Insulin-like Growth Factor 1) and alter its target AKT-mTOR pathway. This pathway is associated with skeletal muscle hypertrophy during periods of upregulation and atrophy during downregulation [48]. SIRT6 also helps to maintain muscle mass by regulating the expression of myostatin, a dominant-negative regulator of muscle mass and its activin receptor type 2b, blocking myostatin signaling [49]. SIRT6 knockout mice results in increased skeletal muscle fibrosis and degeneration associated with higher myostatin levels [49]. SIRT6 is also involved in DNA repair efficiency, and SIRT6 knockout mice demonstrated increased cellular senescence [50, 51]. In an another study involving SIRT6 knockout mice, AMP-activated protein kinase (AMPK) activity was downregulated, resulting in a reduction in fatty acid oxidation, mitochondrial oxidative phosphorylation, and glucose and lipid uptake in muscle cells suggesting SIRT6 also plays a role in energy metabolism [52].
SIRT1, SIRT3 change the mitochondrial function in a similar manner. SIRT3, being localized within the mitochondria, regulates several mitochondrial proteins such as succinate-dehydrogenase (SDH), FOXO3, manganese-dependent superoxide dismutase (Mn-SOD), and acetyl-CoA synthase [53]. During periods of fasting, mitochondrial oxidative phosphorylation is increased in part due to the expression of Sirt3, where SIRT3 is also suggested to regulate oxidative muscle fiber formation [53, 54]. Absence of SIRT3 in mice is linked with direct downregulatory effects on PGC-1α and effect mitochondrial biogenesis [55]. SIRT4 is detected in lower levels within skeletal muscle as compared to SIRT3 but thought to play a role in mitochondrial oxidative metabolism, where it acts as a negative regulator [56]. Overall, the sirtuins functioning is regulated by NAD+ and changes in the levels of NAD+ can influence SIRT activity in skeletal muscle tissue [57].
Along with sirtuins, NAD+ is also necessary for the proper functioning of PARPs, and CD38 [58, 59]. Poly (ADP)-ribosylation is a post-translational modification catalyzed by PARPs that involves the consecutive addition of ADP-ribose moieties from NAD+ to amino acids [60, 61]. PARPs are a family of proteins that partake in an array of cellular processes such as DNA repair and programmed cell death [61]. The role of PARPs in DNA repair helps to explain why PARP levels are elevated during aging, that is accompanied by increased rates of DNA damage. CD38 is a glycoprotein detected on the surface of many white blood cells or immune cells. CD38 has been described to play many roles such as regulating metabolism, immune response, inflammation, and cancer [62, 63].
Metabolism
NAD+ plays a central role in metabolism and acts as a substrate for signaling enzymes. The redox couple of NAD+/NADH is required by enzymes within the cytosol and mitochondria, serving as a regulator for the processes of glycolysis and mitochondrial oxidative phosphorylation (Fig. 2A). The process of glycolysis is performed within the cytosol during which, glucose is converted into glyceraldehyde 3-phosphate (G3P), and NAD+ is reduced to NADH by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in order to convert G3P into 1,3-bisphosphate [64, 65], and this process continues to generate pyruvate. In anaerobic conditions, pyruvate can be reduced to lactate by the enzyme lactate dehydrogenase (LDH), which oxidizes NADH to NAD+ [65]. Pyruvate that is not converted into lactate can be shuttled into the mitochondria. Once in the mitochondria, pyruvate dehydrogenase (PDH) utilizes NAD+ to convert pyruvate into acetyl-CoA, which is then oxidized to form adenosine triphosphate (ATP) within the tricarboxylic acid (TCA) cycle [64].
Figure 2. NAD+/NADH in skeletal muscle glycolysis.
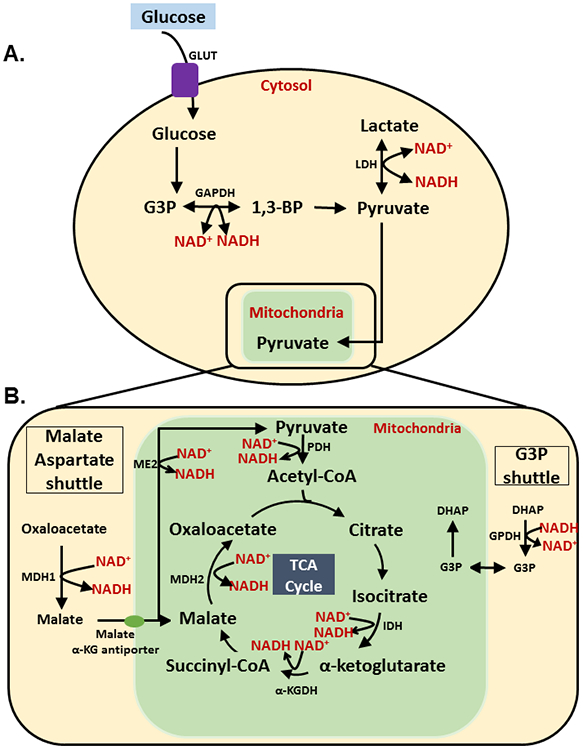
(A) Glucose enters cells using GLUT 4 transporters. In the cytosol, glucose is further converted and processed by enzymes: hexokinase, phosphoglucose isomerase, phosphofructokinase, and aldolase in step-wise manner and converted into G3P. At this juncture, NAD+ is utilized by GAPDH to convert it into 1,3-BP (1,3-Bisphosphoglyceric acid). 1,3-BP is converted into pyruvate via four additional steps, each step involving the action of a unique enzyme. Pyruvate can then be reduced to form lactate by LDH (lactate dehydrogenase). LDH uses NADH and regenerates NAD+. Lower NAD+ levels that are accompanied with aging affect the production of 1,3-BP (1,3-bisphosphoglycerate), and consequently the production of pyruvate as an energy source to be used in the TCA cycle [64, 65]; (B) Cytosolic pyruvate enters the mitochondria where it is oxidized into acetyl-CoA by PDH (pyruvate dehydrogenase). PDH reduces NAD+ into NADH. Acetyl-CoA enters the TCA cycle and is converted into citrate. Citrate is converted into isocitrate by the enzyme aconitase. Isocitrate is oxidized by isocitrate dehydrogenase (IDH), reducing NAD+ into NADH and forming α-ketoglutorate. During this process NAD+ is reduced into its NADH form, by α-ketoglutorate dehydrogenase (α-KGDH), which converts α-ketoglutorate into succinyl-CoA. As shown in Figure 2, Succinyl-CoA then forms malate by enzymes succinyl-CoA synthetase, succinic dehydrogenase and fumarate. NAD+ is reduced to NADH to convert malate into oxaloacetate by malate dehydrogenase 2 (MDH2). Cytosolic oxaloacetate is then converted into malate by the enzyme malate dehydrogenase 1 (MDH1), which oxidizes NADH to NAD+. Cytosolic malate enters the mitochondria through the malate α-ketoglutorate antiporter and is used in the TCA cycle. Cytosolic DHAP (Dihydroxyacetone phosphate) is then converted into G3P by cytosolic GPDH, which oxidizes NADH to NAD+. G3P then enters the mitochondria and is converted into DHAP. This shuttling of G3P into the mitochondria is referred to as the G3P shuttle. Lower levels of NAD+ inhibit acetyl-CoA formation from pyruvate, α-ketoglutorate from isocitrate, succinyl-CoA from α-ketoglutorate, and oxaloacetate from malate. The inhibition of these steps prevents the formation of NADH and consequently effects redox reactions in the electron transport chain. Redox reactions are necessary to produce the proton gradient that is used to phosphorylate ADP (Adenosine diphosphate) into ATP and generate the energy that is required for the proper functioning of skeletal muscle [66, 68-70].
Within the TCA cycle, NAD+ is required in the conversion of D-isocitrate to α-ketoglutorate, α-ketoglutorate to succinyl-CoA, and malate to oxaloacetate by the enzymes isocitrate dehydrogenase, α-ketoglutorate dehydrogenase and malate dehydrogenase, respectively [66]. Malate, a precursor in this cycle, can be converted back to pyruvate by the malic enzyme (ME2) [67], which is NAD-dependent; however, it is expressed in low levels in skeletal muscle and may be involved in metabolism. Glutamate dehydrogenase 1 is a NAD+-dependent mitochondrial enzyme that converts glutamate to α-ketoglutarate and ammonia [68]. Similar to ME2, this enzyme is also expressed at low levels in skeletal muscle.
The malate aspartate shuttle and glyceraldehyde 3-phosphate shuttle both utilize NAD+/NADH to shuttle necessary metabolic substrates (Fig. 2B). In the malate aspartate shuttle, cytosolic oxaloacetate or oxalacetic acid (OAA) is reduced to malate by malate dehydrogenase (MDH1), which utilizes and oxidizes NADH to NAD+ [69]. Malate is shuttled into the mitochondria and converted back into OAA by malate dehydrogenase 2 (MDH2), and in this shuttling process NAD+ is utilized and reduced back into NADH. In the G3P shuttle, the glycolytic intermediate dihydroxyacetone phosphate (DHAP) is reduced to G3P by glycerol-3-phosphate dehydrogenase (GPDH) [70] and this conversion oxidizes NADH into NAD+. G3P can then be shuttled into the mitochondria and partake in other metabolic conversions, such as the conversion back to DHAP.
The processes of glycolysis and oxidative phosphorylation are necessary to generate ATP in the mitochondria, which is required for the healthy contraction and functioning of skeletal muscle. Thus proper functioning of skeletal muscle requires a high turnover of ATP and is dependent on the abundance of NAD+, due to its utilization in the processes of glycolysis and oxidative phosphorylation [71]. Without a sufficient supply of NAD+ that is needed in the processes of glycolysis and oxidative phosphorylation, these pathways that are necessary to produce cellular energy are disrupted. Nowhere is this more relevant than in skeletal muscle, which accounts for more than 70% usage of circulating insulin and glucose [72]. Research has demonstrated a hallmark of aging to be a progressive loss in skeletal muscle mass and function, potentially developing into sarcopenia. Investigation into the ageing population, including sarcopenia patients demonstrated a significant decrease in skeletal muscle biopsy NAD+ levels with a negative correlation with age [36, 73]. NAD+ levels strongly correlated with functional changes, including appendicular lean mass (ALM) index, grip strength and gait speed in sarcopenia patients [73]. Further investigation demonstrated a significant decrease in many mitochondrial genes, including those forming the electron transport chain, Complex I-V in the sarcopenia muscle biopsies [73]. Figure 3 demonstrates decreased Nampt activity and thereby decreased NAD+ biosynthesis during aging and in muscle diseases, including inflammation, which results in increased DNA damage and altered mitochondrial biogenesis. In addition, sex differences exist with relation to NAD+ levels in animal models both with baseline and during injury induced conditions such as stroke. The NAD+ levels were found to be lower in females compared with males or ovariectomized mice. Cerebral ischemia decreased the NAD+ levels, and were linked with PARP-1 and metabolic changes, which were altered in a sex-specific manner [74]. However, the NAD+ levels in blood plasma were not affected with age or sex in humans [75] suggesting the tissue-specific NAD+ biosynthetic activities.
