

Abstract
The SAR at adenosine (P1) and ATP (P2) receptors is reviewed, with emphasis on recently developed selective agonists and antagonists. These include partial (e.g., N6-ethyl-8-cyclopentylaminoadenosine) and full A1 agonists (e.g., NNC 21-0136, 2-chloro-N6-[(R)-(benzothiazolylthio-2-propyl]adenosine), A2 antagonists (e.g., the non-xanthines: SCH58261, 5-amino-7-(phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine and ZM241385, 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3a][1,3,5]triazinyl-amino]ethyl)-phenol; and the 1-propargyl-8-styrylxanthines), and A3 agonists (e.g., CI-IB-MECA, 2-chloro-N6-(3-iodobenzyl)-adenosine-5’-N-methyluron-amide). Novel adenosine receptor antagonists (e.g., BTH4, ethyl 3-benzylthio-4,5,6,7-tetrahydro-benzo[c]thiophen-4-one-1-carboxylate) have been discovered through screening libraries of natural products and heterocyclic derivatives. The first A3 selective antagonists to be identified include derivatives of flavones (MRS 1067), 1,4-dihydropyridines (MRS 1097), triazolonaphthyridine (L-249313), and thiazolopyrimidine (L-268605). Potent P2 receptor agonists are known. For example, 2-HexylthioAMP is a highly potent agonist at the yet uncloned P2Y receptor in C6 glioma cells. Suramin is a weak and non-selective P2 blocker, while a truncated derivative, NF023, appears to be selective for P2X receptors. More selective P2 antagonists are under development, with the cloning of these receptors. [35S]ATP-γS has been used as a radioligand for the direct labeling of several subtypes of cloned P2X receptors (P2X1-P2X4).
Keywords: agonists, antagonists, adenosine, ATP, structure activity relationships, xanthines
INTRODUCTION
Medicinal chemists are currently developing potential therapeutic agents that interact selectively with receptors for adenosine (termed A1, A2A, A2B, and A3 subtypes) or receptors for purine and pyrimidine nucleotides (belonging to P2X and P2Y superfamilies).1 Many highly selective ligands for adenosine A1 and A2A receptors [Jacobson et al., 1992a] have been designed, while the first selective agonists at A3 receptors were only recently introduced [Jacobson et al., 1995]. Agonists and antagonists at receptors for purine and pyrimidine nucleotides (P2) are much less developed than those at adenosine (P1) receptors, in terms of potency, selectivity, and envisioned therapeutic modalities. Numerous new subtypes of ATP receptors, including both ionotropic P2X and metabotropic P2Y receptors, are now cloned [Filtz et al., 1997], and this diversity will present a challenge to medicinal chemists for years to come.
Adenosine agonists [Jacobson et al, 1992a, 1995], which are almost exclusively derivatives of adenosine (Fig. 1), have been sought as potential anti-arrhythmic, cerebroprotective, and cardioprotective agents (via A1), and as hypotensive and anti-psychotic agents (via A2A). Adenosine receptor antagonists, of which xanthines and numerous classes of fused heterocyclic compounds (Fig. 2) are representative, have been under development as anti-asthmatic [Francis et al., 1988], anti-depressant [Sarges et al., 1990], anti-arrhythmic [Belardinelli et al., 1995], renoprotective [Suzuki et al., 1992], anti-Parkinson’s [Shimada et al., 1992], and cognition enhancing [Schingnitz et al., 1991] drugs. A3 receptor agonists and/or antagonists have potential as prophylactic cerebroprotective agents [Jacobson et al., 1995] and possibly in modulating immune function and in treating inflammation. Selective agents acting at P2 receptors may have potential in the treatment of pain, urinary incontinence, and other conditions [Burnstock, 1996].
Figure 1.
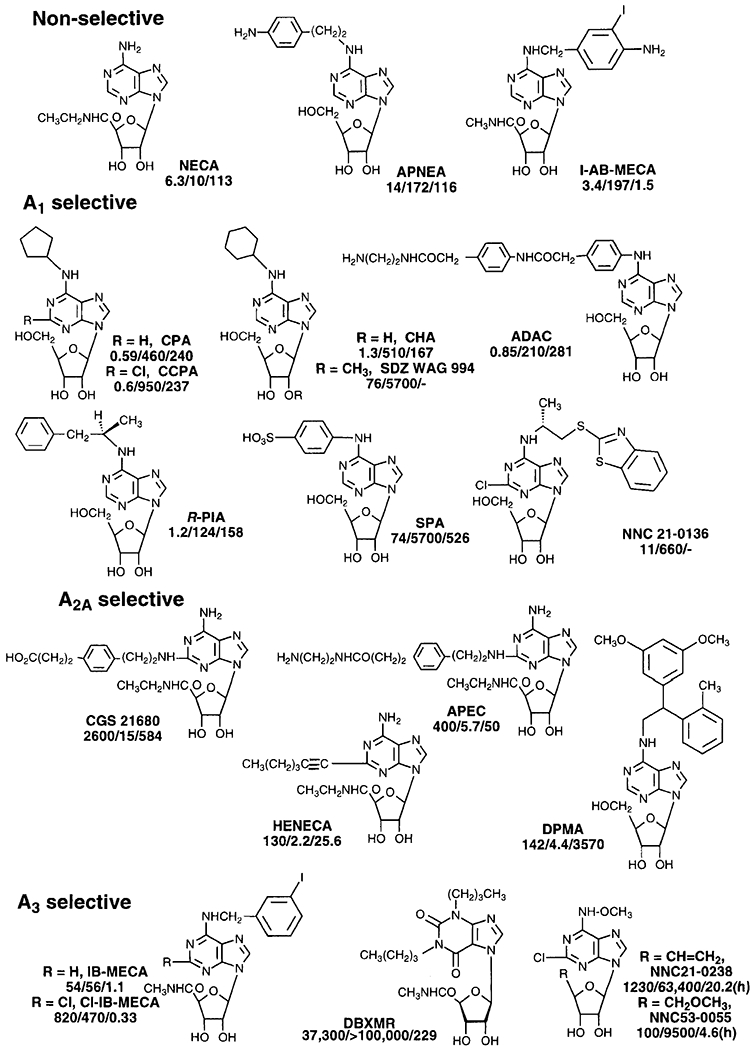
Adenosine agonists (affinity at A1/A2A/A3 receptors in nM rat, except as indicated, h = human).
Figure 2.
