

Abstract
We investigated the role of the A3 adenosine receptor in cells of the astroglial lineage (both rat primary astrocytes and human astrocytoma ADF cells) by means of the selective A3 agonists N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide (IB-MECA) and CI-IB-MECA, and by utilizing the selective A3 receptor antagonist MRS1191. Exposure of ADF cells to μM concentrations of either agonist resulted in reduction of cell number, likely due to cell death. In both rat astrocytes and human astrocytoma cells, at concentrations 2–3 orders of magnitude lower (which were not associated with cytotoxicity), these same agonists induced a marked reorganization of the cytoskeleton, with appearance of stress fibers and numerous cell protrusions. Functionally, these morphological changes were associated with cell protection, as demonstrated by a significant reduction of spontaneous apoptosis in A3 agonist-treated cells. To confirm a role for the A3 receptor in this effect, MRS1191 completely counteracted CI-IB-MECA-induced reduction of spontaneous apoptosis. In ADF cells, A3 agonists also induced changes in the intracellular distribution of the anti-apoptotic protein Bcl-XL, which became localized in cell protrusions. Also, this effect was specifically antagonized by MRS1191. These dual actions of A3 agonists in vitro may have important in vivo implications. For example, a robust and acute activation of the A3 receptor following massive adenosine release during ischemia may contribute to brain cell death; conversely, a subthreshold activation of this receptor prior to ischemia may trigger protective mechanisms (i.e., induction of stress fibers and of a Bcl-XL-dependent reorganization of cytoskeleton) making the brain more resistant to subsequent insults (“ischemic tolerance”).
Keywords: adenosine A3 receptors, astrocytes, rat, human, cell survival
INTRODUCTION
Adenosine regulates various physiological functions through four distinct receptors, the A1, A2A, A2B, and A3 subtypes [Fredholm et al., 1994]. In the central nervous system, adenosine participates in physiological neurotransmission and also acts as an emergency signal under pathological conditions [Rudolphi and Schubert, 1995]. Following ischemia, brain concentrations of adenosine are rapidly increased from 50–300 nM to 10–50 μM, and activation of the A1 receptor under these conditions results in potent neuroprotection [Rudolphi and Schubert, 1995]. The A2 receptor (particularly the A2A subtype) has been implicated in regulation of motor functions [Fredholm et al., 1994].
On the other hand, very little is known about the A3 receptor, initially identified by molecular cloning and whose pharmacological and molecular characterization has revealed unexpected findings for an adenosine receptor [Jacobson et al., 1995]. The A3 receptor has a unique structure–activity relationship profile, tissue distribution, and effector coupling. Although there may be species differences [Linden, 1994; Ji et al., 1994], actions at this receptor are relatively insensitive to xanthine derivatives [Zhou et al., 1992; van Galen et al., 1994], which behave as antagonists at the A1 and A2 subtypes [Jacobson et al., 1992]. In the last few years, insights into the physiopathological role of the A3 receptor have come from studies employing the first really selective A3 agonists [Gallo-Rodriguez et al., 1994; Kim et al., 1994] and antagonists [Karton et al., 1996; Jiang et al., 1996; Kim et al., 1996; Jacobson et al., 1996]. In humans, A3 receptors have been found in the lung, liver, and kidney, with a lower density in brain and testes [Linden, 1994]. Surprisingly, when examining the effects mediated by the A3 receptor on cell viability, both protective and lethal effects have been discovered [for review, see Jacobson, 1998].
The first study suggesting multiple effects of the A3 receptor on cell viability dates back to 1994. Von Lubitz and coworkers [1994] reported that the in vivo chronic administration of the A3 receptor agonist N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide [IB-MECA; Gallo-Rodriguez et al., 1994] significantly reduced ischemia-associated neurodegeneration in gerbils. Opposite effects, i.e., enhanced mortality and extensive brain damage, were observed after a single IB-MECA administration immediately prior to induction of ischemia [von Lubitz et al., 1994]. After this study, a number of other reports suggested dual and opposite effects mediated by the A3 receptor, i.e., either cytoprotection or cytotoxicity depending on the level of receptor activation and the paradigm studied.