Figure 3. Skeletal muscle inflammation results in decreased Nampt and NAD+ biosynthesis with increased DNA damage and altered mitochondrial biogenesis.

DMD is linked with a reduction in NAMPT activity and subsequent NAD+ biosynthesis. The DNA damage present in patients with DMD leads to heightened PARP activity. PARPs and SIRT1 both compete for NAD+, leading to a decrease in SIRT1 activity due to increased PARP activation. The reduction in SIRT1 activity results in decreased mitochondrial biogenesis, due to inhibition of the SIRT1/PGC-1α pathway. Inflammation of muscle and the consequent production of inflammatory cytokines in muscle diseases leads to reductions in NAMPT. Similarly, the inflammation associated with aging, also known as “inflamm-aging” leads to further reductions in NAMPT as more inflammatory cytokines are seen to circulate in the skeletal muscle. NAMPT regulates NAD+ levels and downstream activity of PARPs and sirtuins [166].
NAD+ in skeletal muscle disease
Muscular Dystrophies
Inherited muscle diseases show effects of decreased muscle performance, mass, and function not attributed to aging [76]. Muscular dystrophies are defined as genetic alterations that result in progressive skeletal muscle weakness and deterioration. Duchenne’s muscular dystrophy (DMD) is the most common type of muscular dystrophy, and as such has the most strides being made in terms of its treatment. DMD is characterized by the degeneration of skeletal muscle due to the absence of the protein dystrophin [77], has a co-occurrence of low muscle NAD+ abundance [78]. Major consumers of NAD+ such as PARPs and N-methyltransferase (NNMT) are elevated in DMD patients and other muscular diseases. Further, in the muscles of mdx mouse model of human DMD, the levels of NAD+ were significantly reduced along with a reduction in NAMPT activity and increase in PARP activity [79]. In mdx mice, treatment with resveratrol, a SIRT1 activator, helped to relieve the pathophysiology of this muscular dystrophy [80]. Given the role of NAD+ in skeletal muscle functioning and aging, it is hopeful to think that the effects of NAD+ biosynthesis in DMD animal models could be extended to other muscular dystrophies and muscle diseases as a therapy.
Metabolic diseases
Skeletal muscle is a major site of energy expenditure and metabolic functioning. Metabolic disorder and disease such as obesity and type 2 diabetes mellitus (T2DM) affect skeletal muscle, with far reaching inflammatory effects that lead to reductions in the amount of available NAD+ [81]. A major cause of skeletal muscle metabolic disorder is due to high-fat and high-sugar diet, leading to obesity. Skeletal muscle samples from mice fed a high-fat diet (HFD) displayed high levels of PARP-2, a NADase enzyme in the PARP protein family. These mice also showed decreased activation of the SIRT1/PGC-1α pathway, a pathway that is important in mitochondrial biogenesis and function [82]. Further delineation is observed in diabetes-induced muscle injury with type 1 diabetes demonstrating a decline in NAD+ levels likely the result of increased activation of PARP consequence of low NAD+ levels remain unknown, however, ATP levels were significantly decreased [83].
Given the roles of NAD+ in metabolic regulation and sirtuin activity, it is reasonable to hypothesize if metabolic health could be improved by NAD+ supplementation, and if NAD-dependent proteins like PARP could be inhibited to elevate the NAD+ levels. Surprisingly, in a study involving the overexpression of NAMPT, which resulted in a 50% increase in mouse skeletal muscle NAD+ levels did not alleviate the negative effects of a HFD or induce mitochondrial biogenesis [84]. This raises questions as to the tissues in which NAD+ levels to be increased, and contribute to positive effects within skeletal muscle.
NAD+ in skeletal muscle aging
Inflammation
Insulin resistance is known to co-occur with the aging process in humans [85] including inflammation. The relationship between inflammation and insulin resistance is cyclic, in which inflammation can cause insulin resistance and insulin resistance leads to inflammation [86] (Fig. 4). Skeletal muscle inflammation has emerged as a player in the development of T2DM [87, 88]. Skeletal muscle inflammation is the result of multiple causes, such as obesity. With obesity levels increasing with age, chronic low grade inflammation is also on the rise [89]. Adipose tissue and skeletal muscle both secrete inflammatory cytokines, such as IL-1β, IL-6, and TNF- α [90].
Figure 4. The cyclic effects of inflammation and insulin resistance in skeletal muscle involving NAD+ biosynthesis.

Skeletal muscle inflammation results in the production of inflammatory cytokines. Inflammatory cytokines can lead to a reduction in NAD+ synthesis by downregulating NAD+ salvage enzymes. A reduction of NAD+ biosynthesis results in impaired sirtuin signaling and function, which is involved in insulin resistance. Insulin resistance has inflammatory effects that creates a cyclic effect, promoting further insulin resistance [90].
The inflammatory cytokines and oxidative stress decreases NAD+ biosynthesis by the intracellular nicotinamide phosphoribosyl transferase (iNAMPT) [81]. The skeletal muscle-specific NAMPT deficient mice resulted in a dramatic reduction of NAD+ levels along with fiber degeneration and muscle weakness [91]. Whereas, overexpression of NAMPT in aging mice helped to preserve NAD+ levels, as well as muscle mass and function [91]. In an another study, involving the reduction of NAMPT in mice resulted in a metabolic shift from oxidative to glycolytic, which was associated with acetylation of the SIRT6 targets within the SIRT6/HIF1α axis [38].
A metabolic shift away from glucose oxidation is positively correlated with insulin resistance [92, 93]. It is thought that mitochondrial dysfunction could contribute to age-related insulin resistance, however, the mechanisms by which mitochondria effect insulin resistance are not fully understood [87, 92, 94]. In mice that had genetic ablation of SIRT2, the insulin-induced glucose uptake occurring in skeletal muscle was decreased compared to control groups [95]. This result implicates SIRT2, as a cytosolic and mitochondrial deacetylase, with a possible role in the mitochondrial dysfunction resulting in insulin sensitivity.
Changes in metabolism
SIRT1 is a predominant sirtuin isoform that has been studied well for its effects on mitochondrial functioning. SIRT1 directly affects the activity of an important regulator of mitochondrial biogenesis, PGC-1α [46]. PGC-1α is a coactivator of multiple transcription factors that work together to increase mitochondrial biogenesis. The activation of SIRT1 by resveratrol deacetylates PGC-1α, enhancing its transcriptional activity with increased mitochondrial biogenesis in skeletal muscle [96].
The activation of sirtuin is negatively correlated to the activity of NADases. CD38 is a NADase that actively increases with aging and is thought to lead to mitochondrial dysfunction in part due to changes in SIRT3 activity [97]. Mice that are deficient in CD38 demonstrate higher metabolic rates with reduced risk of obesity, which is attributed to the enhanced mitochondrial function through the upregulation of sirtuin activity [98, 99]. While the observed decline in skeletal muscle-specific SIRT3 with age is due to higher acetylation levels and decreased mitochondrial functioning [100, 101]. Within the mitochondria are enzymes and proteins that can be acetylated and regulate mitochondrial biogenesis through their activation or inhibition.
Mitochondrial dysfunction is thought to be involved in the muscle aging process [102, 103]. There are multiple factors playing into the deterioration of mitochondrial function, implicating NAD+-dependent protein acetylation/deacetylation as a cause [100, 104]. In mice and humans, higher acetylation levels within skeletal muscle mitochondria were associated with lower exercise capacity and increased acetylation of fatty acid β-oxidation enzymes [105]. Acetylation of these enzymes are related to reductions in the capacity of the mitochondria to carry out processes of fatty-acid oxidation within skeletal muscle [105, 106]. The results from this study implicated SIRT3 in the regulation of acetylation levels, however, the abundance of NAD+ and NAD+/NADH were not shown to differ significantly between the experimental and control groups [105].
Muscle damage and repair
The genetic ablation of SIRT1 in mouse muscle stem cells impaired muscle function [107]. However, the study was not able to directly link SIRT1 levels to muscle structure, function, and recovery following injury. Whereas, the skeletal-muscle specific SIRT1-null mice showed muscular fragility attributed to impaired membrane resealing and repair [108]. Increased DNA damage occurs with aging and results in either cell death or cellular senescence through mechanisms in healthy cells that prevent unchecked cell growth [109]. A senescence-associated secretory phenotype is observed in senescent cells, which activates PARP-1 and CD38 [58]. While aging is reported to upregulate SIRTs, there is a decline in NAD+ abundance seen partly due to competition from other NAD+-dependent enzymes, including CD38 and PARP-1 [58, 59]. A major cause for the decline in NAD+ levels during aging is CD38 [110]. Aging is linked to elevated levels of CD38, but not PARP1 or SIRT1, indicating CD38 as a major culprit contributing to NAD+ deficiencies [97]. However, SIRT1 regulates cell survival through the interaction of DNA damage proteins, including Ku70, FOXL2, NBS1 (Nibrin), WRN (Werner Syndrome ATP-dependent Helicase), and XPC (Xeroderma Pigmentosa Complementation Group C) [111]. SIRT1 also helps to modulate apoptosis through the interaction of FOXO proteins, demonstrated specifically in the activation of FOXO3 that can potentiate its ability to resist oxidative stress [112].