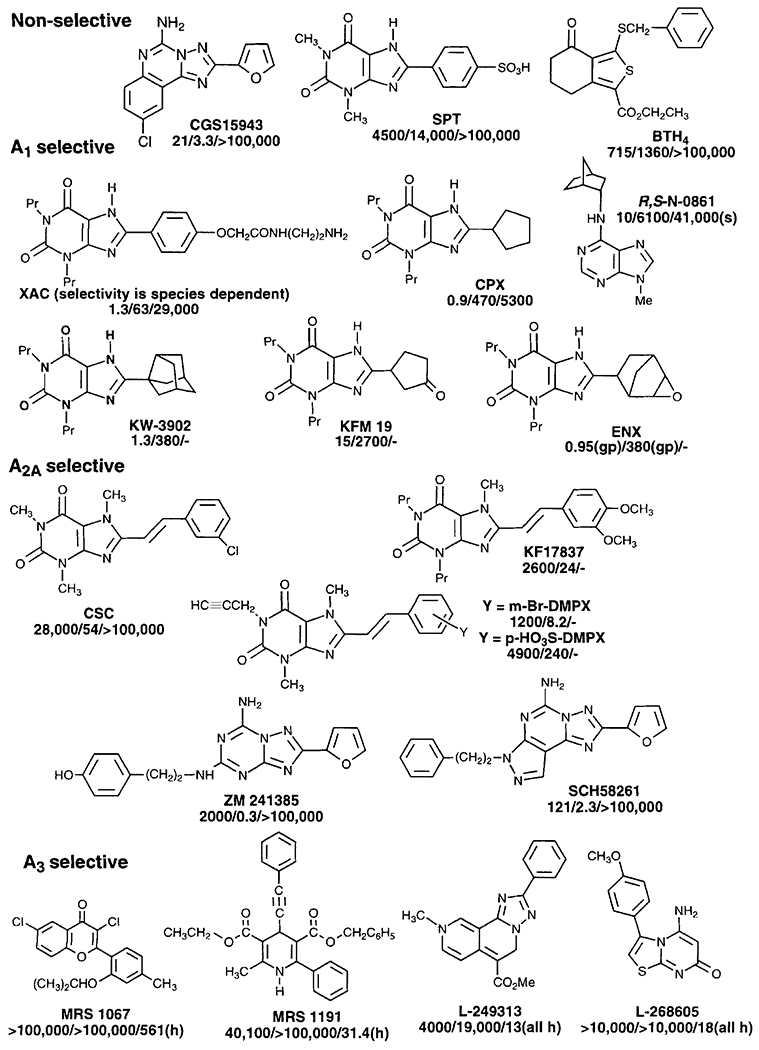
Adenosine antagonists (affinity at A1/A2A/A3 receptors in nM in rat, except as indicated, h = human, s = sheep, gp = guinea pig).
P1 Receptor Agonists
In general, for adenosine agonists (Fig. 1), numerous modifications of the N6-position with hydrophobic moieties (such as CPA, N6-cyclopentyladenosine; CHA, N6-cyclohexyladenosine; and R-PIA, (R)-N6-phenylisopropyladenosine) provide selectivity for A1 receptors. The 2-chloro analogue CCPA (2-chloro-N6-cyclopentyladenosine) [Klotz et al., 1989] has slightly greater A1 receptor selectivity than the parent CPA. The affinities of these N6-substituted adenosine derivatives at A3 receptors are often intermediate between their respective A1 and A2A affinities. SPA (N6-p-sulfophenyladenosine), which is fully negatively charged at physiological pH, is an A1 agonist somewhat selective for peripheral receptors [Jacobson et al., 1992b]. NNC 21-0136 (2-chloro-N6-[(R)-(benzothiazolylthio-2-propyl]adenosine) is somewhat selective for central vs. peripheral A1 receptors and is under development as a neuro-protectant agent [Knutsen et al., 1997]. Both classical medicinal chemical approaches and a functionalized congener approach [Jacobson et al., 1992a] have been utilized in purinoceptor ligand development efforts. By the latter approach, a chemically functionalized chain is incorporated at a specific site on a pharmacophore (e.g., the primary amine functionalized agonist ADAC). The site of attachment must correspond to a region of relaxed steric requirements at or near the receptor binding site, and targeting accessory sites of favorable interaction on the receptor may enhance the affinity and/or selectivity of the ligands.
Although most N6-substituted adenosine agonists are A1-selective, the agonist DPMA (N6-[2-(3,5-dimethoxyphenyl)-1-(2-methylphenyl)ethyl] adenosine) as the racemate was found to be 30-fold selective for the A2A-receptor [Bridges et al., 1988]. Among ring nitrogen substituted derivatives of adenosine, only 1-deazaadenosines retain high affinity for adenosine receptors. Similarly, few ribose modifications are tolerated; however, the 2’-methyl ether of CHA, SDZ WAG 994 [Wagner et al., 1995], was found to be an orally active agonist and surprisingly potent in vivo. This compound has been examined for potential as an anti-lipolytic agent for the treatment of diabetes.
N6,C8-Disubstituted derivatives of adenosine, such as N6-ethyl-8-cyclopentylaminoadenosine (ECPA), have been investigated by IJzerman and colleagues [Roelen et al., 1996] as potential partial agonists that are selective for A1 adenosine receptors. Radioligand binding vs. the tritiated antagonist CPX (Fig. 2) was carried out in the presence and absence of 1 mM GTP. The ratios of Ki values determined in this manner (GTP shift) indicated partial agonism when the value was significantly lower than that for the prototypic full agonist, CPA (6.0). Compounds with ratios of between 1.1 and 3.8 were found to have lower intrinsic activities in decreasing heart rate in conscious, normotensive rats. Preliminary studies indicated that the same compounds showed full intrinsic activity as inhibitors of lipolysis in the same strain of rats, suggesting a gain in tissue selectivity.
Among ribose modifications, amide substitution at the 5’-position as in NECA (adenosine-5’-N-ethyluronamide) provides increased potency at A2A receptors. NECA is also among the most potent agonists at the A2B receptor [Hide et al., 1992]. Thus NECA is a highly potent but non-selective agonist. A2A-selective adenosine agonists derived from NECA have also been developed. Evaluation of 2-position modifications of NECA led to the identification of CGS 21680 (2-[4-[(2-carboxyethyl)phenyl]ethylamino]-5’-N-ethylcarboxamidoadenosine), which is 140-fold selective for A2A vs. A1 receptors [Hutchison et al., 1989], with a Ki value of 15 nM at A2A receptors. The related ethylenediamine conjugate APEC, which has been elaborated chemically as an amine functionalized congener, can be radioiodinated to obtain the conjugate iodo-PAPA-APEC (Jacobson et al., 1992a]. Unlike CGS 21680, which apparently does not cross the blood brain barrier, APEC is highly potent as a centrally acting locomotor depressant when administered peripherally. Both CGS 21680 and APEC are inactive at A2B receptors [Hide et al., 1992 ]. The SAR (structure activity relationship) at the 2-position of such adenosine-5’-uronamide derivatives has also been extensively probed, leading to such derivatives as the 2-alkynyl-5’-uronamide derivative, HENECA [Cristalli et al., 1995], which is a very potent A2A agonist (Ki = 2.2 nM). The SAR of various 2-alkenyl derivatives of NECA has been explored [Vittori et al., 1996b].