Micromolar concentrations of either IB-MECA or its 2-chloro-derivative CI-IB-MECA [Kim et al., 1994] are toxic for human eosinophils [Kohno et al., 1996a], cells of the lymphoid lineage [Kohno et al., 1996b; Yao et al., 1997; Barbieri et al., 1998], rat cardiac myocytes [Shneyvais et al., 1998] and rat cerebellar granule cells [Sei et al., 1997; see also Jacobson, 1998]. In most cases, cell death occurs through induction of apoptosis (although enhancement of glutamate-induced necrosis seems to be a major cause of death in the case of cerebellar granules). In CHO cells transfected with the human A3 receptor, exposure to A3 agonists resulted in inhibition of cell growth, likely due to block of cells at the S-phase of the cell cycle [Brambilla et al., 1998]. The different outcome of A3 agonist-induced effect in this experimental model (i.e., inhibition of cell growth against cell death) may simply depend on the expression of different target proteins downstream of the A3 receptor, or to insufficient expression in the host system of key proteins involved in the triggering of cell death (i.e., inhibition of cell growth may simply precede induction of apoptosis). Nanomolar concentrations of the same A3 agonists tend instead to protect cells. This has been shown to be the case for human leukemia cells [Yao et al., 1997] and chick ventricular myocytes [Stambaugh et al., 1997]. To confirm a role for the adenosine A3 receptor in such protective effects, transfection of the human A3 receptor into atrial myocytes has recently been shown to confer protection against ischemia-associated cell death [Liang and Jacobson, 1998]. In certain cultured cell lines, both protective and cell-death-inducing effects have been demonstrated. For example, in HL-60 cells and U937 human lymphoma cells, low A3 agonist concentrations have been shown to protect against toxicity induced by A3 antagonists alone [Yao et al., 1997]. This suggested that there may exist a tonic state of activation of the A3 receptor conferring protection; the apoptotic effect of antagonists alone may simply be explained on the basis of a block of a protective action induced by endogenous adenosine [Yao et al., 1997].
As previously mentioned, regulation of cell viability by the A3 receptor may play a crucial role in the development of ischemic damage in the brain, as well as in chronic neurological conditions characterized by neurodegenerative events. In brain, A3 receptors are found in both neurones [Dunwiddie et al., 1997] and astrocytes [Abbracchio et al., 1997]. Hence, based on the importance of astroglial cells in brain repair and regeneration in both acute and chronic neurological diseases [Ridet et al., 1997] and also based on preliminary results suggesting a dual regulation of astrocytic cell viability by the A3 receptor [Ceruti et al., 1996], a specific aim of this study has been the characterization of the effects induced by the A3 selective agonists IB-MECA and CI-IB-MECA on cells belonging to the astroglial lineage.
MATERIALS AND METHODS
Cell Culture and Treatments
Astroglial primary cultures were prepared from 7-day-old Sprague Dawley rats (Charles River, Como, Italy) and maintained in culture as previously described [Abbracchio et al., 1994]. Human astrocytoma ADF cells were grown at 37°C in a humidified atmosphere as previously described [Malorni et al., 1994]. After 24 h in culture to allow attachment to the substratum, cultures were then exposed to the various pharmacological agents and after 24–72 h fixed with 4% paraformaldehyde in PBS (pH 7.4) for 20 min at room temperature. Nuclear chromatin was then stained by using the fluorescent dye Hoechst 33258 (10 μg/ml, 20 min at room temperature) and cell number was evaluated by using a fluorescence microscope (Zeiss, Germany) equipped with a UV filter and by counting cells contained in an identical area for each coverslip. Results represent the mean of 10–12 different replications from at least three independent experiments. The A3 selective agonists IB-MECA and CI-IB-MECA and antagonist MRS1191 (3-ethyl 5-benzyl-2-methyl-6-phenyl-4-phenylethynyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate) were synthesized as previously described [Gallo-Rodriguez et al., 1994; Kim et al., 1994; Jiang et al., 1996]. Due to its different affinity for the rat and human receptor, MRS1191 was tested at a 10 μM concentration on rat primary astrocytes and at a 10 nM concentration on human ADF cells.
Evaluation of Spontaneous Apoptosis
Primary cultures were grown for 48 h in the absence or presence of 100 nM CI-IB-MECA alone or in combination with 10 μM MRS1191. At the end of the incubation period, culture media were collected, centrifuged, and pellets fixed with 4% paraformaldehyde in PBS; cells were then allowed to adhere to polylysine-coated glass coverslips and nuclear chromatin stained as described above. Apoptotic cells (i.e., showing chromatin aggregation, fragmentation, or clumping) were detected and counted under a fluorescence microscope equipped with a UV filter.