NAD+-dependent epigenetic modifications of DNA and RNA in musculoskeletal disease and aging
NAD+-dependent acetylation and deacetylation
Although, there are several NAD+-dependent enzymes in the cellular milieu, sirtuins represent a unique and important class of histone modifiers/erasers known as HDACs (Histone Deacetylases) contributing to the epigenetic control of gene expression. Sirtuins erase or remove acetyl groups from the specific amino acids of histone tails. The acetylation and deacetylation of lysine residues at the N-terminal tail of histone proteins has gene regulatory effects. Acetylation neutralizes the positive charge of lysine, reducing the electrostatic interactions and results in a less densely packed form of chromatin. This form of chromatin is referred to as euchromatin and is often associated with active transcription. Whereas, the tightly packed DNA, known as heterochromatin is associated with deacetylation and lower levels of transcription or gene silencing. Histone proteins are acetylated and deacetylated typically by HATs (Histone Acetyl Transferases) and HDACs, respectively.
Aging is known to coincide with a decrease in histone acetylation and there are four different groups of HDACs classified thus far, playing various roles in skeletal muscle development. Class I enzymes contain HDACs 1-3, and 8, Class II contain HDACs 4-7, 9 and 10, Class III HDACs contain Sirtuins (SIRT) 1-7 in mammals and Sir2 in yeast, and Class IV contain HDAC 11 [22]. Class I and II HDACs utilize Zn as a cofactor to catalyze deacetylation of histone/non-histone substrates. Whereas, Class III HDACs or sirtuins utilize NAD+ as a cofactor for deacetylation of their substrates. Although, there are several class III HDACs, their activity is regulated or restricted to their subcellular localization. SIRT 1, 2, 6 and 7 are predominantly nuclear, and because NAD+ is relatively a small molecule, it can shuttle in and out of the nucleus. This shuttling integrates the availability of NAD+ through metabolic processes and deacetylation of histone, which will turn off gene expression [113, 114].
NAD+ is an important cofactor playing a key role as a cellular rheostat in enzyme catalysis and whose levels dictate the global acetylation or deacetylation patterns brought about by sirtuins, relating metabolic flux and gene expression. Sirtuins interact with not only histones, but also with several other important transcriptional factors and proteins involved in cellular signaling networks. They can also remove several other acyl groups from histones (Lysine-K) like benzoyl, propionyl, butyryl, etc. [115, 116]. The roles of these covalent modifications and the consequence of acyl groups removal by sirtuins remains to be unexplored from a normal cellular physiology or pathology point of view.
Sirtuins also regulate the activity of several transcriptional factors such as NF-kB, FOXO1 and other important proteins involved in transcriptional, developmental and signaling cascades [117-120]. The protein type and subcellular localization are factors that affect whether the removal of acetyl groups from specific amino acids of proteins, which will lead to either loss-of-function or gain-of-function. Table 2 shows the list of various histone and non-histone substrates of sirtuins, and their corresponding cellular outcomes.
Table 2.
Substrates of sirtuins and their corresponding cellular outcomes.
| NAD-dependent Sirtuins |
Epigenetic modification -deacetylation / deacylation - Histone substrates |
Role in gene regulation | References |
|---|---|---|---|
| SIRT1 | H3K9ac, H3K14ac, H1K26ac, H4K16ac, H3K4ac, H3K56ac Kpr, Lys propionylation Kbu, Lys butyrylation Kcr, Lys crotonylation |
Repression Unknown function |
[115, 116, 153-156] |
| NF-kB | Inhibits NF-kB-mediated transcription | [117] | |
| FOXO1 | Promotes cell survival | [118] | |
| PTEN | Repression of PTEN and downstream effector signaling cascades | [119, 120] | |
| p65K310, NF-kB | Inhibits transcription of NF-kB target genes | [117] | |
| SIRT2 | H4K16ac, H3K56ac H2AK5 debenzoylation Kpr, Lys propionylation Kbu, Lys butyrylation Kcr, Lys crotonylation |
Repression Unknown function |
[115, 116, 157-159] |
| p53 | Repression | [160] | |
| α-Tubulin | Microtubule depolymerization | [161, 162] | |
| SIRT3 | H4K16ac Kpr, Lys propionylation Kbu, Lys butyrylation Kcr, Lys crotonylation |
Repression Unknown function |
[115, 116] |
| SIRT5 | Kma, Lys malonylation Ksucc,Lys succinylation Kglu, Lys glutarylation |
Unknown function | [116] |
| SIRT6 | H3K9ac, H3K56ac, H3K18ac | Repression / prevents DNA erosion | [49, 163, 164] |
| SIRT7 | H3K18ac Non-histone substrates |
Repression Cellular function |
[165] |
Association between NAD+ and DNA methylation
DNA methylation occurs at the 5’-C position in a CpG (5’-Cytosine-phosphate-Guanine-3’) dinucleotide and is generated and maintained by DNMTs (DNA methyl transferases). There are four DNMTs found in eukaryotes, including DNMT1 and DMNT3s (DNMT3A, DNMT3B, and DNMT3L). DNMT1 is primarily active during cell division, maintaining DNA methylation, while DNMT3s are responsible for de novo cytosine methylation and maintenance [121]. Promoter hypermethylation typically occurs at the CpG islands and causes gene repression. Within the skeletal muscle tissue, hypermethylation and consequent gene repression are reported to increase with age [122].
Methylated Cytosines in CpG islands are recognized by methyl CpG binding proteins (CBP), and these further recruit histone deacetylases (HDACs/Sirtuins), histone methyl transferases (HMTs), and chromatin remodeling complexes to co-repress transcription, ultimately leading to gene repression (Fig. 5A). This process occurs in an opposite direction where promotor hypomethylation cause gene activation. 5-methylcytosine and 5-hydroxymethylcytosine is called the 5th and 6th base, respectively apart from the normal Adenine, Thymine, Guanine and Cytosine nucleotide bases, since they play a role in gene regulation mediated by the action of DNMTs and TET (ten-eleven-translocase) enzymes. The 5-methylcytosine and 5-hydroxymethylcytosine play an important role in connecting epigenetic control of gene expression and metabolism.
Figure 5. Epigenetic regulation and metabolic links to NAD.

(A) The NAD-dependent epigenetic modifications of the DNA. The heterochromatin is associated with histones deacetylation and lower levels of gene transcription or gene silencing. (B) RNA modifications involved in mRNA splicing, translation, stability, and transport. The RNA methylases (Mettle3, and Mettle14 interact with WTAP), and demethylases (FTO and ALKBH5) are involved in the most commonly observed m6A mRNA modifications.
Aging is a complex process and involves gradual but substantial changes in gene expression and metabolic pathways mainly dependent on environmental factors like food (folate intake), physical exercise, etc. Dietary folate, choline, and vitamin B12 are important methyl donors and regulate DNA and histone methylation. In addition, α-ketoglutarate is essential for the action of TET enzymes and links the TCA cycle to DNA methylation and gene expression. As shown in Figure 2B, the reduction of NAD+ is required to generate α-ketoglutarate from isocitrate. Without adequate supply of NAD+, the TCA cycle will not be able to run efficiently, limiting the amounts of α-ketoglutarate formed from this pathway and hindering TET activity [123].
The genome-wide association studies (GWAS) performed in humans surveying the DNA methylation status in skeletal muscle DNA display the role of epigenetics during normal aging. In a study performed on post-mitotic skeletal muscle tissue of 48 healthy males, half of the individuals aged between 18-27 years and the other half aged between 68-89 years showed differences in the differentially methylated CpG nucleotides[124]. This study identified over 2,114 genes within the aged skeletal muscle having at least one dmCpG site located intragenically. Within the genes observed to be differentially methylated in aging individuals was Tubulin-folding co-factor D (TBCD), containing the highest number of intragenic dmCpG sites. TBCD is a component of microtubules that can result in the disruption of microtubule pathways, if altered, affecting motor neuron’s function. The NFATC1 (Nuclear Factor of Activated T Cells 1) gene was also identified to be of interest, having a large number of intragenic dmCpG sites and playing a role in skeletal muscle transcription regulation [124]. A similar study was performed on the whole blood of 1,550 female twins aged between 17-82 years in an effort to identify the relationship between DNA methylation levels and the skeletal muscle mass variations with age. Seven regions were identified to be of importance due to their location in or near genes previously noted in muscle-related studies. These genes being DNAH12 (Dynein Axonemal Heavy Chain 12), CAND1 (Cillin-Associated NEDD8-Dissociated Protein 1), CYP4F29P (Cytochrome P450 Family 4 Subfamily F Member 29, pseudogene), and ZFP64 (Zinc Finger Protein 64) [125].
The effect of methylation patterns within skeletal muscle stem cell on aging was also investigated in a clinical study. This study used muscle stem cells obtained from 22 adults, 7 young and 14 aged individuals, demonstrates that DNA methylation is affected by age and regulates genes controlling cell quiescence [126]. In the muscle samples from aged individuals, methylation of SPRY1 (Sprouty RTK Signaling Antagonist 1) negatively impacted the abundance of the stem cell pool, caused by a failure of re-quiescence in the activated stem cells. It was shown that the myogenic potential is preserved in these stem cells, however, the ability to return to quiescence is impaired after the activation process is triggered. Overall, the suppression of SPRY1 by DNA methylation leads to fewer reserve cells and impairs muscle stem cells self-renewal [126].
RNA modifications
Eukaryotic messenger RNA (mRNA) contains several methylation modifications reported with modifications that include N7-methylguanosine (m7G) at the cap, internal 5-methylcytosine (m5C) and N6-methyladenosine (m6A), N6-methyl-2'-O-methyladenosine (m6Am), and 2’-O-methylation (Nm) at internal positions and within the cap (Fig. 5B) [127]. The m7G cap is known to play significant roles in mRNA translation, stability, and nuclear export. Whereas m6A modifications regulates mRNA splicing, translation, stability, and transport among other processes [127]. Thus, the significance of many RNA methylations remains to be determined. However, the most observed mRNA modification is m6A [128]. Methyltransferases (METTL3, METTL14, and WTAP) and demethylases (ALKBH5 and FTO) can modulate the m6A modification [127, 129-131]. Low levels of FTO (fat mass and obesity-associated protein) increased m6A methylation levels and decreased lipid content in skeletal muscle [132]. The obese ob/ob mice are reported with larger amounts of lipid deposition within the skeletal muscle, implicating higher levels of FTO [132]. In addition, the m6A methylation is also implicated in cell senescence [133]. In mice containing low levels of METTL3 in skeletal muscle tissue, MyoD RNA was downregulated and associated with a decrease in processed mRNA [134]. This demonstrated the role of METTL3 in stabilizing MyoD mRNA levels and promoting skeletal muscle differentiation, as reducing METTL3 levels led to lower levels of m6A methylation and premature differentiation of C2C12 myoblasts [134]. Thus, RNA modifications are implicated in the metabolic disorders, including obesity, cell senescence and myogenesis, however, the significance of NAD+ is not fully evaluated and therefore remains unclear.