The cloning of the A3 adenosine receptor [Zhou et al., 1992; Salvatore et al., 1993] has opened new therapeutic vistas in the adenosine field. The A3 receptor has a unique SAR profile, tissue distribution, and effector coupling. Activation of A3 receptors requires relatively high, i.e., pathological, concentrations of adenosine; the Ki value of adenosine at the rat A3 receptor has been estimated to be ~1 μM vs. 10 and 30 nM at rat A1 and A2A receptors, respectively [Jacobson et al., 1995]. Thus, the physiological role of A3 receptors may be very different from that of A1 and A2A subtypes.
The first A3 selective agonists have been reported very recently [Jacobson et al., 1995]. Previously, N6-p-aminophenylethyladenosine (APNEA) had been used as an agent to activate A3 receptors in the presence of non-A3 antagonists, although APNEA is actually 8-fold selective for A1 receptors [Kim et al., 1994b]. One principle of achieving true A3 selectivity among adenosine derivatives is the combination of optimal substitutions at the N6- and 5’-positions of adenosine. Specifically, among N6-substituents, a benzyl group is favored, due to its diminished potency at A1 and A2A receptors, and these A3-selectivity enhancing effects are additive with the A3-affinity enhancing effects of the 5’-uronamido group. Empirical and QSAR studies [Siddiqi et al., 1995] of substituent effects on the N6-benzyl group have shown that substitution at the 3-position of the phenyl ring with stericallv bulky groups, such as the iodo group, is optimal, leading to the development of the highly potent A3 agonist N6-(3-iodobenzyl)-adenosine-5’-N-methyluronamide (IB-MECA) which is 50-fold selective [Gallo-Rodriguez et al., 1994] for A3 vs either A1 or A2A receptors in vitro and appears to be highly A3 selective in vivo. The radioligand [125I]I-AB-MECA is widely used as a high affinity radio-ligand for A3 receptors [Olah et al., 1994], although it is not as selective for A3 vs. A1 receptors as IB-MECA. [125I]I-AB-MECA bound to cloned human A3 receptors expressed in HEK-293 cells with a Kd value of 0.59 nM [Ji et al., 1996]. Substitution at the 2-position in combination with modifications at N6 and 5’-positions was found to further enhance A3 selectivity [Kim et al., 1994b]. Cl-IB-MECA (2-chloro-N6(3-iodobenzyl)-adenosine-5’-N-methyluronamide), which displayed a Ki value of 0.33 nM at A3 receptors, was selective for A3 vs. A1 and A2A receptors by 2,500- and 1,400-fold, respectively.
Nearly all of the adenosine agonists yet reported are primary or secondary amine derivatives at the 6-position. Unexpectedly, acylation of the exocyclic amine of NECA, to form urea derivatives and certain amide derivatives, was shown to lead to potent agonists, some of which displayed A3 selectivity [Baraldi et al., 1996a]. For example, the 3-chlorophenylaminocarbonyl derivative of NECA is 10-fold selective for rat A3 vs. A1 receptors (Ki = 4.4 nM), while the 4-phenylbenzoyl derivative is 165-fold selective for rat A1 vs. A3 receptors (Ki = 5.9 nM). Knutsen and coworkers have also explored 5’-vinylic (NNC 21-0238) and 5’-methyloxy (NNC 53-0055) analogues of N6-alkoxyadenosines, which also proved to be A3 selective [Bowler et al., 1996].
A3 agonists have been shown to both inhibit adenylyl cyclase and activate phosphatidylinositol-4,5-bisphosphate-specific phospholipase C, with a rank order of potency that reflects the order of affinity determined in binding assays but at generally higher concentrations than the Ki values in competition of binding [Kim et al., 1994b; Abbracchio et al., 1995]. A3 receptor activation has also been shown to be coupled to activation of phospholipase D [Ali et al., 1996]. There may be differences in the relative activation of various second messenger systems based on the concentration of the A3 agonists. Low concentrations of A3 agonists such as C1-IB-MECA (<100 nM, Fig. 1) have been shown to antagonize the A1 receptor mediated inhibition of EPSPs in hippocampal slices [Dunwiddie et al., 1996]. High concentrations of A3 agonists have been found to induce apoptosis [Kohno et al., 1996], thus intense activation of the A3 receptor may have a damage-inducing, rather than a protective role.
P1 Receptor Antagonists
Theophylline (1,3-dimethylxanthine) and caffeine (1,3,7-trimethylxanthine) are non-selective adenosine antagonists (A1 and A2) of low affinity (Ki ≥ 10 μM). Selective antagonists for A1 receptors (Fig. 2) include principally many 8-aryl and 8-cycloalkyl xanthine derivatives, such as the xanthine amine congener XAC (8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine), which is selective for rat but not human or rabbit A1 receptors, and CPX (1,3-dipropyl-8-cyclopentylxanthine). CPX (also known as DPCPX) is ~500-fold selective for A1 vs. A2A receptors generally across species [Jacobson et al., 1992a]. Recently, ENX (1,3-dipropyl-8-[2-(5,6-epoxy)norbornyl]xanthine) was found to be extremely selective for A1 receptors in binding and functional assays [Belardinelli et al., 1995]. Other 8-substituted xanthines include KW-3902, 1,3-dipropyl-8-noradamantylxanthine, which has potent diuretic properties and is in Phase II clinical trials [Suzuki et al., 1992], 8-(dicyclopropylmethyl)-1,3-dipropylxanthine(KF15372), which is even more potent and A1-selective than CPX in guinea-pig forebrain [Suzuki et al., 1992], and KFM-19 ((±)8-(3-oxocyclopentyl)-1,3-dipropylxanthine), a potent A1-selective compound with sufficient aqueous solubility to display good bioavailability [Schingnitz et al., 1991]. The S-isomer of KFM-19, known as BIIP20, is the more potent enantiomer. KF15372 and KFM-19 are currently under development as cognition enhancers. 8-p-Sulfophenyltheophylline (8-SPT) is useful as a peripheral acting antagonist, but is non-selective. The A1 receptor has considerably more bulk tolerance than the A2A receptor for substitution at the N3-position of xanthines.
The parallel in A1 affinity between 8-substituted xanthines and similarly N6-substituted adenosines, suggests a common mode of binding of xanthines and adenosines in the receptor binding site. A computer-generated model of the antagonist binding site of the adenosine receptor assumes that N6-substituents of agonists and C8-substituents of xanthine antagonists bind to the same region of the receptor [van der Wenden et al., 1995].
Selectivity for A2A receptors in xanthines has been more difficult to achieve. However, such selectivity is observed for 7-methyl-8-styrylxanthines, such as KF17837 [Shimada et al., 1992], and CSC (8-(3-chlorostyryl)caffeine) [Jacobson et al., 1993], which displayed selectivity for A2A vs. A1 receptors of 62-fold and 520-fold, respectively. Styrylxanthines in dilute solution, however, suffer from sensitivity to photoisomerization. 3,7-Dimethyl-l-propargylxanthine (DMPX) is a weak and slightly A2A selective antagonist. Müller et al. [1996] have introduced hybrids of the styrylxanthines and 1-propargylxanthines, to provide novel A2A receptor selective antagonists (Fig. 2). Sulfonate groups have been introduced in the styryl ring to enhance water solubility.