Analytical Cytology
For evaluation of cytoskeleton organization, primary cultures were grown on glass coverslips in 35 mm petri dishes, fixed, permeabilized, and incubated with fluorescein-phalloidin as previously described [Abbracchio et al., 1997]. For detection of Bcl-XL protein, ADF cells were grown on glass coverslips, fixed, permeabilized, and immunostained with a Bcl-XL polyclonal antibody as previously described [Abbracchio et al., 1997].
RESULTS
Exposure of either rat primary astrocytes (data not shown) or human astrocytoma ADF cells (Fig. 1) to 10–60 μM CI-IB-MECA resulted in a concentration-dependent reduction of cell number, likely due to induction of cell death (Fig. 1). Similar effects were obtained with IB-MECA (data not shown). This effect was not antagonized by either the mixed A1/A2 antagonist XAC (xanthine-amine-congener) or the selective A2A antagonist SCH58261 (5-amino-7-(phenylethyl)-2-(2-furyl)-pyrazolo [4,3-e] 1,2,4-triazolo [1,5-c] pyrimidine) or the selective A2A and A2B antagonist ZM 241385 (4-(2-[7-amino-2-(2-furyl) [1,2,4] triazolo [2,32] [1,3,6] triazinyl-amino]ethyl)-phenol). Preliminary data would also suggest insensitivity to several selective A3 receptor antagonists [Jacobson and Suzuki, 1996] (data not shown), suggesting a nonadenosine receptor-mediated mechanism at these concentrations.
Fig. 1.
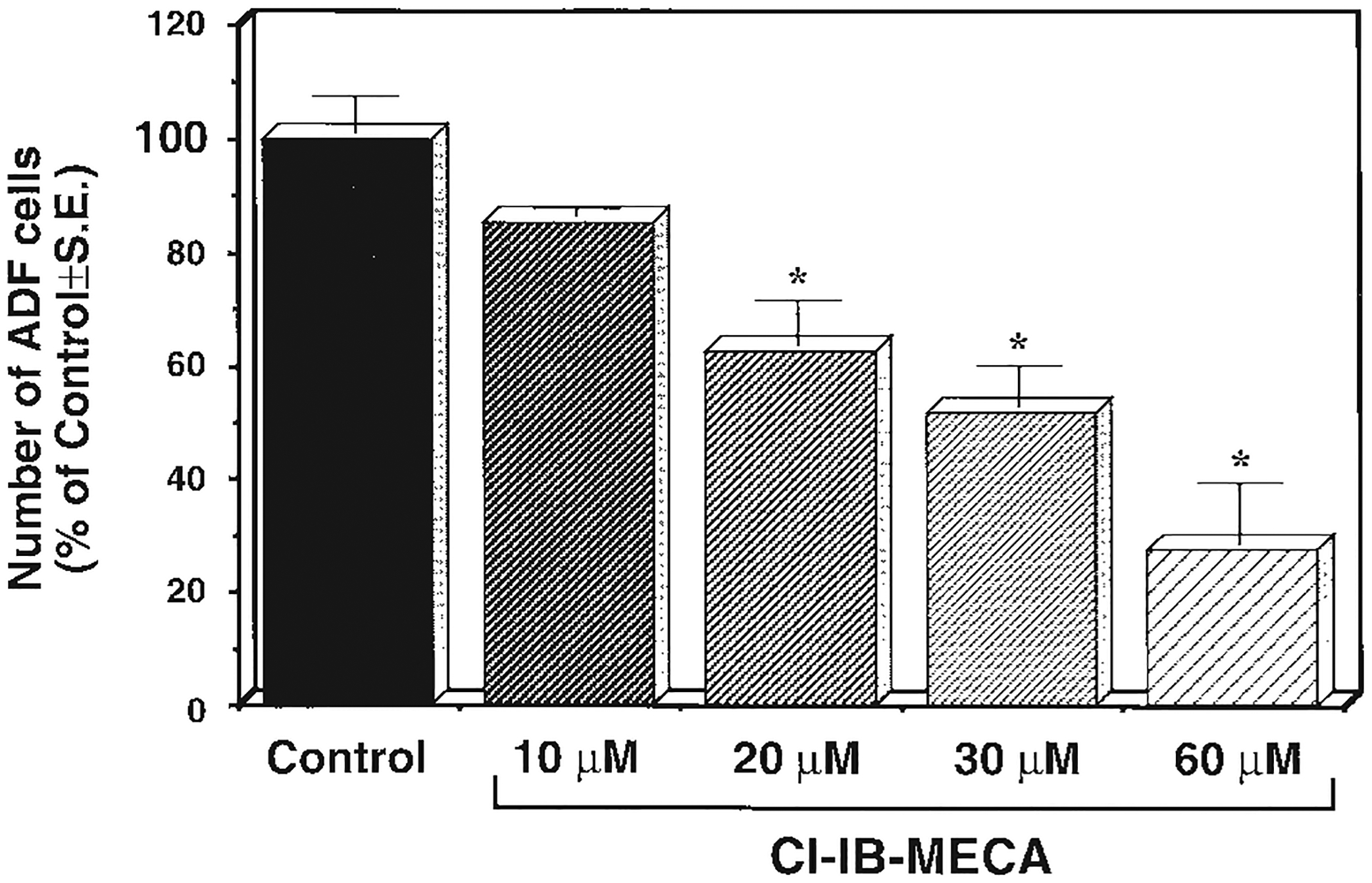
Reduction of cell number induced by μM concentrations of CI-IB-MECA in human ADF astrocytoma cells. Cells were grown for 48 h in the absence (Control) or presence of the indicated CI-IB-MECA concentrations. At the end of the incubation period, the number of adhering cells was evaluated as described in Materials and Methods. *P < 0.05 with respect to Control, one way ANOVA (Scheffe’s F-test).
At concentrations 2–3 orders of magnitude lower (10–100 nM), both in rat primary astrocytes and in human ADF cells A3 agonists promoted marked morphogenic changes. In ADF cells, a striking alteration of cell shape and morphology, with an increase in both the number and length of cellular processes, could be detected after exposure to 100 nM CI-IB-MECA or IB-MECA [Abbracchio et al., 1997]. Marked changes of the actin cytoskeleton were observed in rat primary astrocytic cultures. Figure 2 shows fluorescence staining of actin microfilaments with fluorescein phalloidin in control cultures (Fig. 2A,B) and in cultures exposed for 48 h to 100 nM of either A3 agonist. Both CI-IB-MECA (Fig. 2C,D) and IB-MECA (Fig. 2E,F) produced a remarkable increase of stress fibers and elongation of cell