Treatments and Interventions
Dietary Supplements and Pharmacological Treatments
The baseline requirement of NAD+ is mainly met by the dietary tryptophan or niacin, which serves as the precursor of NAD+ synthesis. The other dietary NAD+ precursors such as NAM and NR are also incorporated into the intracellular NAD+ pool [135]. Increasing the NAD+ biosynthesis and availability more than that met by our daily dietary intake is considered beneficial for human health, especially in metabolic and muscle diseases such as obesity, diabetes, muscular dystrophies, and other neurodegenerative diseases [135]. The pharmacological use of NR has advantages over the use of other precursors such as NA or NAM. NR appears to be more effective in increasing the intracellular NAD+ levels with less side effects than the NA or NAM precursors [135]. A study utilizing younger mice fed with NR demonstrated improved endurance suggesting enhanced mitochondrial functioning resulting in a higher oxidative capacity as compared with the vehicle treated group of mice [136]. These studies demonstrate that NR supplementation increased the pool of NAD+ and led to a significant increase in the activity of SIRT1 and SIRT3 within skeletal muscle. Furthermore, sirtuin activity increased the expression of PGC-1α, thereby increasing the mitochondrial biogenesis. In a separate study involving aged men given oral NR supplementation there was an increase in the muscle NAD+ metabolome, as evidenced by a two-fold increase in muscle nicotinic acid adenine dinucleotide (NaAD), a biomarker of NAD+ synthesis [137], however, NAD+ levels were not increased. Interestingly, the NR supplementation was also associated with the downregulation of genes involved in glycolysis, TCA cycle and mitochondrial energy metabolism within muscle. Further analysis of these results did not show increased mitochondrial copy number, respiration, or citrate synthase activity. NR-mediated changes were not observed in sirtuin functioning, as changes in the acetylation levels of muscle proteins were not detected. While these results did not demonstrate enhanced sirtuin activity, the study provided insights into decreased inflammatory effects that were observed with NR supplementation. In addition, NR supplementation led to a reduction in the levels of circulating inflammatory cytokine such as IL-2, IL-5, IL-6, and TNF-α [137].
Recent reports utilizing NR supplementation in the ageing population sought to identify skeletal muscle content in metabolic disease, including obese and insulin-resistant men. Similar results were observed with NR supplementation having little to no effect on skeletal muscle mitochondria. Interestingly, NR-supplementation resulted in a significant decrease in NAMPT skeletal muscle protein expression [138]. Further, NAMPT expression correlated with key mitochondrial proteins, including the electron transport complex (ETC) I, succinate dehydrogenase, and sirtuin 3.
Metabolic enzyme activity profiling of mice suggested that NAMPT and NRK1/2-mediated salvage pathways are the main biosynthetic routes within skeletal muscle to replenish NAD+ pools [27]. NMN and NR are the substrates and product of these two enzymes, respectively. Supplementation using these NAD+ precursors was seen to result in a 2-fold increase of NAD+ biosynthesis within the skeletal muscle myotubes. NAMPT knockout mice treated with NAD+ supplements showed high levels of NAD+, confirming that the NAD+ precursors of NMN and NR can be salvaged independently of NAMPT. This suggests the potential of NRK2 as a skeletal muscle-specific target to improve NAD+ abundance and has provided new context for the role of NAMPT as a rate-limiting step of NAD+ salvage. NRK2 knockout in mice had no effect on the NAD+ abundance, demonstrating redundancy within the NAD+ salvage pathway [27]. In a 12-month long NMN supplementation using mice as subjects, enhanced energy metabolism, suppressed weight gain, and increased physical activity and insulin sensitivity were observed [139]. Overtime, the NAD+ levels were decreased in skeletal muscle tissue, however, the mitochondrial respiration rate in skeletal muscle was enhanced. Furthermore, a 10-week treatment of NMN supplementation in obese prediabetic women enhanced skeletal muscle insulin sensitivity and signaling [135]. These results show promise in the potential use of NRK2 as a skeletal muscle-specific target in conjunction with NMN and NR supplementation to improve NAD+ abundance.
As an important enzyme in the NAD+ salvage pathway, NAMPT has been investigated as a target to increase the abundance of viable NAD+ within skeletal muscle cells. In mice that had muscle-specific overexpression of NAMPT, NAD+ levels were consequently higher but were not seen to regulate any targets of oxidative metabolism, however, an enhanced exercise capacity was observed [84, 91]. NAMPT and NRK2 both function within the NAD+ salvage pathway. Whereas, the Preiss-Handler pathway is another biosynthetic pathway for the generation of NAD+ in cells. In both these pathways NMNAT3 is present as a common enzyme (Figs. 1A, B). In mice, the administration of the Nampt activator P7C3 (Fig. 1C), a small molecule neuroprotective compound belonging to the class of aminopropyl carbazole has also led to increased NAD+ biosynthesis and availability [140-143]. Recently, we demonstrated that administration of P7C3 in the diabetic db/db mice increased insulin sensitivity and improved muscle function via. Nampt activation [30].
NMNAT3 has been established as the only enzyme in the NAD+ synthesis pathway that is localized in the mitochondria. This brings into question whether NMNAT3 could be seen as a target to increase NAD+ abundance within the mitochondria, however, NMNAT3 requires the import of cytosolic NMN for NAD+ production [144]. Despite this limitation, NMNAT3-overexpressing mice had significantly increased mitochondrial NAD+ levels within skeletal muscle tissue, accompanied by increased fatty acid oxidation [145]. Inconsistent with these results is the demonstration that NMNAT3 knockout in mice does not result in a significant difference in mitochondrial NAD+ levels [145]. This could be due to a redundancy in the enzymatic activity. More work needs to be done on NMNAT3 expression to determine its viability as a pharmacological target in humans to increase NAD+ biosynthesis.
Exercise
There are many different pathways that can be affected by exercise and is considered as the gold standard to improve the aging muscle function. AMPK plays a key role in the regulation of ATP levels within the cell through the upregulation or downregulation of anabolic and catabolic pathways, respectively [146]. Because AMPK acts as an energy sensor and exercise utilizes ATP (the bodies energy currency), it is clear that exercise would result in increased AMPK activation [147]. AMPK signaling was seen to indirectly activate SIRT1 by altering intracellular NAD+/NADH ratio. The effects of AMPK on NAD+ levels help to explain the increased mitochondrial biogenesis that is observed along with increased oxidative capacity of skeletal muscle and reduced signs of skeletal muscle fragility [148, 149].
Sirtuin activity is demonstrated to be enhanced with exercise, unsurprisingly given the elevated effects exercise is reported to have on intracellular NAD+ levels [57, 150]. It has also been suggested that SIRT1 can be activated by an acute load of exercise, while exercise training can activate both SIRT1 and SIRT3 [57]. In human subjects of 65 years of age and older, master athletes in this age group had higher protein levels of SIRT1 and SIRT3 [151].
NAMPT decreases with age in skeletal muscle, helping to explain the decline in NAD+ levels that is also seen during aging. Recent studies demonstrate that distinct modes of exercise can impact the NAD+ salvage capacity in the skeletal muscle. Aerobic and resistance exercise training was compared in a study involving young and aged individuals to assess whether these exercise modes have varying effects on NAD+ salvage pathways [152]. The increase in NAMPT levels was positively correlated with VO2 peak and lean body mass. At the end of 12 weeks, aerobic exercise training increased NAMPT levels by 28% in older individuals and just 12% in younger individuals. Resistance exercise training was seen to increase NAMPT levels by 30% in older individuals and 28% in younger individuals [152]. While these numbers demonstrate aerobic and resistance training modes have similar impacts in aged individuals, what is interesting is the difference that is seen in the effects of the younger participants. If resistance exercise can improve NAMPT levels more strikingly than aerobic exercise in the younger participants, it suggests that resistance exercise has the potential to be used in younger populations as a method to prevent or prolong the effects of NAD+ deficiencies later in life. It would be interesting to follow the impact of resistance exercise from young to old age to study the viability of this exercise mode in preventing NAD-deficiency-mediated aging compared to aerobic exercise groups.
Summary and future perspectives
NAD+ is an essential substrate for many enzymes involved in the metabolic processes. The present review provides an overview of the involvement on NAD+ in aging-related musculoskeletal diseases and inflammation. A decrease in intracellular NAD+ level is seen in various tissues and stem cells during aging, and in metabolic and muscle diseases. While advanced clinical care and research in geroscience successfully extends the lifespan of the individuals, additional research focused on epigenetics and molecular mechanisms are required to understand the process of aging along with the pharmacological ways of using small molecules to restore function. Metabolic links between glucose utilization, diabetes, and fatty acid metabolism provide insights into skeletal muscle function and mitochondrial cellular pathways that may be utilized to rescue the dysfunction in musculoskeletal disorders during aging.
Various genetic and biochemical approaches were utilized to restore or increase the intracellular NAD+ levels during aging and in muscle diseases. The NAD+ centric roles are provided in the present review with links to epigenetics, metabolic changes, and pathological states. The strategies for modulating the intracellular NAD+ levels via supplementation of the NAD+ precursors such as NR, NMN, NA, and NAM or by increasing the enzymatic activity of the enzymes involved in the NAD+ biosynthetic pathways along with exercise provide beneficial effects in repair and reversing the muscle injury during aging, and in metabolic diseases. The therapeutic advantage of Nampt activation as the key rate limiting enzyme to ameliorate muscle disease and improve muscle function is interesting but it is not tested in humans, yet. Overall, it will be interesting to combine the beneficial effects of the dietary supplementation of NAD+ precursors and small molecule based enzymatic activation with moderate exercise as an effective combination strategy to circumvent the aging-associated skeletal muscle pathophysiologies, and muscle diseases. However, more studies are needed to validate their use in human as a therapeutic option to increase the lifespan.