The A3 adenosine receptor cloned from rat [Zhou et al., 1992] was shown to be unique among the subtypes in that agonist action is not antagonized by xanthines, such as theophylline. A3 selective antagonists for general use in pharmacological studies across species are currently under development. Typical Ki values at rat A3 receptors of roughly 100 μM have been determined for many xanthines that have nearly nanomolar potency at the A1 or A2A subtypes [Kim et al., 1994a], and these xanthines do not effectively antagonize A3 agonist-elicited inhibition of adenylyl cyclase. An anionic group attached to the xanthine tended to diminish the affinity at A1 and A2A receptors. The A3 receptor affinity of most xanthines is highly species dependent. XAC, for example, is more potent in binding to the human (Ki = 71 nM) and sheep (Ki = 180 nM) homologues [Salvatore et al., 1993] of the A3 receptor than to the rat A3 receptor (Ki = 29 μM). XAC has been used in pharmacological experiments in vitro and in vivo in rodents for distinguishing A3 receptors from A1 and A2A receptors, at which it is much more potent.
Kim et al. [1994c] have “anchored” xanthines in the A3 binding site by adding a sugar moiety at the N7-position to form xanthine-7-ribosides. The presence of the ribose moiety enhances affinity of xanthines at rat A3 receptors, while at A1 receptors the xanthine-7-riboside derivatives are, as a rule, less potent than the parent xanthines. Further structural modification, based on parallels in SAR between adenosine derivatives and the xanthine-7-ribosides, increased the potency and selectivity of the xanthine-7-ribosides at A3 receptors. This approach has led, for example, to 1,3-dibutylxanthine-7-riboside-5’-N-methylcarboxamide, DBXRM (Fig. 1), which has a Ki value of 229 nM at A3 receptors and is 160-fold and >400-fold selective for A3 vs. A1 and A2A receptors, respectively. Although a xanthine derivative, DBXRM appeared to be a full agonist in the inhibition of adenylate cyclase via rat A3 receptors.
Numerous structurally diverse non-xanthine antagonists (Fig. 2) have also been identified, many of which are not well defined in terms of SAR. Among the first classes of heterocycles found to antagonize the effects of adenosine agonists were the “tricyclic” non-xanthine antagonists, including the triazoloquinazolines [Francis et al., 1988], the triazoloquinoxalines [Sarges et al., 1990], and the imidazoquinolines [van Galen et al., 1991].
Other potent and A1-selective antagonists have been derived from adenine and include the N6-substituted 9-methyladenines [Thompson et al., 1991], such as (R/S)N-0861 ((±)N6-endonorbornyl-9-methyladenine; Ki at A1 10 nM; at A2A: 6,100 nM in bovine brain). An attempt to design A3 selective adenine derivatives by applying the structural principles of recognition derived from adenosine agonists resulted in loss of potency and selectivity [Jacobson et al., 1995]. Vittori et al. [1996a] have further explored the affinity of adenine derivatives and found that the presence of 2-aralkylamino or 2-alkynyl groups enhanced affinity for A1 and A2A subtypes, and a 2- or 8-alkynyl group resulted in A3 receptor affinity in the low micromolar range.
The traizoloquinazoline, CGS 15943 (9-chloro-2-(2-furyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine), is a potent adenosine receptor antagonist with only 7-fold selectivity for A2A receptors vs. A1 receptors, and an IC50 of 3 nM at rat A2A receptors [Francis et al., 1988], 5-Amino-7-(phenylethyl)-1-(2-furyl)-pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine (SCH58261) [Baraldi et al., 1995, 1996b] and 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3a][1,3,5]triazinylamino]ethyl)-phenol (ZM241385) [Poucher et al., 1995] are highly potent and selective non-xanthine A2A receptor antagonists (Fig. 2) that appear to have very favorable pharmacological properties, both as the radiolabeled and the unlabeled form. ZM241385 is the most selective A2A antagonist (6,800-fold) yet reported. Radioiodination of ZM241385 has provided a highly potent and selective A2A antagonist radioligand [Palmer et al., 1995]. The SAR at the N7-substituent of the pyrazole ring of SCH 58261 was explored by Baraldi et al. [1996b]. SCH 633990, the corresponding phenylpropyl homologue, was found to have a Ki value of 2.4 nM at A2A receptors with an enhanced selectivity of 210-fold vs. A1 receptors.
Recently, the Molecular Recognition Section at NIH has identified novel adenosine receptor ligands as a result of screening libraries of natural products and various heterocyclic derivatives [Siddiqi et al., 1996]. These novel non-xanthine adenosine antagonists includes both nitrogen-containing heterocycles and several classes of non-nitrogen heterocycles. For example, van Rhee et al. [1996a] recently reported that tetrahydrobenzothiophenones, e.g., ethyl 3-benzylthio-4,5,6,7-tetrahydrobenzo[c]thiophen-4-one-l-carboxylate (Fig. 2, BTH4), bind to adenosine receptors in the micromolar range. Diverse structures which have displayed some degree of selectivity in binding to rat A3 receptors [Siddiqi et al., 1996] include folic acid, a pyridopyrimidinone, cytochalasin H, 11-hydroxytetracarbazolenine, dipyridamole, and certain sulfonylpiperazines (e.g., 1-(5-isoquiniolinesulfonyl)-piperazine dihydrochloride, HA-100).
A broad screening of phytochemicals has demonstrated that certain flavone, flavonol, and flavanone derivatives have micromolar affinity at adenosine receptors [Ji et al., 1996]. Galangin and other flavonoids display micromolar affinity at A3 receptors, suggesting that there may be a wide range of phenolic adenosine receptor-modulating substances in the human diet. Also, dihydropyridine calcium channel blockers were found to bind to human A3 receptors with relatively high affinity [van Rhee et al., 1996b]. The structures of both the dihydropyridines and the flavonoids have been modified to achieve clear selectivity in binding to cloned human A3 receptors. The flavonoid 3,6-dichloro-2’-(isopropoxy)-4’-methylflavone (MRS1067) [Karton et al., 1996] and the dihydropyridine 3,5-diethyl 2-methyl-4-trans-styryl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (MRS1097) [van Rhee et al., 1996b] were both relatively potent, with Ki values of 560 and 110 nM, respectively, and highly selective (~200-fold) for human A3 vs. human A1 receptors. MRS1097 is inactive at L-type calcium channels. These derivatives effectively antagonized the inhibitory effects of an A3 receptor agonist on adenylate cyclase. MRS1222 (the p-nitro derivative of the styryl ring of MRS1097) is even more A3 selective, by a factor of > 1,700-fold, vs. rat A1 receptors. A 4-phenylethynyl dihydropyridine derivative, MRS1191 [Jiang et al., 1996], which is 1,300-fold selective for human A3 vs. rat A1 receptors, was shown to selectively antagonize A3 receptors in the rat hippocampus [Dunwiddie et al., 1997]. The most potent A3 receptor antagonist yet reported is MRS1220, an N5-acyl derivative of CGS15943, which has a Ki, value of 0.65 nM at human A3 receptors.