protrusions, suggesting a reorganization of the cytoskeleton and induction of cell differentiation. Functionally, these cytoskeletal changes resulted in a secondary protection of cells. Treatment of rat primary astrocytes with 100 nM CI-IB-MECA for 48 h also significantly reduced the extent of spontaneous apoptosis with respect to control cultures in cells detached from the culture substrate and collected from the culture medium (Fig. 3). This suggests that the cytoskeletal reinforcement induced by the A3 agonist (Fig. 2) may result in a better adherence of cells to the culture substrate and, hence, in reduced susceptibility to detach and undergo spontaneous apoptosis. A specific role for the adenosine A3 receptor in this effect is also confirmed by the ability of the selective A3 receptor antagonist MRS1191 to fully prevent the agonist-induced reduction of spontaneous cell death (Fig. 3).
Fig. 2.

Morphological changes induced by nM adenosine A3 agonist concentrations in rat primary astrocytic cultures. Cultures were grown in the absence (Control, A, B) or presence of either adenosine A3 agonist (100 nM CI-IB-MECA, C, D, or 100 nM IB-MECA, E, F). After 48 h, cells were fixed, incubated with fluorescein-phalloidin to detect actin, and analyzed under a fluorescence microscope (see Materials and Methods). Stress fibers, which were hardly detectable in control cultures (A, B), were markedly increased after exposure to either agonist (e.g., C, D, E). Some cells also showed marked elongation of processes (F). Similar effects were observed in three independent experiments.
Fig. 3.

Reduction of spontaneous apoptosis induced by nM adenosine A3 agonist concentrations in rat primary astrocytic cultures. Cultures were grown in the absence or presence of 100 nM CI-IB-MECA alone or in combination with 10 μM MRS1191. After 48 h, cells floating in the culture supernatants were stained with the Hoechst 33258 chromatin dye as described in Materials and Methods. Detached cells showed signs of apoptosis, such as chromatin aggregation, fragmentation, or clumping (cells indicated with “a” in the micrographs); a lower number of apoptotic cells was found in cultures exposed to CI-IB-MECA. As shown by histograms, this effect was statistically significant (P < 0.0001 with respect to Control, Student’s t-test) and was completely prevented by contemporary exposure to the A3 adenosine antagonist MRS1191 (10 μM; **P < 0.0001 with respect to CI-IB-MECA alone, not statistically different from Control, Student’s t-test). Similar results were obtained in three independent experiments.