Funding
SMT and MB are partially supported by NIH Grants - National Institutes of Diabetes, Digestive, and Kidney NIDDK-R01DK119066 (SMT, MB). National Institutes of Aging (NIA) 2-PO1AG039355 (MB), NIA-R01AG056504 (MB), NIA-R01AG060341 (MB). We are thankful for the generous support from the William Saunders Endowed Chair in geriatric Pharmacotherapy (SMT) at University of South Florida. The George W. and Hazel M. Jay and Evanston Research Endowments (MB), and the University of Texas - Arlington College of Nursing and Health Innovation, Bone-Muscle Research Center (https://www.uta.edu/conhi/research/bmrc/index.php). The opinions expressed in this review are solely the authors’ opinions and conclusions, and not represent the official opinions and/or endorsement from National Institutes of Health, USF, or UTA.
Footnotes
Conflicts of interest
All authors declared that they have no conflict of interest.
Disclosures
US Patent: 19A039PR[143], USF disclosure, 19A039 (patent pending).
Data Availability
All data were included in the manuscript, since the article is a review paper this is not applicable.
References
- 1.Dieleman JL, Squires E, Bui AL, Campbell M, Chapin A, Hamavid H, Horst C, Li Z, Matyasz T, Reynolds A, Sadat N, Schneider MT and Murray CJL (2017) Factors Associated With Increases in US Health Care Spending, 1996-2013. JAMA 318:1668–1678. doi: 10.1001/jama.2017.15927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalbarczyk M and Mackiewicz-Łyziak J (2019) Physical Activity and Healthcare Costs: Projections for Poland in the Context of an Ageing Population. Applied Health Economics and Health Policy 17:523–532. doi: 10.1007/s40258-019-00472-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung E, Wongrakpanich S and Munshi MN (2018) Diabetes Management in the Elderly. Diabetes Spectrum : A Publication of the American Diabetes Association 31:245–253. doi: 10.2337/ds18-0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi P and Tyagi SC (2018) NAD(+) : A big player in cardiac and skeletal muscle remodeling and aging. J Cell Physiol 233:1895–1896. doi: 10.1002/jcp.26014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaku K, Okabe K and Nakagawa T (2018) NAD metabolism: Implications in aging and longevity. Ageing Res Rev 47:1–17. doi: 10.1016/j.arr.2018.05.006 [DOI] [PubMed] [Google Scholar]
- 6.Tur J, Chapalamadagu KC, Manickam R, Cheng F and Tipparaju SM (2021) Deletion of Kvβ2 (AKR6) Attenuates Isoproterenol Induced Cardiac Injury with Links to Solute Carrier Transporter SLC41a3 and Circadian Clock Genes. Metabolites 11. doi: 10.3390/metabo11040201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Bhatnagar A and Tipparaju SM (2017) Kvβ1.1 (AKR6A8) senses pyridine nucleotide changes in the mouse heart and modulates cardiac electrical activity. Am J Physiol Heart Circ Physiol 312:H571–h583. doi: 10.1152/ajpheart.00281.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kilfoil PJ, Chapalamadugu KC, Hu X, Zhang D, Raucci FJ Jr., Tur J, Brittian KR, Jones SP, Bhatnagar A, Tipparaju SM and Nystoriak MA (2019) Metabolic regulation of Kv channels and cardiac repolarization by Kvβ2 subunits. J Mol Cell Cardiol 137:93–106. doi: 10.1016/j.yjmcc.2019.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goody MF and Henry CA (2018) A need for NAD+ in muscle development, homeostasis, and aging. Skelet Muscle 8:9. doi: 10.1186/s13395-018-0154-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spinelli JB and Haigis MC (2018) The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nature cell biology 20:745–754. doi: 10.1038/s41556-018-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osellame LD, Blacker TS and Duchen MR (2012) Cellular and molecular mechanisms of mitochondrial function. Best Practice & Research. Clinical Endocrinology & Metabolism 26:711–723. doi: 10.1016/j.beem.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung AKL (2017) PARPs. Current Biology 27:R1256–R1258. doi: 10.1016/j.cub.2017.09.054 [DOI] [PubMed] [Google Scholar]
- 13.Lee HC (2012) Cyclic ADP-ribose and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) as Messengers for Calcium Mobilization. The Journal of Biological Chemistry 287:31633–31640. doi: 10.1074/jbc.R112.349464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Covarrubias AJ, Perrone R, Grozio A and Verdin E (2021) NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 22:119–141. doi: 10.1038/s41580-020-00313-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eleazer R and Fondufe-Mittendorf YN (2021) The multifaceted role of PARP1 in RNA biogenesis. Wiley Interdiscip Rev RNA 12:e1617. doi: 10.1002/wrna.1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A and Bruzzone S (2013) CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem 288:25938–25949. doi: 10.1074/jbc.M113.470435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaku K, Okabe K, Hikosaka K and Nakagawa T (2018) NAD Metabolism in Cancer Therapeutics. Front Oncol 8:622. doi: 10.3389/fonc.2018.00622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czura AW and Czura CJ (2006) CD38 and CD157: biological observations to clinical therapeutic targets. Molecular medicine (Cambridge, Mass.) 12:309–311. doi: 10.2119/2007-00006.czura [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higashida H, Hashii M, Tanaka Y, Matsukawa S, Higuchi Y, Gabata R, Tsubomoto M, Seishima N, Teramachi M, Kamijima T, Hattori T, Hori O, Tsuji C, Cherepanov SM, Shabalova AA, Gerasimenko M, Minami K, Yokoyama S, Munesue SI, Harashima A, Yamamoto Y, Salmina AB and Lopatina O (2019) CD38, CD157, and RAGE as Molecular Determinants for Social Behavior. Cells 9. doi: 10.3390/cells9010062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerdts J, Brace EJ, Sasaki Y, DiAntonio A and Milbrandt J (2015) SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348:453–7. doi: 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A and Altucci L (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clinical Epigenetics 8. doi: 10.1186/s13148-016-0224-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh ME and Van Remmen H Emerging roles for histone deacetylases in age-related muscle atrophy. Nutrition and Healthy Aging 4:17–30. doi: 10.3233/NHA-160005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarino M and Dufour J-F (2019) Nicotinamide and NAFLD: Is There Nothing New Under the Sun? Metabolites 9. doi: 10.3390/metabo9090180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen SH and Yu X (2019) Human DNA ligase IV is able to use NAD+ as an alternative adenylation donor for DNA ends ligation. Nucleic Acids Res 47:1321–1334. doi: 10.1093/nar/gky1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bird JG, Zhang Y, Tian Y, Panova N, Barvík I, Greene L, Liu M, Buckley B, Krásný L, Lee JK, Kaplan CD, Ebright RH and Nickels BE (2016) The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature 535:444–7. doi: 10.1038/nature18622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson S and Imai Si (2018) NAD + biosynthesis, aging, and disease. F1000Research 7. doi: 10.12688/f1000research.12120.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva Xavier G, Garten A, Elhassan YS, Redpath P, Migaud ME, Philp A, Brenner C, Canto C and Lavery GG (2017) Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Molecular Metabolism 6:819–832. doi: 10.1016/j.molmet.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verdin E (2015) NAD+ in aging, metabolism, and neurodegeneration. Science 350:1208–1213. doi: 10.1126/science.aac4854 [DOI] [PubMed] [Google Scholar]
- 29.Yang Y and Sauve AA (2016) NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochimica et biophysica acta 1864:1787–1800. doi: 10.1016/j.bbapap.2016.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manickam R, Tur J, Badole SL, Chapalamadugu KC, Sinha P, Wang Z, Russ DW, Brotto M and Tipparaju SM (2022) Nampt activator P7C3 ameliorates diabetes and improves skeletal muscle function modulating cell metabolism and lipid mediators. J Cachexia Sarcopenia Muscle. doi: 10.1002/jcsm.12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talbot J and Maves L (2016) Skeletal muscle fiber type: using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley interdisciplinary reviews. Developmental biology 5:518–534. doi: 10.1002/wdev.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourdeau Julien I, Sephton CF and Dutchak PA (2018) Metabolic Networks Influencing Skeletal Muscle Fiber Composition. Frontiers in Cell and Developmental Biology 6. doi: 10.3389/fcell.2018.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manickam R and Wahli W (2017) Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 136:42–48. doi: 10.1016/j.biochi.2016.11.010 [DOI] [PubMed] [Google Scholar]
- 34.Purves-Smith FM, Sgarioto N and Hepple RT (2014) Fiber Typing in Aging Muscle. Exercise and Sport Sciences Reviews 42:45–52. doi: 10.1249/JES.0000000000000012 [DOI] [PubMed] [Google Scholar]
- 35.Wang Y and Pessin JE (2013) Mechanisms for fiber-type specificity of skeletal muscle atrophy. Current opinion in clinical nutrition and metabolic care 16:243–250. doi: 10.1097/MCO.0b013e328360272d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B and Guillemin GJ (2012) Age-Associated Changes In Oxidative Stress and NAD+ Metabolism In Human Tissue. PLoS ONE 7. doi: 10.1371/journal.pone.0042357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Opitz CA and Heiland I (2015) Dynamics of NAD-metabolism: everything but constant. Biochem Soc Trans 43:1127–32. doi: 10.1042/bst20150133 [DOI] [PubMed] [Google Scholar]
- 38.Agerholm M, Dall M, Jensen BAH, Prats C, Madsen S, Basse AL, Graae A-S, Risis S, Goldenbaum J, Quistorff B, Larsen S, Vienberg SG and Treebak JT (2017) Perturbations of NAD+ salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. American Journal of Physiology-Endocrinology and Metabolism 314:E377–E395. doi: 10.1152/ajpendo.00213.2017 [DOI] [PubMed] [Google Scholar]