The Merck group [M. Jacobson et al., 1996], using high throughput screening of heterocyclic compounds, has identified two antagonists highly selective for human A3 receptors. These novel structures, as shown in Figure 2, are: the triazolonaphthyridine derivative L-249313, which behaves as a non-competitive antagonist, and the thiazolopyrimidine derivative L-268605, which is competitive in binding. L-249313 bound to rat A3 receptors with a Ki of 58 μM.
P2 Receptor Agonists
Various P2 receptor agonists are shown in Figure 3 and all are adenine or uridine nucleotide derivatives. Among metabotropic P2Y receptors, some of the cloned subtypes (e.g., P2Y2, P2Y4, and P2Y6) have been found to respond to uridine nucleotides with the same degree or an even greater degree of potency than the corresponding adenine nucleotides [Filtz et al., 1997]. Thus UTP is as potent as ATP at the P2Y2 (corresponding to the pharmacologically defined P2u) receptor subtype. UTP-γ-S has been introduced as a potent agonist at this subtype that is not readily degraded by nucleotidases [Lazarowski et al., 1996].
Figure 3.

P2 receptor agonists (K0.5 in nM for activation of turkey P2Y1 receptors is shown).
2-MethylthioATP was formerly regarded as selective for P2Y receptors; however, adjusting for its lability in classical smooth muscle assays in which P2X receptors have been assayed, it appears to be highly potent at the P2X superfamily as well [Kennedy and Leff, 1995]. Conversely, α,β-meATP is highly stable under such assay conditions and for this reason was previously described as the most potent P2X receptor agonist. Another potent P2X receptor agonist, 3’-benzylamino-3’-deoxyATP (Fig. 3), was synthesized at NIH [Burnstock et al., 1994].
[35S]ATP-γS has been used as a radioligand [Michel et al., 1996] for the direct labeling of several subtypes of cloned P2X receptors (P2X1–P2X4) expressed in CHO cells by use of the Semliki forest virus expression system. The affinities for antagonists determined in these binding assays were similar to those obtained in functional assays. Among agonist competitors, 2-meSATP was more potent than α,β-meATP at all four subtypes. It appears that the radioligand is labeling a desensitized high affinity state of the receptors, which is subject to positive allosteric modulation by antagonist cibachron blue and negative modulation by d-tubocurarine.
2-HexylthioAMP has proven to be a highly potent agonist (EC50 0.2 nM) at the yet uncloned P2Y receptor in C6 glioma cells that is coupled to inhibition of adenylate cyclase [Boyer et al., 1996]. Other bulky and long chain 2-alkylthio-ether derivatives of ATP [Fischer et al., 1993], such as 2-cycloheylthioATP, combine high potency and stability to ectonucleotidases. This compound was 420-fold selective for P2Y receptor of C6 glioma cells vs. turkey erythrocytes [Boyer et al., 1995]. 2-(4-Aminophenylethylthio)ATP is intended as a substrate for radioiodination and displays a potency of 1.5 nM at turkey erythrocyte P2Y1 receptors.
P2 Receptor Antagonists
The P2-receptor mediated effects of ATP may be antagonized by a number of highly negatively charged organic molecules of high molecular weight (Fig. 4). Among these substances are histochemical dyes, such as reactive blue 2 [Burnstock and Warland, 1987], which is not a chemically pure substance, but rather an isomeric mixture of cibachron blue and basilen blue. Reactive blue 2 is a particularly potent, competitive antagonist (IC50 = 30 nM) at the C6 glioma P2Y receptor [Boyer et al., 1994].
Figure 4.
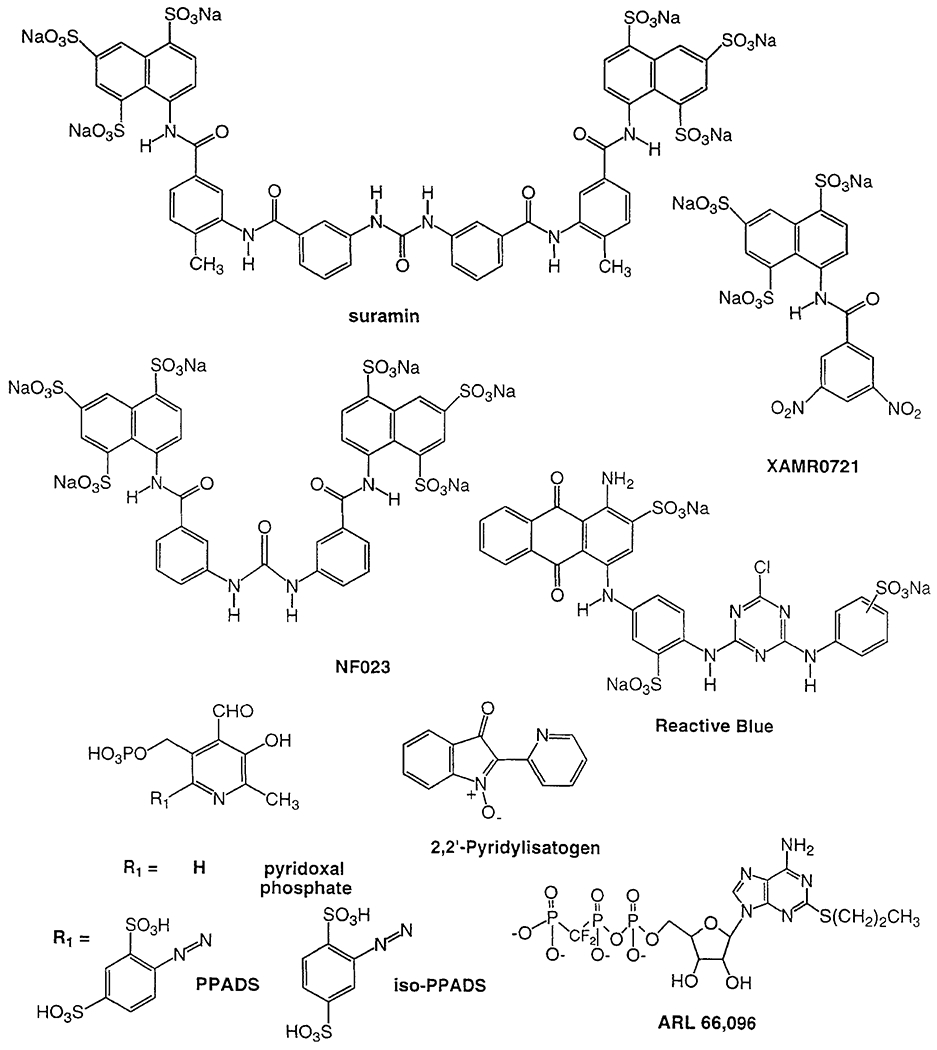
P2 receptor antagonists.