Since proteins belonging to the Bcl-2 family have been implicated in the regulation of cell survival [Brown, 1997], we evaluated the effect of A3 agonists on the expression of the anti-apoptotic proteins Bcl-2 and Bcl-XL in human ADF cells. Preliminary immunoblotting experiments allowed us to rule out significant expression of Bcl-2 in human astrocytoma ADF cells (data not shown). ADF cells instead express significant amounts of Bcl-XL [Cheng et al., 1996; see also below]. Exposure of cells to A3 receptor agonists under experimental conditions that elicited cytoskeletal reinforcement and protection against cell death resulted in no statistically significant changes of the total amount of Bcl-XL [Abbracchio et al., 1997] but instead markedly affected its intracellular organization (Fig. 4). In control adhering cultures, immunoreactivity to the anti-Bcl-XL antibody was mainly located in the cytoplasm (Fig. 4A). In Cl-IB-MECA-treated cultures, immunoreactivity was evident as bright spots positive for the antigen scattered throughout the cell cytoplasm, including cellular protrusions (see arrows in Fig. 4B). In contrast, dying cells floating in the culture medium as well as cells still adhering to the substrate but undergoing detachment were negative for the expression of this molecule (data not shown). A3 agonist-induced changes of intracellular Bcl-XL distribution were specifically mediated by activation of the A3 adenosine receptor, as demonstrated by the ability of the selective A3 antagonist MRS1191 to prevent the accumulation of Bcl-XL in cellular protrusions induced by Cl-IB-MECA (Fig. 4C).
Fig. 4.

Modulation of the intracellular distribution of Bcl-XL by A3 agonists in ADF cells. In the majority of control cells (A), immunoreactivity is located in the cytoplasm. No immunoreactivity for the anti-Bcl-XL antibody was found in apoptotic cells floating in the culture substrate and suspended in the culture medium (data not shown). After exposure of cultures to 100 nM CI-IB-MECA for 48 h, positivity to the anti-Bcl-XL antibody is also found as very bright spots that are particularly abundant in cell protrusions (see arrows in B). Similar data were obtained after a 48-h treatment with 100 nM IB-MECA (data not shown). The concomitant exposure of cultures to 10 nM MRS1191 counteracted agonist-induced accumulation of Bcl-XL in cellular protrusions (C). Cultures exposed to MRS1191 alone showed no differences with respect to control cultures. Note that all micrographs were taken at the same high magnification (1,800×) in order to compare the biological effect and to better point out intracellular labeling.
DISCUSSION
The present results suggest that selective A3-receptor agonists exert dual actions on cells belonging to the astroglial lineage, depending on their concentrations. In particular, we have shown that 1) at μM concentrations adenosine A3 agonists reduce the number of human astrocytoma ADF cells in culture, and 2) at nM concentrations (which are closer to the Ki values demonstrated by these compounds in binding studies to the transfected receptor and are not associated with induction of cell death), these same agonists induce a dramatic reorganization of the cytoskeleton, with the appearance of stress fibers, elongation of cellular processes, and marked intracellular redistribution of the Bcl-2-related protein Bcl-XL, which in these cells becomes strictly associated with cellular processes.
Although the effect induced by μM A3 agonist concentrations on ADF cell number is consistent with previous data [Kohno et al., 1996a,b; Yao et al., 1997; see also Jacobson, 1998 for review], the role of adenosine receptors in such an effect is still uncertain. Preliminary data would indeed suggest that reduction of ADF cell number by μM A3 agonist concentrations cannot be reversed by either xanthine- and nonxanthine adenosine receptor antagonists. However, in cells transfected with the human A3 receptor, μM A3 agonist concentrations also induced a reduction of cell number which could be fully antagonized by nM concentrations of the selective A3 receptor antagonists MRS1220, MRS1191, and L249313 [Brambilla et al., 1998]. Hence, it may simply be that an atypical adenosine A3 receptor characterized by insensitivity to the currently available antagonists is involved in the effects induced by μM A3 agonist concentrations in astroglial cells. Future studies aimed at testing the effects of various novel A3 receptor antagonists may clarify this important aspect.