- 39.White AT and Schenk S (2012) NAD+/NADH and skeletal muscle mitochondrial adaptations to exercise. American Journal of Physiology-Endocrinology and Metabolism 303:E308–E321. doi: 10.1152/ajpendo.00054.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ and Kraus WL (2018) Metabolic Regulation of Transcription Through Compartmentalized NAD+ Biosynthesis. Science (New York, N.Y.) 360. doi: 10.1126/science.aan5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mumford PW, Osburn SC, Fox CD, Godwin JS and Roberts MD (2020) A Theacrine-Based Supplement Increases Cellular NAD(+) Levels and Affects Biomarkers Related to Sirtuin Activity in C2C12 Muscle Cells In Vitro. Nutrients 12. doi: 10.3390/nu12123727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basse AL, Agerholm M, Farup J, Dalbram E, Nielsen J, Ørtenblad N, Altıntaş A, Ehrlich AM, Krag T, Bruzzone S, Dall M, de Guia RM, Jensen JB, Møller AB, Karlsen A, Kjær M, Barrès R, Vissing J, Larsen S, Jessen N and Treebak JT (2021) Nampt controls skeletal muscle development by maintaining Ca(2+) homeostasis and mitochondrial integrity. Mol Metab 53:101271. doi: 10.1016/j.molmet.2021.101271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M and Sartorelli V (2015) The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell stem cell 16:171–183. doi: 10.1016/j.stem.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL and Sartorelli V (2003) Sir2 Regulates Skeletal Muscle Differentiation as a Potential Sensor of the Redox State. Molecular Cell 12:51–62. doi: 10.1016/S1097-2765(03)00226-0 [DOI] [PubMed] [Google Scholar]
- 45.Zhang N and Sauve AA (2018) Regulatory Effects of NAD(+) Metabolic Pathways on Sirtuin Activity. Prog Mol Biol Transl Sci 154:71–104. doi: 10.1016/bs.pmbts.2017.11.012 [DOI] [PubMed] [Google Scholar]
- 46.Pan H and Finkel T (2017) Key proteins and pathways that regulate lifespan. The Journal of Biological Chemistry 292:6452–6460. doi: 10.1074/jbc.R116.771915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zullo A, Simone E, Grimaldi M, Musto V and Mancini FP (2018) Sirtuins as Mediator of the Anti-Ageing Effects of Calorie Restriction in Skeletal and Cardiac Muscle. International Journal of Molecular Sciences 19. doi: 10.3390/ijms19040928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC and Glass DJ (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. NATURE CELL BIOLOGY, pp. 1014. [DOI] [PubMed] [Google Scholar]
- 49.Samant SA, Kanwal A, Pillai VB, Bao R and Gupta MP (2017) The histone deacetylase SIRT6 blocks myostatin expression and development of muscle atrophy. Scientific Reports 7. doi: 10.1038/s41598-017-10838-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lombard DB (2009) Sirtuins at the Breaking Point: SIRT6 in DNA Repair. Aging (Albany NY) 1:12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, Bohr VA and Chua KF (2009) SIRT6 stabilizes DNA-dependent Protein Kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 1:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui X, Yao L, Yang X, Gao Y, Fang F, Zhang J, Wang Q and Chang Y (2017) SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. American Journal of Physiology-Endocrinology and Metabolism 313:E493–E505. doi: 10.1152/ajpendo.00122.2017 [DOI] [PubMed] [Google Scholar]
- 53.Lin L, Chen K, Khalek WA, Ward JL, Yang H, Chabi B, Wrutniak-Cabello C and Tong Q (2014) Regulation of Skeletal Muscle Oxidative Capacity and Muscle Mass by SIRT3. PLoS ONE 9. doi: 10.1371/journal.pone.0085636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Siahpirani AF, Kemmerer ZA, Prolla TA, Roy S, Coon JJ and Denu JM (2015) SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell metabolism 21:637–646. doi: 10.1016/j.cmet.2015.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Iii JLW, Goodyear LJ and Tong Q (2009) Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1α in skeletal muscle. Aging (Albany NY) 1:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, Ren X, Wu Z, Streeper RS and Bordone L (2010) SIRT4 Regulates Fatty Acid Oxidation and Mitochondrial Gene Expression in Liver and Muscle Cells. The Journal of Biological Chemistry 285:31995–32002. doi: 10.1074/jbc.M110.124164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vargas-Ortiz K, Pérez-Vázquez V and Macías-Cervantes MH (2019) Exercise and Sirtuins: A Way to Mitochondrial Health in Skeletal Muscle. International Journal of Molecular Sciences 20. doi: 10.3390/ijms20112717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeo D, Kang C and Ji LL (2020) Aging alters acetylation status in skeletal and cardiac muscles. GeroScience 42:963–976. doi: 10.1007/s11357-020-00171-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohamed JS, Wilson JC, Myers MJ, Sisson KJ and Alway SE (2014) Dysregulation of SIRT-1 in aging mice increases skeletal muscle fatigue by a PARP-1-dependent mechanism. Aging (Albany NY) 6:820–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang B-H, Tseng W-L, Li H-Y, Wang M-L, Chang Y-L, Sung Y-J and Chiou S-H (2015) Poly(ADP-Ribose) Polymerase 1: Cellular Pluripotency, Reprogramming, and Tumorogenesis. International Journal of Molecular Sciences 16:15531–15545. doi: 10.3390/ijms160715531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amé J-C, Spenlehauer C and Murcia Gd (2004) The PARP superfamily. BioEssays 26:882–893. doi: 10.1002/bies.20085 [DOI] [PubMed] [Google Scholar]
- 62.Chini EN, Chini CCS, Netto JME, de Oliveira GC and van Schooten W (2018) The Pharmacology of CD38/NADase: An emerging target for cancer and aging diseases. Trends in pharmacological sciences 39:424–436. doi: 10.1016/j.tips.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T, Malavasi F and Ausiello CM (2006) CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 107:2392–9. doi: 10.1182/blood-2005-07-2913 [DOI] [PubMed] [Google Scholar]
- 64.Komanetsky SM, Hedrick V, Sobreira T, Aryal UK, Kim SQ and Kim K-H (2020) Proteomic identification of aerobic glycolysis as a potential metabolic target for methylglyoxal in adipocytes. Nutrition Research 80:66–77. doi: 10.1016/j.nutres.2020.06.009 [DOI] [PubMed] [Google Scholar]
- 65.Han H-S, Kang G, Kim JS, Choi BH and Koo S-H (2016) Regulation of glucose metabolism from a liver-centric perspective. Experimental & Molecular Medicine 48:e218. doi: 10.1038/emm.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brière J-J, Favier J, Gimenez-Roqueplo A-P and Rustin P (2006) Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. American Journal of Physiology-Cell Physiology 291:C1114–C1120. doi: 10.1152/ajpcell.00216.2006 [DOI] [PubMed] [Google Scholar]
- 67.Lee W-C, Ji X, Nissim I and Long F (2020) Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Reports 32:108108. doi: 10.1016/j.celrep.2020.108108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith HQ, Li C, Stanley CA and Smith TJ (2019) Glutamate Dehydrogenase, a Complex Enzyme at a Crucial Metabolic Branch Point. Neurochemical research 44:117–132. doi: 10.1007/s11064-017-2428-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Minárik P, Tomášková N, Kollárová M and Antalík M Malate Dehydrogenases – Structure and Function. Structure and Function:9. [PubMed] [Google Scholar]
- 70.Larsson C, Påhlman I-L, Ansell R, Rigoulet M, Adler L and Gustafsson L (1998) The importance of the glycerol 3-phosphate shuttle during aerobic growth of Saccharomyces cerevisiae. Yeast 14:347–357. doi: [DOI] [PubMed] [Google Scholar]
- 71.Stein LR and Imai S-i (2012) The dynamic regulation of NAD metabolism in mitochondria. Trends in endocrinology and metabolism: TEM 23:420–428. doi: 10.1016/j.tem.2012.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jensen J, Rustad PI, Kolnes AJ and Lai Y-C (2011) The Role of Skeletal Muscle Glycogen Breakdown for Regulation of Insulin Sensitivity by Exercise. Frontiers in Physiology 2. doi: 10.3389/fphys.2011.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Migliavacca E, Tay SKH, Patel HP, Sonntag T, Civiletto G, McFarlane C, Forrester T, Barton SJ, Leow MK, Antoun E, Charpagne A, Seng Chong Y, Descombes P, Feng L, Francis-Emmanuel P, Garratt ES, Giner MP, Green CO, Karaz S, Kothandaraman N, Marquis J, Metairon S, Moco S, Nelson G, Ngo S, Pleasants T, Raymond F, Sayer AA, Ming Sim C, Slater-Jefferies J, Syddall HE, Fang Tan P, Titcombe P, Vaz C, Westbury LD, Wong G, Yonghui W, Cooper C, Sheppard A, Godfrey KM, Lillycrop KA, Karnani N and Feige JN (2019) Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nature Communications 10. doi: 10.1038/s41467-019-13694-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siegel CS and McCullough LD (2013) NAD+ and nicotinamide: sex differences in cerebral ischemia. Neuroscience 237:223–231. doi: 10.1016/j.neuroscience.2013.01.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwarzmann L, Pliquett RU, Simm A and Bartling B (2021) Sex-related differences in human plasma NAD+/NADH levels depend on age. Biosci Rep 41. doi: 10.1042/bsr20200340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Echaniz-Laguna A, Mohr M, Lannes B and Tranchant C (2010) Myopathies in the elderly: A hospital-based study. Neuromuscular Disorders 20:443–447. doi: 10.1016/j.nmd.2010.05.003 [DOI] [PubMed] [Google Scholar]
- 77.Pandey SN, Kesari A, Yokota T and Pandey GS (2015) Muscular Dystrophy: Disease Mechanisms and Therapies. BioMed Research International 2015. doi: 10.1155/2015/456348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP and Bohr VA (2017) NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends in molecular medicine 23:899–916. doi: 10.1016/j.molmed.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mázala DAG, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Cantó C, Conley KE, Walter L, Lovering RM, Chin ER, Jasmin BJ, Marcinek DJ, Menzies KJ and Auwerx J (2016) NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Science translational medicine 8:361ra139. doi: 10.1126/scitranslmed.aaf5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sebori R, Kuno A, Hosoda R, Hayashi T and Horio Y (2018) Resveratrol Decreases Oxidative Stress by Restoring Mitophagy and Improves the Pathophysiology of Dystrophin-Deficient mdx Mice. Oxidative Medicine and Cellular Longevity 2018. doi: 10.1155/2018/9179270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Imai S-i J (2013) The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and aging. Diabetes, obesity & metabolism 15. doi: 10.1111/dom.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mohamed JS, Hajira A, Pardo PS and Boriek AM (2014) MicroRNA-149 Inhibits PARP-2 and Promotes Mitochondrial Biogenesis via SIRT-1/PGC-1α Network in Skeletal Muscle. Diabetes 63:1546–1559. doi: 10.2337/db13-1364 [DOI] [PubMed] [Google Scholar]