The symmetrical and polysulfonated, trypanocidal drug suramin is a weak inhibitor of the action of P2 agonists, and has been used in many studies of P2 receptors. At P2Y receptors [Charlton et al., 1996], the sensitivity to suramin occurs in the rank order of P2Y1 > P2Y2 > P2Y4. The suramin analogue XAMR0721, was found to antagonize P2 receptors without inhibiting ectonucleotidases [van Rhee et al., 1994]. Another truncated derivative of suramin, NF023, however, appears to be selective for P2X receptors and antagonizes effects at these receptors in rat, hamster, and rabbit isolated blood vessels [Ziyal et al., 1996]. Unfortunately suramin suffers from a lack of both selectivity and competitive antagonism at P2 receptors. The affinity of both suramin and NF023 in inhibiting G proteins was reported to be submicromolar, i.e., at concentrations lower than those normally used for blocking P2 receptors [Freissmuth et al., 1996].
A diazo derivative of the coenzyme pyridoxal phosphate, pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic acid (PPADS), was recently shown to be an ATP antagonist at smooth muscle P2X and P2Y receptors [Windscheif et al., 1995]. PPADS was found to be 10- to 20-fold selective for P2X receptors. PPADS was demonstrated to antagonize ATP-agonist effects at several P2Y receptors, including P2Y1 receptors and the cAMP-inhibitory receptor on rat C6 glioma cells [Boyer et al., 1994]. Initially, no distinction was made between PPADS and isoPPADS (pyridoxalphosphate-6-azophenyl-2’ ,5’-disulfonic acid); however, they have distinct pharmacological profiles [e.g., Connolly, 1995].
ARL-66,096 was found to be an inhibitor of the P2T receptor found on platelets and is in clinical development as an anti-thrombotic agent [Humphries et al., 1996]. This yet uncloned receptor is generally activated by ADP derivatives and antagonized by ATP derivatives [Cusack and Hourani, 1990]. 2,2’-Pyridylisatogen [PIT, Spedding et al., 1996] has been found to have both P2-antagonistic (at concentrations > 1 μM) and positive allosteric properties at P2Y1 receptors expressed in xenopus oocytes (at concentrations > 0.1 μM).
Footnotes
The P1 receptors include exclusively all of the A1–3 adenosine receptor subtypes. P2 receptors, activated by either or both adenine and uridine nucleotide derivatives and previously termed purinoceptors, are distinguished using two separate nomenclature systems. The pharmacologically defined subtypes are termed P2X, P2Y, P2U, P2t, P2Z, and P2d. Many subtypes of P2 receptors are now cloned, and these are termed either P2X1-7 (for ligand-gated ion channels) or P2Y1-7 (for G protein-coupled) receptors. The correspondence of the cloned to the pharmacologically defined P2 subtypes is only partially elucidated.
REFERENCES
- Abbracchio MP Brambilla R, Ceruti S, Kim HO, von Lubitz DKJE, Jacobson KA, Cattabeni F (1995): G-protein-dependent activation of phospholipase-C by adenosine A3 receptors in rat-brain. Mol Pharmacol 48:1038–1045. [PubMed] [Google Scholar]
- Ali II, Choi OH, Fraundorfer PF Yamada K, Gonzaga HMS, Beaven M (1996): Sustained activation of phospholipase D via adenosine A3 receptors is associated with enhancement of antigen- and Ca2+-ionophore-induccd secretion in a rat mast cell line. J Pharmacol Exp Ther 276:837–845. [PubMed] [Google Scholar]
- Baraldi PG, Cacciari B, Spalluto G, Borioni A, Viziano M, Dionisotti S, Ongini E (1995): Current developments of A2a adenosine receptor antagonists. Curr Med Chem 2:707–722. [Google Scholar]
- Baraldi PG, Cacciari B, Spalluto G, Ji XD, Olah ME, Stiles G, Dionisotti S, Zocchi C, Ongini E, Jacobson KA (1996a): Novel N6-(substituted-phenylcarbamoyl)adenosine-5’-uronamides as potent agonists for A3 adenosine receptors. J Med Chem 39:802–806. [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Cacciari B, Spalluto G, Pineda de Las Infantas MJ, Zocchi C, Dionisotti S, Ongini E (1996b): Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives. Potent and selective A2A adenosine antagonists. J Med Chem 39:1164–1171. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Zhang Y, Scammells PJ, Olsson R, Dennis D, Milner B Pfister J, Baker SP (1995): 1,3-dipropyl-8-[2-(5,6-epoxy)norbornyl]xanthine, a potent, specific and selective A1 adenosine receptor antagonist in the guinea-pig heart and brain and in DDT1MF-2 cells. J Pharmacol Exp Ther 275:1167–1176. [PubMed] [Google Scholar]
- Bowler AN, Olsen UB, Thomsen C, Knutsen LJS (1996): New adenosine A3 ligands controlling cytokines. Drug Dev Res 37:173. [Google Scholar]
- Boyer JL, Zohn IE, Jacobson KA, Harden TK (1994): Differential effects of P2-purinoceptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y-purinoceptors. Br J Pharmacol 113:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, O’Tuel J, Fischer B, Jacobson KA, Harden TK (1995): Potent agonist action of 2-thioether derivatives of adenine nucleotides at adenylyl cyclase-linked P2Y purinoceptors. Brit J Pharmacol 116:2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Siddiqi S, Fischer B, Romera-Avila T, Jacobson KA, Harden TK (1996): Identification of potent P2Y purinoceptor agonists that are derivatives of adenosine 5’-monophosphate. Br J Pharmacol 118:1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AJ, Bruns RF Ortwine DF Priebe SR, Szotek DL, Trivedi BK (1988): N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl]-adenosine and its uronamide derivatives. Novel adenosine agonists with both high affinity and high selectivity for the adenosine A2 receptor. J Med Ghem 31:1282–1285. [DOI] [PubMed] [Google Scholar]
- Burnstock G, (1996) A unifying purinergic hypothesis for the initiation of pain. Lancet 347:1604–1605. [DOI] [PubMed] [Google Scholar]
- Burnstoek G, Warland JJ (1987): P2-purinoccptors of two sulrtypes in the rabbit mesenteric artery: Reactive Blue 2 selectively inhibits responses mediated via the P2Y- but not the P2X-purinoceptor. Br J Pharmacol 90:383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G, Fischer B, Hoyle CHV, Maillard M, Ziganshin AU, Brizzolara AL, von Isakovics A, Boyer JL, Harden TK, Jacobson KA (1994): Structure-activity-relationships for derivatives of adenosines-5’-triphosphate as agonists at P2 purinoceptors: Heterogeneity within P2X and P2Y subtypes. Drug Dev Res 31:206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton S, Brown CA, Boarder MR (1996): Suramin and PPADS antagonists at transfected P2Y1, P2Y2, P2Y3, and P2Y4 receptors. Drug Dev Res 37:113. [Google Scholar]