On the contrary, the effects induced by nM A3 agonist concentrations (cytoskeleton reinforcement, reorganization of intracellular Bcl-XL distribution, and protection against spontaneous apoptosis) are clearly mediated by the A3 adenosine receptor, since they could be antagonized by the selective A3 receptor antagonist MRS1191. Such protective effects are consistent with similar effects reported for myocardial cells [Liang and Jacobson, 1998], suggesting that activation of the A3 adenosine receptor may result in cell protection in both heart and brain. A3 agonist-induced cell protection seems to involve the intracellular redistribution of the anti-apoptotic Bcl-XL protein and its association with cytoskeletal elements in long dendritic-like cell protrusions. The latter finding is consistent with other recent findings that proteins belonging to the Bcl-2 family may play key roles in crucial functions other than cell survival or in functions that may only indirectly affect cell viability. For example, Bcl-2 was shown to promote growth and regeneration of retinal axons [Chen et al., 1997]. It may be hypothesized that Bcl-XL would associate with the cytoskeleton in order to improve some important cell function such as heterotypic cell adhesion, finally resulting in a secondary increased resistance to cell detachment and death. Consistent with this hypothesis, no immunostaining for Bcl-XL was found in apoptotic cells floating in the culture medium or in cells about to detach from the culture substrate, suggesting that expression of this protein may be suppressed before cells detach and undergo cell death. This is in agreement with previous data obtained by our group on adhering cells of different origin (i.e., epithelial cells), indicating a relationship between Bcl-XL turning off and loss of cell−substrate contacts [Matarrese et al., 1998].
The opposing actions induced in astroglial cells by nM and μM A3 agonist concentrations may be, at least in part, the basis of the strikingly different ischemic outcomes observed in vivo after either acute or chronic administration of IB-MECA [von Lubitz et al., 1994]. In that study, administration of this A3 agonist immediately prior to the induction of ischemia in gerbils was shown to dramatically worsen both animal survival and the extent of cellular damage in the ischemic hippocampus. However, since adenosine receptors are expressed by a variety of different cell types [Dalziel and Westfall, 1994], it was not evident whether the effects induced by IB-MECA were due to indirect rather than direct mechanisms (for example, it is known that, besides modulating neuronal and glial function, adenosine also regulates cerebral blood flow [von Lubitz et al., 1994]). The present results suggest that activation of A3 receptors located on astroglial cells may represent a mechanism at the basis of the in vivo effects of IB-MECA. We speculate that a robust and acute activation of these receptors during ischemia as a consequence of massive release of adenosine (see Introduction) may contribute to the development of ischemic damage. It could be hypothesized that A3 receptor-mediated apoptosis of astroglial cells may result in a reduced survival rate of neuronal cells. Of course, this does not rule out that possible direct effects of adenosine A3 receptors located on other cell types (e.g., on either neurons or vasal cells) may also contribute to ischemia-induced damage. In von Lubitz et al. [1994], it was also shown that strikingly opposite effects (i.e., a marked cerebroprotection associated with increased survival rate of the animals) could be obtained if ischemia was induced after a subchronic treatment with IB-MECA. Desensitization of central A3 receptors as a consequence of prolonged agonist exposure was hypothesized to reduce the putative contribution of this receptor to ischemic damage. The present data show that, under certain experimental conditions, A3 agonists can indeed activate cell protection mechanisms (e.g., changes of the structural components of the cytoskeleton and of Bcl-XL intracellular distribution, leading to enhanced resistance to subsequent insults). However, since these beneficial effects are induced at nM agonist concentrations, molecular mechanisms other than agonist-induced receptor desensitization are likely involved. A subthreshold stimulation of the A3 receptor prior to the induction of ischemia may result in activation of protective mechanisms that make the brain less sensitive to a subsequent ischemic insult (“ischemic tolerance,” by analogy to preconditioning of the heart [Strickler et al., 1996]). In this respect, it was previously shown that in gerbils mild ischemic treatments (e.g., a 2-min carotid occlusion) induce tolerance to a subsequent, and what would be lethal, ischemic stress [Kitagawa et al., 1990].