- 83.Fan L, Cacicedo JM and Ido Y (2020) Impaired nicotinamide adenine dinucleotide (NAD+) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. Journal of Diabetes Investigation 11:1403–1419. doi: 10.1111/jdi.13303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frederick DW, Davis JG, Dávila A, Agarwal B, Michan S, Puchowicz MA, Nakamaru-Ogiso E and Baur JA (2015) Increasing NAD Synthesis in Muscle via Nicotinamide Phosphoribosyltransferase Is Not Sufficient to Promote Oxidative Metabolism. The Journal of Biological Chemistry 290:1546–1558. doi: 10.1074/jbc.M114.579565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Supale S, Li N, Brun T and Maechler P (2012) Mitochondrial dysfunction in pancreatic β cells. Trends in endocrinology and metabolism: TEM 23:477–487. doi: 10.1016/j.tem.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 86.Shoelson SE, Lee J and Goldfine AB (2006) Inflammation and insulin resistance. Journal of Clinical Investigation 116:1793–1801. doi: 10.1172/JCI29069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA and Sowers JR (2008) Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 294:R673–R680. doi: 10.1152/ajpregu.00561.2007 [DOI] [PubMed] [Google Scholar]
- 88.Wu H and Ballantyne CM Skeletal muscle inflammation and insulin resistance in obesity. The Journal of Clinical Investigation 127:43–54. doi: 10.1172/JCI88880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jura M and Kozak LP (2016) Obesity and related consequences to ageing. Age 38. doi: 10.1007/s11357-016-9884-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burhans MS, Hagman DK, Kuzma JN, Schmidt KA and Kratz M (2018) Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Comprehensive Physiology 9:1–58. doi: 10.1002/cphy.c170040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frederick DW, Loro E, Liu L, Davila A, Chellappa K, Silverman IM, Quinn WJ, Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, Redpath P, Migaud ME, Nakamaru-Ogiso E, Rabinowitz JD, Khurana TS and Baur JA (2016) Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell metabolism 24:269–282. doi: 10.1016/j.cmet.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JRB, Newgard CB, Lopaschuk GD and Muoio DM (2008) Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metabolism 7:45–56. doi: 10.1016/j.cmet.2007.10.013 [DOI] [PubMed] [Google Scholar]
- 93.Abdul-Ghani MA and DeFronzo RA (2010) Pathogenesis of Insulin Resistance in Skeletal Muscle. Journal of Biomedicine and Biotechnology 2010. doi: 10.1155/2010/476279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meo SD, Iossa S and Venditti P (2017) Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. Journal of Endocrinology 233:R15–R42. doi: 10.1530/JOE-16-0598 [DOI] [PubMed] [Google Scholar]
- 95.Lantier L, Williams AS, Hughey CC, Bracy DP, James FD, Ansari MA, Gius D and Wasserman DH (2018) SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PLoS ONE 13. doi: 10.1371/journal.pone.0208634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P and Auwerx J (2006) Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 127:1109–1122. doi: 10.1016/j.cell.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 97.Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A and Chini EN (2016) CD38 dictates age-related NAD decline and mitochondrial dysfunction through a SIRT3-dependent mechanism. Cell metabolism 23:1127–1139. doi: 10.1016/j.cmet.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P and Chini EN (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. The FASEB Journal 21:3629–3639. doi: 10.1096/fj.07-8290com [DOI] [PubMed] [Google Scholar]
- 99.Wang LF, Miao LJ, Wang XN, Huang CC, Qian YS, Huang X, Wang XL, Jin WZ, Ji GJ, Fu M, Deng KY and Xin HB (2018) CD38 deficiency suppresses adipogenesis and lipogenesis in adipose tissues through activating Sirt1/PPARγ signaling pathway. Journal of Cellular and Molecular Medicine 22:101–110. doi: 10.1111/jcmm.13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Carrico C, Meyer JG, He W, Gibson BW and Verdin E (2018) The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, Metabolic and Disease Implications. Cell metabolism 27:497–512. doi: 10.1016/j.cmet.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hebert SL, Marquet-de Rougé P, Lanza IR, McCrady-Spitzer SK, Levine JA, Middha S, Carter RE, Klaus KA, Therneau TM, Highsmith EW and Nair KS (2015) Mitochondrial Aging and Physical Decline: Insights From Three Generations of Women. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 70:1409–1417. doi: 10.1093/gerona/glv086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boengler K, Kosiol M, Mayr M, Schulz R and Rohrbach S (2017) Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. Journal of Cachexia, Sarcopenia and Muscle 8:349–369. doi: 10.1002/jcsm.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Picca A, Calvani R, Bossola M, Allocca E, Menghi A, Pesce V, Lezza AMS, Bernabei R, Landi F and Marzetti E (2018) Update on mitochondria and muscle aging: all wrong roads lead to sarcopenia. Biological Chemistry 399:421–436. doi: 10.1515/hsz-2017-0331 [DOI] [PubMed] [Google Scholar]
- 104.van de Ven RAH, Santos D and Haigis MC (2017) Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends in molecular medicine 23:320–331. doi: 10.1016/j.molmed.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsuda M, Fukushima A, Matsumoto J, Takada S, Kakutani N, Nambu H, Yamanashi K, Furihata T, Yokota T, Okita K, Kinugawa S and Anzai T (2018) Protein acetylation in skeletal muscle mitochondria is involved in impaired fatty acid oxidation and exercise intolerance in heart failure. Journal of Cachexia, Sarcopenia and Muscle 9:844–859. doi: 10.1002/jcsm.12322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang T, Cao Y, Zheng Q, Tu J, Zhou W, He J, Zhong J, Chen Y, Wang J, Cai R, Zuo Y, Wei B, Fan Q, Yang J, Wu Y, Yi J, Li D, Liu M, Wang C, Zhou A, Li Y, Wu X, Yang W, Chin YE, Chen G and Cheng J (2019) SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Molecular Cell 75:823–834.e5. doi: 10.1016/j.molcel.2019.06.008 [DOI] [PubMed] [Google Scholar]
- 107.Myers MJ, Shepherd DL, Durr AJ, Stanton DS, Mohamed JS, Hollander JM and Alway SE (2019) The role of SIRT1 in skeletal muscle function and repair of older mice. Journal of Cachexia, Sarcopenia and Muscle 10:929–949. doi: 10.1002/jcsm.12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fujiwara D, Iwahara N, Sebori R, Hosoda R, Shimohama S, Kuno A and Horio Y (2019) SIRT1 deficiency interferes with membrane resealing after cell membrane injury. PLoS ONE 14. doi: 10.1371/journal.pone.0218329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hitomi K, Okada R, Loo TM, Miyata K, Nakamura AJ and Takahashi A (2020) DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. International Journal of Molecular Sciences 21. doi: 10.3390/ijms21103720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schultz MB and Sinclair DA (2016) Why NAD+ Declines during Aging: It’s Destroyed. Cell metabolism 23:965–966. doi: 10.1016/j.cmet.2016.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT and Tschöop MH (2012) SIRTUIN 1 AND SIRTUIN 3: Physiological modulators of metabolism. Physiological reviews 92:1479–1514. doi: 10.1152/physrev.00022.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hwang J-w, Yao H, Caito S, Sundar IK and Rahman I (2013) Redox regulation of SIRT1 in inflammation and cellular senescence. Free radical biology & medicine 0:95–110. doi: 10.1016/j.freeradbiomed.2013.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fan J, Krautkramer KA, Feldman JL and Denu JM (2015) Metabolic regulation of histone post-translational modifications. ACS chemical biology 10:95–108. doi: 10.1021/cb500846u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakahata Y, Sahar S, Astarita G, Kaluzova M and Sassone-Corsi P (2009) Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science (New York, N.Y.) 324:654–657. doi: 10.1126/science.1170803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Seto E and Yoshida M (2014) Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harbor Perspectives in Biology 6. doi: 10.1101/cshperspect.a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sabari BR, Zhang D, Allis CD and Zhao Y (2017) Metabolic regulation of gene expression through histone acylations. Nature reviews. Molecular cell biology 18:90–101. doi: 10.1038/nrm.2016.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA and Mayo MW (2004) Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. The EMBO Journal 23:2369–2380. doi: 10.1038/sj.emboj.7600244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Horst A, Tertoolen LGJ, de Vries-Smits LMM, Frye RA, Medema RH and Burgering BMT (2004) FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). The Journal of Biological Chemistry 279:28873–28879. doi: 10.1074/jbc.M401138200 [DOI] [PubMed] [Google Scholar]
- 119.Ikenoue T, Inoki K, Zhao B and Guan K-L (2008) PTEN acetylation modulates its interaction with PDZ domain. Cancer Research 68:6908–6912. doi: 10.1158/0008-5472.CAN-08-1107 [DOI] [PubMed] [Google Scholar]
- 120.Yao X-H and Nyomba BLG (2008) Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 294:R1797–1806. doi: 10.1152/ajpregu.00804.2007 [DOI] [PubMed] [Google Scholar]
- 121.Gujar H, Weisenberger DJ and Liang G (2019) The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 10. doi: 10.3390/genes10020172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Turner DC, Gorski PP, Maasar MF, Seaborne RA, Baumert P, Brown AD, Kitchen MO, Erskine RM, Dos-Remedios I, Voisin S, Eynon N, Sultanov RI, Borisov OV, Larin AK, Semenova EA, Popov DV, Generozov EV, Stewart CE, Drust B, Owens DJ, Ahmetov II and Sharples AP (2020) DNA methylation across the genome in aged human skeletal muscle tissue and muscle-derived cells: the role of HOX genes and physical activity. Scientific Reports 10. doi: 10.1038/s41598-020-72730-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scourzic L, Mouly E and Bernard OA (2015) TET proteins and the control of cytosine demethylation in cancer. Genome Medicine 7. doi: 10.1186/s13073-015-0134-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zykovich A, Hubbard A, Flynn JM, Tarnopolsky M, Fraga MF, Kerksick C, Ogborn D, MacNeil L, Mooney SD and Melov S (2014) Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 13:360–366. doi: 10.1111/acel.12180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Livshits G, Gao F, Malkin I, Needhamsen M, Xia Y, Yuan W, Bell CG, Ward K, Liu Y, Wang J, Bell JT and Spector TD (2016) Contribution of Heritability and Epigenetic Factors to Skeletal Muscle Mass Variation in United Kingdom Twins. The Journal of Clinical Endocrinology and Metabolism 101:2450–2459. doi: 10.1210/jc.2016-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bigot A, Duddy WJ, Ouandaogo ZG, Negroni E, Mariot V, Ghimbovschi S, Harmon B, Wielgosik A, Loiseau C, Devaney J, Dumonceaux J, Butler-Browne G, Mouly V and Duguez S (2015) Age-Associated Methylation Suppresses SPRY1, Leading to a Failure of Re-quiescence and Loss of the Reserve Stem Cell Pool in Elderly Muscle. Cell Reports 13:1172–1182. doi: 10.1016/j.celrep.2015.09.067 [DOI] [PubMed] [Google Scholar]