- Connolly GP (1995) Differentiation by pyridoxal 5-phosphate, PPADS and isoPPADS between responses mediated by UTP and those evoked by α,β-methylene-ATP on rat sympathetic-ganglia. Br J Pharmacol 114:727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristalli G, Camaioni E, Vittori S, Volpini R, Borea PA, Conti A, Dionisotti S, Ongini E, Monopoli A (1995): 2-Aralkynyl and 2-heteroalkynyl derivatives of adenosine-5’-N-ethyluronamide as selective A2a adenosine receptor agonists. J Med Chem 38:1462–1472. [DOI] [PubMed] [Google Scholar]
- Cusack NJ, Hourani SMO (1990): Structure activity relationships for adenine nucleotide receptors on mast cells, human platelets, and smooth muscle. In Jacobson KA, Daly JW, Manganiello V (eds): Purines in Cellular Signalling: Targets for New Drugs. New York: Springer, pp. 254–259. [Google Scholar]
- Dunwiddie TV, Diao L, Kim HO, Jacobson KA (1996): Activation of hippocampal adenosine A3 receptors produces a heterologous desensitization of A1 receptor mediated responses in rat hippocampus. J Neurosci (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filtz TM, Harden TK, Nicholas RA (1997): Structure, pharmacological selectivity and second messenger properties of G protein coupled P2 purinergic receptors. In Jacobson KA, Jarvis MF (eds): Purinergic Approaches in Experimental Therapeutics. New York: Wiley; (in press). [Google Scholar]
- Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight CE, Zimmet J, Burnstock G, Harden TK, Jacobson KA (1993): Identification of potent, selective P2Y-purinoceptor agonists: Structure-activity-relationships for 2-thioether derivatives of adenosine 5’-triphosphate. J Med Chem 36:3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P Friedmann RC, Atkins C, Warren V Furness P Hyun JL, et al. (1988): Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J Med Chem 31:1014–1020. [DOI] [PubMed] [Google Scholar]
- Freissmuth M, Boehm S, Beindl W, Nickel P IJzerman AP Hohenegger M, Nanoff C (1996): Suramin analogues as subtype selective G protein inhibitors. Mol Pharmacol 49:602–611. [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA (1994): Structure-activity-relationships of N6-benzyladenosine-5’-uronamides as A3-selective adenosine agonists. J Med Chem 37:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hide I, Padgett WL, Jacobson KA, Daly JW (1992): A2A adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: Characterization with radioligand binding and by activation of adenylate cyclase. Mol Pharmacol 41:352–359. [PMC free article] [PubMed] [Google Scholar]
- Humphries RG, Leff P Robertson MJ (1996): P2T-purinoceptor antagonists: A novel class of anti-thrombotic agents. Drug Dev Res 37:175. [Google Scholar]
- Hutchison AJ, Webb RL, Oei HH, Ghai GR, Zimmerman MB, Williams M (1989): CGS 21680C, an A2 selective adenosine receptor agonist with preferential hypotensive activity. J Pharmacol Exp Ther 251:47–55. [PubMed] [Google Scholar]
- Jacobson KA, van Galen PJM, Williams M (1992a): Perspective. Adenosine receptors: Pharmacology, structure-activity relationships and therapeutic potential. J Med Chem 35:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Ji X-D, Berkich DA, Eveleth D, Dean RL, Hiramatsu KI, Kassell NF van Galen PJM, Lee KS, Bartus R, Daly JW, LaNoue KF Maillard M (1992b): Synthesis and biological activity of N6-p-sulfophenylalkyl- and derivatives of adenosine: Water soluble and peripherally selective adenosine agonists. J Med Chem 35:4143–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gallo-Rodriguez C, Melman N, Fischer B, Maillard M, van Bergen A, van Galen PJM, Karton Y (1993): Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J Med Chem 36:1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kim HO, Siddiqi SM, Olah ME, Stiles G, von Lubitz DKJE (1995): A3 adenosine receptors: Design of selective ligands and therapeutic prospects. Drugs Future 20:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M, Chakravarty PK, Johnson RG, Norton R (1996): Novel selective non-xanthine A3 adenosine receptor antagonists. Drug Dev Res 37:131. [Google Scholar]
- Ji XD, Melman N, Jacobson KA (1996): Interactions of flavonoids and other phytochemicals with adenosine receptors. J Med Chem 39:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JL, van Rhee AM, Melman N, Ji XD, Jacobson KA (1996) 6-Phenyl-1,4-dihydropyridine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem 39:4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karton Y, Jiang JL, Ji XD, Melman N, Olah ME, Stiles GL, Jacobson KA (1996): Synthesis and biological activities of flavonoid derivatives as A3 adenosine receptor antagonists. J Med Chem 39:2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy C, Leff P (1995): How should P2X purinoceptors be classified pharmacologically. Trends Pharmacol Sci 16:168–174. [DOI] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Melman N, Olah ME, Stiles GL, Jacobson KA (1994a): Structure-activity-relationships of 1,3-dialkylxanthine derivatives at rat A3 adenosine receptors. J Med Chem 37:3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA (1994b): 2-Substitution of N6-benzyladenosine-5’-uronamides enhances selectivity for A3 adenosine receptors. J Med Chem 37:3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Melman N, Olah ME, Stiles GL, Jacobson KA (1994c): Selective ligands for rat A3 adenosine receptors: Structure-activity-relationships of 1,3-dialkylxanthine 7-riboside derivatives. J Med Chem 37:4020–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Ji XD, Jacobson KA (1996): Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39:4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz KN, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M (1989): 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]CCPA): A high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedeberg Arch Pharmacol 340:679–683. [DOI] [PubMed] [Google Scholar]
- Knutsen LJS, Lau J, Petersen HJ, Thomsen C, Weis JU, Shalmi M, Judge ME, Hansen J, Sheardown MJ (1997): Novel neuroprotective adenosine agonists with diminished hypotensive effects. J Med Chem (in press). [DOI] [PubMed] [Google Scholar]
- Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA (1996): Induction of apoptosis in HL-60 human promyelocytic leukemia cells by selective adenosine A3 receptor agonists. Biochem Biophys Res Commun 219:904–910. Correction in Biochem Biophys Res Commun (1996) 221:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowski ER, Watt WC, Stutts MJ, Brown HA, Boucher RC, Harden TK (1996): Enzymatic-synthesis of UTP-γ-S, a potent hydrolysis resistant agonist of P2u-purinoceptors. Br J Pharmacol 117:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Miller KJ, Buell G, Lundström K, Humphrey PPA (1996): Direct labeling of P2X purinoceptor subtypes. Drug Dev Res 37:113. [Google Scholar]
- Müller CE, Hipp J, Knoblauch B, Schobert U, Sauer R, Ceis U (1996): DMPX (3,7-dimethyl-l-propargylxanthine) derivatives: Structure activity relationships of potent selective A2a-adenosine receptor antagonists. Drug Dev Res 37:112. [Google Scholar]
- Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL (1994): 125I-4-aminobenzyl-5’-N-methylcarboxamido-adenosine, a high-affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol 45:978–982. [PMC free article] [PubMed] [Google Scholar]
- Palmer TM, Poucher SM, Jacobson KA, Stiles GL (1995): 125I-4-(2-[7-amino-2-(2-furyl)(1,2,4)triazolo(2,3-a)(1,3,5)triazin-5-yl-amino]ethyl)phenol, a high-affinity antagonist radiolig and selective for the A2a, adenosine receptor. Mol Pharmacol 48:970–974. [PMC free article] [PubMed] [Google Scholar]
- Poucher SM, Keddie JR, Singh P Stoggall SM, Caulkett PWR, Jones G, Collis MG (1995): The in-vitro pharmacology of ZM241385, a potent, nonxanthine, A2a selective adenosine receptor antagonist. Br J Pharmacol 115:1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelen H, Veldman N, Spek AL, von Frijtag Drabbe Künzel J, Mathot RAA, IJzerman AP (1996): N6,C8-Disubstituted adenosine derivatives as partial agonists for adenosine A1 receptors. J Med Chem 39:1463–1471. [DOI] [PubMed] [Google Scholar]
- Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG (1993): Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci USA 90:10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK (1990): 4-Amino[1,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J Med Chem 33:2240–2254. [DOI] [PubMed] [Google Scholar]
- Schingnitz G, Küfner-Mühl U, Ensinger H, Lehr E, Kuhn FJ (1991): Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides 10:1067–1076. [Google Scholar]
- Shimada J, Suzuki F, Nonaka H, Ishii A, Ichikawa S (1992): (E)-1,3-Dialkyl-7-methyl-8-(3,4,5-Trimethoxystyryl)xanthines: Potent and selective adenosine-A2 antagonists. J Med Chem 35:2342–2345. [DOI] [PubMed] [Google Scholar]
- Siddiqi SM, Pearlstein RA, Sanders LH, Jacobson KA (1995): Comparative molecular-field analysis of selective A3 adenosine receptor agonists. Bioorg Med Chem 3:1331–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi SM, Ji XD, Melman N, Olah ME, Jain R, Evans P Glashofer M, Padgett WL, Cohen LA, Daly JW, Stiles GL, Jacobson KA (1996): A survey of nonxanthine derivatives as adenosine receptor ligands. Nucleosides Nucleotides 15:693–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedding M, King BF Dacquet C, Vanhoutte PM, Burnstock G (1996): Specific modulation of P2 receptors by 2,2’-pyridylisatogen (PIT). Drug Dev Res 37:174. [Google Scholar]
- Suzuki F Shimada J, Mizumoto H, Karasawa A, Kubo K, Nonaka H, Ishii A, Kawakita T (1992): Adenosine-A1 antagonists. 2. Structure-activity relationships on diuretic activities and protective effects against acute renal failure. J Med Chem 35:3066–3075. [DOI] [PubMed] [Google Scholar]
- Thompson RD, Secunda S, Daly JW, Olsson RA (1991): N6,9-Disubstituted adenines: Potent, selective antagonists at the A1-adenosine receptor. J Med Chem 34:2877–2882. [DOI] [PubMed] [Google Scholar]
- van der Wenden EM, Price SL, Apaya RR IJzerman AP Soudijn W (1995): Relative binding orientations of adenosine-A1 receptor ligands: A test-case for distributed multipole analysis in medicinal chemistry. J Comp Aided Mol Design 9:44–54. [DOI] [PubMed] [Google Scholar]
- van Galen PJM, Nissen P van Wijngaarden I, IJzerman AP Soudijn W (1991): 1H-imidazo[4,5-c]quinolin-4-amines: Novel non-xanthine adenosine antagonists. J Med Chem 34:1202–1206. [DOI] [PubMed] [Google Scholar]
- van Rhee AM, van der Heijden MPA, Beukers MW, IJzerman AP Soudijn W, Nickel P (1994): Novel competitive antagonists for P2 purinoceptors. Eur J Pharmacol 268:1–7. [DOI] [PubMed] [Google Scholar]
- van Rhee AM, Siddiqi SM, Melman N, Shi D, Padgett WL, Daly JW, Jacobson KA (1996a): Tetrahydrobenzothiophenone derivatives as a novel class of adenosine receptor antagonists. J Med Chem 39:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhee AM, Jiang J-I, Melman N, Olah ME, Stiles GL, Jacobson KA (1996b): Interaction of 1,4-dihydropyridine and pyridine derivatives with adenosine receptors: Selectivity for A3 receptors. J Med Chem 39:2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittori S, Camaioni E, Di Francesco E, Volpini R, Klotz KN, Lohse MJ, Cristalli G (1996a): Adenosine receptor antagonists: New purine derivatives. Drug Dev Res 37:113. [Google Scholar]
- Vittori S, Camaioni E, Di Francesco E, Volpini R, Monopoli A, Dionisotti S, Ongini E and Cristalli G (1996b): 2-Alkenyl and 2-alkyl derivatives of adenosine and adenosine-5’-N-ethyluronamide: Different affinity and selectivity of E- and Z-diastereomers at A2A adenosine receptors. J Med Chem 39:4211–4217. [DOI] [PubMed] [Google Scholar]
- Wagner H, Milaveckrizman M, Gadient F Menninger K, Schoeffter P Tapparelli C, Pfannkuche HJ, Fozard JR (1995): General pharmacology of SDZ WAG-994, a potent selective and orally-active adenosine A1 receptor agonist. Drug Dev Res 34:276–288. [Google Scholar]
- Windscheif U, Pfaff O, Ziganshin AU, Hoyle CHV, Bäumert HG, Mutschler E, Burnstock G, Larnbrecht G (1995): Inhibitory action of PPADS on relaxant responses to adenine-nucleotides or electrical-field stimulation in guinea-pig taenia-coli and rat duodenum. Br J Pharmacol 115:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O (1992): Molecular cloning and characterization of an adenosine receptor: The A3 adenosine receptor. Proc Natl Acad Sci USA 89:7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyal R, Ralevic V, Ziganshin AU, Nickel P Adanuy U, Mutschler E, Lambrecht G, Burnstock G (1996): NF023, a selective P2X-purinoeeptor antagonist in rat hamster and rabbit isolated blood vessels. Drug Dev Res 37:113. [Google Scholar]