If confirmed, this hypothesis may have intriguing implications for the pharmacological manipulation of brain damage in both ischemia and in neurodegenerative diseases and suggests that the A3 receptor may represent an interesting target for the development of novel pharmacological agents of potential therapeutic value in these central nervous system disorders.
ACKNOWLEDGMENTS
M.P.A., S.C., R.B. and F.C. have been involved in the concerted action ADEURO (EU, BIOMED1) (ADEURO is the acronym for the concerted action “Physiology and pharmacology of brain adenosine receptors—implications for the rational design of neuroactive drugs” supported by the European Union within the Biomedical and Health Research Programme).
Contract grant sponsors:
AIRC (Associazione Italiana Ricerca sul Cancro); the Italian Ministero dell’Universita’ e Ricerca Scientifica e Tecnologica (1201008 MURST to MPA); the Consiglio Nazionale delle Ricerche (98.01047.CTOU to MPA).
Footnotes
CI-IB-MECA was kindly provided by Research Biochemical International as part of the Chemical Synthesis Program of the National Institute of Mental Health, Contract No. 01MH30003.
REFERENCES
- Abbracchio MP, Saffrey MJ, Hopker V, Burnstock G. 1994. Modulation of astroglial cell proliferation by analogues of adenosine and ATP in primary cultures of rat striatum. Neuroscience 59:67–76. [DOI] [PubMed] [Google Scholar]
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W. 1997. The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton and distribution of Bcl-XL. Studies in human astroglioma cells. Biochem Biophys Res Commun 241:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri D, Abbracchio MP, Salvioli S, Monti D, Cossarizza A, Ceruti S, Brambilla R, Cattabeni F, Jacobson KA, Franceschi C. 1998. Apoptosis by 2-chloro-2′-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem Int 32:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceschi C, Kim YC, Jacobson KA, Casavola V, Reshkin SJ, Klotz K-N, Lohse MJ, Abbracchio MP. 1998. Activation of the human A3 adenosine receptor in CHO transfected cells results in cytosolic acidification and block of cells at the S phase. Drug Dev Res 43:13. [Google Scholar]
- Brown R 1997. The bcl-2 family of proteins. Br Med Bull 53:451–465. [DOI] [PubMed] [Google Scholar]
- Ceruti S, Barbieri D, Franceschi C, Giammarioli AM, Rainaldi G, Malorni W, Kim HO, von Lubitz DKJE, Jacobson KA, Cattabeni F, Abbracchio MP. 1996. Effects of adenosine A3 receptor agonists on astrocytes: Induction of cell protection at low and cell death at high concentrations. Drug Dev Res 37:177. [Google Scholar]
- Chen DF, Schneider GE, Martinou J-C, Tonegawa S. 1997. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385:434–439. [DOI] [PubMed] [Google Scholar]
- Cheng EHY, Levine B, Boise LH, Thompson CB, Hardwick JM. 1996. Bax-independent inhibition of apoptosis by Bcl-XL. Nature 379:554–556. [DOI] [PubMed] [Google Scholar]
- Dalziel HH, Westfall DP. 1994. Receptors for adenine nucleotides and nucleosides: Subclassification, distribution, and molecular characterization. Pharmacol Rev 46:449–466. [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Kim HO, Jiang J-L, Jacobson KA. 1997. Activation of hippocampal adenosine A3 receptors produces a heterologous desensitization of A1 receptor mediated responses in rat hippocampus. J Neurosci 17:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. 1994. Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Pu Q, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. 1994. Structure-activity-relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem 37:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA. 1998. Adenosine A3 receptors: Novel ligands and paradoxical effects. Trends Pharmacol Sci 19:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Suzuki F. 1996. Recent developments in selective agonists acting at purine and pyrimidine receptors. Drug Dev Res 39:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Ji X-D, Berkich DA, Eveleth D, Dean RL, Hiramatsu K-I, Kassell NF, van Galen PJM, Lee KS, Bartus RT, Daly JW, LaNoue KF, Maillard M. 1992. Synthesis and biological activity of N6-p-sulfophenylalkyl and derivatives of adenosine: Water soluble and peripherally selective adenosine agonists. J Med Chem 35:4143–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kim HO, Siddiqi SM, Olah ME, Stiles G, von Lubitz DKJE. 1995. A3 adenosine receptors: Design of selective ligands and therapeutic prospects. Drugs Future 20:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M, Chakravarty PK, Johnson RG, Norton R. 1996. Novel selective non-xanthine A3 adenosine receptor antagonists. Drug Dev Res 37:131. [Google Scholar]