- 127.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W and He C (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nature chemical biology 10:93–95. doi: 10.1038/nchembio.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pan Y, Ma P, Liu Y, Li W and Shu Y (2018) Multiple functions of m6A RNA methylation in cancer. Journal of Hematology & Oncology 11. doi: 10.1186/s13045-018-0590-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, Zhao X, Li A, Yang Y, Dahal U, Lou X-M, Liu X, Huang J, Yuan W-P, Zhu X-F, Cheng T, Zhao Y-L, Wang X, Danielsen JMR, Liu F and Yang Y-G (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Research 24:177–189. doi: 10.1038/cr.2014.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X and Semenza GL (2016) Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proceedings of the National Academy of Sciences of the United States of America 113:E2047–E2056. doi: 10.1073/pnas.1602883113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur J-J, Rentmeister A, Gross SS, Pellizzoni L, Debart F, Goodarzi H and Jaffrey SR (2019) FTO controls reversible m6Am RNA methylation during snRNA biogenesis. Nature chemical biology 15:340–347. doi: 10.1038/s41589-019-0231-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu W, Feng J, Jiang D, Zhou X, Jiang Q, Cai M, Wang X, Shan T and Wang Y (2017) AMPK regulates lipid accumulation in skeletal muscle cells through FTO-dependent demethylation of N6-methyladenosine. Scientific Reports 7. doi: 10.1038/srep41606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K and Gorospe M (2019) Transcriptome signature of cellular senescence. Nucleic Acids Research 47:7294–7305. doi: 10.1093/nar/gkz555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gheller BJ, Blum JE, Fong EHH, Malysheva OV, Cosgrove BD and Thalacker-Mercer AE (2020) A defined N6-methyladenosine (m6A) profile conferred by METTL3 regulates muscle stem cell/myoblast state transitions. Cell Death Discovery 6. doi: 10.1038/s41420-020-00328-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yoshino M, Yoshino J, Kayser BD, Patti GJ, Franczyk MP, Mills KF, Sindelar M, Pietka T, Patterson BW, Imai SI and Klein S (2021) Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 372:1224–1229. doi: 10.1126/science.abe9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA and Auwerx J (2012) The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet induced obesity. Cell metabolism 15:838–847. doi: 10.1016/j.cmet.2012.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Elhassan YS, Kluckova K, Fletcher RS, Schmidt MS, Garten A, Doig CL, Cartwright DM, Oakey L, Burley CV, Jenkinson N, Wilson M, Lucas SJE, Akerman I, Seabright A, Lai Y-C, Tennant DA, Nightingale P, Wallis GA, Manolopoulos KN, Brenner C, Philp A and Lavery GG (2019) Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD+ Metabolome and Induces Transcriptomic and Anti-inflammatory Signatures. Cell Reports 28:1717–1728.e6. doi: 10.1016/j.celrep.2019.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dollerup OL, Chubanava S, Agerholm M, Søndergård SD, Altıntaş A, Møller AB, Høyer KF, Ringgaard S, Stødkilde-Jørgensen H, Lavery GG, Barrès R, Larsen S, Prats C, Jessen N and Treebak JT (2020) Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. The Journal of Physiology 598:731–754. doi: 10.1113/JP278752 [DOI] [PubMed] [Google Scholar]
- 139.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J and Imai S-i (2016) Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell metabolism 24:795–806. doi: 10.1016/j.cmet.2016.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM and McKnight SL (2014) P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 158:1324–1334. doi: 10.1016/j.cell.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Loris ZB, Pieper AA and Dietrich WD (2017) The neuroprotective compound P7C3-A20 promotes neurogenesis and improves cognitive function after ischemic stroke. Exp Neurol 290:63–73. doi: 10.1016/j.expneurol.2017.01.006 [DOI] [PubMed] [Google Scholar]
- 142.Hua X, Sun DY, Zhang WJ, Fu JT, Tong J, Sun SJ, Zeng FY, Ouyang SX, Zhang GY, Wang SN, Li DJ, Miao CY and Wang P (2021) P7C3-A20 alleviates fatty liver by shaping gut microbiota and inducing FGF21/FGF1, via the AMP-activated protein kinase/CREB regulated transcription coactivator 2 pathway. Br J Pharmacol 178:2111–2130. doi: 10.1111/bph.15008 [DOI] [PubMed] [Google Scholar]
- 143.TIPPARAJU S M, BADOLE S L, CHAPALAMADUGU K C and TUR J (2017) METHODS AND USES OF NAMPT ACTIVATORS FOR TREATMENT OF DIABETES, CARDIOVASCULAR DISEASES, AND SYMPTOMS THEREOF. [Google Scholar]
- 144.Nikiforov A, Dölle C, Niere M and Ziegler M (2011) Pathways and Subcellular Compartmentation of NAD Biosynthesis in Human Cells. The Journal of Biological Chemistry 286:21767–21778. doi: 10.1074/jbc.M110.213298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gulshan M, Yaku K, Okabe K, Mahmood A, Sasaki T, Yamamoto M, Hikosaka K, Usui I, Kitamura T, Tobe K and Nakagawa T (2018) Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell 17. doi: 10.1111/acel.12798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kjøbsted R, Hingst JR, Fentz J, Foretz M, Sanz MN, Pehmøller C, Shum M, Marette A, Mounier R, Treebak JT, Wojtaszewski JFP, Viollet B and Lantier L (2018) AMPK in skeletal muscle function and metabolism. Faseb j 32:1741–1777. doi: 10.1096/fj.201700442R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gowans GJ and Hardie DG (2014) AMPK – a cellular energy sensor primarily regulated by AMP. Biochemical Society transactions 42:71–75. doi: 10.1042/BST20130244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, Wang Y, Subramaniam S, Chien S and Shyy JY (2017) AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal 10. doi: 10.1126/scisignal.aaf7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thomson DM (2018) The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. International Journal of Molecular Sciences 19. doi: 10.3390/ijms19103125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, Nyakas C and Radak Z (2010) Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mechanisms of ageing and development 131:21–28. doi: 10.1016/j.mad.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Koltai E, Bori Z, Osvath P, Ihasz F, Peter S, Toth G, Degens H, Rittweger J, Boldogh I and Radak Z (2018) Master athletes have higher miR-7, SIRT3 and SOD2 expression in skeletal muscle than age-matched sedentary controls. Redox Biology 19:46–51. doi: 10.1016/j.redox.2018.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.de Guia RM, Agerholm M, Nielsen TS, Consitt LA, Søgaard D, Helge JW, Larsen S, Brandauer J, Houmard JA and Treebak JT (2019) Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiological Reports 7. doi: 10.14814/phy2.14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L and Reinberg D (2007) SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 450:440–444. doi: 10.1038/nature06268 [DOI] [PubMed] [Google Scholar]
- 154.Bosch-Presegué L and Vaquero A (2015) Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. The FEBS journal 282:1745–1767. doi: 10.1111/febs.13053 [DOI] [PubMed] [Google Scholar]
- 155.Alves-Fernandes DK and Jasiulionis MG (2019) The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. International Journal of Molecular Sciences 20. doi: 10.3390/ijms20133153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rifaï K, Judes G, Idrissou M, Daures M, Bignon Y-J, Penault-Llorca F and Bernard-Gallon D (2018) SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 9:30661–30678. doi: 10.18632/oncotarget.25771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Serrano L, Martínez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, Fondevila D, Muñoz P, Krüger M, Tischfield JA and Vaquero A (2013) The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes & Development 27:639–653. doi: 10.1101/gad.211342.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Das C, Lucia MS, Hansen KC and Tyler JK (2009) CBP / p300-mediated acetylation of histone H3 on lysine 56. Nature 459:113–117. doi: 10.1038/nature07861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huang H, Zhang D, Wang Y, Perez-Neut M, Han Z, Zheng YG, Hao Q and Zhao Y (2018) Lysine benzoylation is a histone mark regulated by SIRT2. Nature Communications 9. doi: 10.1038/s41467-018-05567-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Luo J, Nikolaev AY, Imai S-i, Chen D, Su F, Shiloh A, Guarente L and Gu W (2001) Negative Control of p53 by Sir2α Promotes Cell Survival under Stress. Cell 107:137–148. doi: 10.1016/S0092-8674(01)00524-4 [DOI] [PubMed] [Google Scholar]
- 161.Gu W and Roeder RG (1997) Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 90:595–606. doi: 10.1016/S0092-8674(00)80521-8 [DOI] [PubMed] [Google Scholar]
- 162.Glozak MA, Sengupta N, Zhang X and Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23. doi: 10.1016/j.gene.2005.09.010 [DOI] [PubMed] [Google Scholar]
- 163.Pan H, Guan D, Liu X, Li J, Wang L, Wu J, Zhou J, Zhang W, Ren R, Zhang W, Li Y, Yang J, Hao Y, Yuan T, Yuan G, Wang H, Ju Z, Mao Z, Li J, Qu J, Tang F and Liu G-H (2016) SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Research 26:190–205. doi: 10.1038/cr.2016.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tasselli L, Xi Y, Zheng W, Tennen RI, Odrowaz Z, Simeoni F, Li W and Chua KF (2016) SIRT6 deacetylates H3K18Ac at pericentric chromatin to prevent mitotic errors and cell senescence. Nature structural & molecular biology 23:434–440. doi: 10.1038/nsmb.3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K, Struhl K, Garcia BA, Gozani O, Li W and Chua KF (2012) SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487:114–118. doi: 10.1038/nature11043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Imai S-i and Guarente L (2016) It takes two to tango: NAD + and sirtuins in aging/longevity control. npj Aging and Mechanisms of Disease 2:1–6. doi: 10.1038/npjamd.2016.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data were included in the manuscript, since the article is a review paper this is not applicable.