- Ji X-D, von Lubitz DKJE Olah ME, Stiles GL Jacobson KA. 1994. Species-differences in ligand affinity at central A3-adenosine receptors. Drug Dev Res 33:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J-L, van Rhee AM, Melman N, Ji X-D, Jacobson KA. 1996. 6-phenyl-1,4-dihydropyridine derivates as potent and selective A3 adenosine receptor antagonist. J Med Chem 39:4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karton Y, Jiang J-I, Ji X-D, Melman N, Olah ME, Stiles GL, Jacobson KA. 1996. Synthesis and biological activities of flavonoid derivatives as A3 adenosine receptor antagonists. J Med Chem 39:2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji K-D, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 1994. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3-adenosine receptors. J Med Chem 37:3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-C, Ji X-D, Jacobson KA. 1996. Derivatives of the triazolo-quinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39:4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, Kamada T. 1990. “Ischemic tolerance” phenomenon found in the brain. Brain Res 528:21–24. [DOI] [PubMed] [Google Scholar]
- Kohno Y, Ki X-D, Mawhorter SD, Koshiba M, Jacobson KA. 1996a. Activation of A3 adenosine receptors on human eosinophils elevates intracellular calcium. Blood 88:1–6. [PMC free article] [PubMed] [Google Scholar]
- Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA. 1996b. Induction of apoptosis in HL-60 human promyelocytic leukemia cells by adenosine A3 receptor agonists. Biochem Biophys Res Commun 219:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang BT, Jacobson KA. 1998. A physiological role of the adenosine A3 receptor: Sustained cardioprotection. Proc Natl Acad Sci USA 95:6995–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden J 1994. Cloned adenosine A3 receptors-pharmacological properties, species-differences and receptor functions. Trends Pharmacol Sci 15:298–306. [DOI] [PubMed] [Google Scholar]
- Malorni W, Rainaldi G, Rivabene R, Santini MT. 1994. Different susceptibilities to cell death induced by t-butylhydroperoxide could depend upon cell histotype-associated growth features. Cell Biol Toxicol 10:207–218. [DOI] [PubMed] [Google Scholar]
- Matarrese P, Giandomenico V, Fiorucci G, Rivabene R, Straface E, Romeo G, Affabris E, Malorni W. 1998. Antiproliferative activity of interferon A and retinoic acid in SiHa carcinoma cells: The role of cell adhesion. Int J Cancer 76:531–540. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. 1997. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci 20:570–577. [DOI] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P. 1995. Adenosine and brain ischemia. In: Belardinell L, Pelleg A, editors. Adenosine and ademime nucleotides: from molecular biology to integrative physiology. Kluwer Norwell. p 391–396. [Google Scholar]
- Sei Y, von Lubitz DKJE, Abbracchio MP, Ji X-D, Jacobson KA. 1997. Adenosine A3 receptor agonist-induced neurotoxicity in rat cerebellar granule neurons. Drug Dev Res 40:267–273. [Google Scholar]
- Shneyvais V, Jacobson KA, Shainberg A. 1998. Induction of apoptosis in cultured cardiac myocytes by adenosine A3 receptor agonist. Exp Cell Res, in press. [DOI] [PubMed] [Google Scholar]
- Stambaugh K, Jacobson KA, Jiang JL, Liang B. 1997. A novel cardioprotective function of adenosine A1 and A3 receptor during prolonged simulated ischemia. Am J Physiol 273:H501–H505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler J, Jacobson KA, Liang BT. 1996. Direct preconditioning of cultured chick ventricular myocytes. Novel functions of cardiac A2A and A3 receptors. J Clin Invest 98:1773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Olah ME, Ijzerman AP, Stiles GL, Jacobson KA. 1994. A binding-site model and structure-activity-relationships for the rat A3-adenosine receptor. Mol Pharmacol 45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- von Lubitz DKJE, Lin RC-S, Popik P, Carter MF, Jacobson KA. 1994. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Sei Y, Abbracchio MP, Jiang J-L, Kim YC, Jacobson KA. 1997. Adenosine A3 receptor agonists protect HL-60 cells and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun 232:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q-Y, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O. 1992. Molecular cloning and characterization of an adenosine receptor—the A3 adenosine receptor. Proc Natl Acad Sci USA 89:7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]