

Abstract
Interleukin 2 (IL)‐2 induces antitumor immunity and clinical responses in melanoma and renal cell carcinoma. However, IL‐2 also increases the number of CD4+CD25+ regulatory T (Treg) cells that suppress antitumor immune responses. The aim of the present study was to elucidate the effect of depletion of Treg cells on IL‐2‐induced antitumor immunity. IL‐2‐transfected mouse colon adenocarcinoma (MC38/IL‐2) cells were implanted subcutaneously or intrahepatically into male C57BL/6 mice, and tumor growth and the proportion of tumor‐infiltrating lymphocytes with Treg‐cell depletion in response to treatment with anti‐CD25 monoclonal antibody (PC61) were determined. In mice treated with phosphate‐buffered saline, 40–60% of MC38/IL‐2 tumors were rejected. In contrast, all MC38/IL‐2 tumors were rejected in mice treated with PC61. The number of tumor‐infiltrating CD8+ T cells in mice treated with PC61 was approximately twice that in mice treated with PBS. The numbers of tumor‐infiltrating CD4+ and natural killer cells were also increased significantly. To test the antimetastatic effects of IL‐2 treatment in combination with Treg‐cell depletion, human recombinant IL‐2 (rIL‐2) and PC61 were administered to mice implanted with MC38/mock cells in the spleen, and hepatic metastasis was investigated. The average liver weight in mice treated with rIL‐2 plus PC61 was 1.04 ± 0.03 g, less than that in mice treated with rIL‐2 (2.04 ± 0.51 g) or PC61 alone (1.81 ± 0.38 g). We conclude that IL‐2‐induced antitumor immunity is enhanced by Treg‐cell depletion and is due to expansion of the tumor‐infiltrating cytotoxic CD8+ T‐cell population. (Cancer Sci 2007; 98: 416–423)
Interleukin‐2 (IL)‐2 was first identified in 1976, and a cDNA clone of IL‐2 was first isolated in 1987.( 1 , 2 ) A number of studies regarding antitumor immunity conferred by IL‐2 have since been conducted. Although it has been shown that IL‐2 can induce activation of lymphokine‐activated killer cells and can activate potent antitumor immunity in mice, high‐dose IL‐2 treatment for patients with melanoma or renal cell carcinoma is limited due to toxicity.( 3 , 4 ) Low‐dose IL‐2 therapy and combination therapies of IL‐2 with chemotherapy or interferon have been attempted for the treatment of colorectal cancer, but no clinical trials have shown increased survival with these therapies compared to that with high‐dose IL‐2 therapy.( 5 , 6 , 7 )
CD4+CD25+ regulatory T (Treg) cells play critical roles in immunological tolerance to self‐antigens and in suppression of antitumor immune responses.( 8 ) A previous study showed that depletion of Treg cells with an anti‐CD25 monoclonal antibody (mAb) resulted in the development of tumor‐specific effector cells and caused tumor regression in mice.( 9 ) Furthermore, established tumors were eliminated in response to a multimodal therapy consisting of Treg‐cell depletion, vaccination and local tumor radiation in a mouse model.( 10 ) In cancer patients, it was reported that Treg‐cell depletion enhanced vaccine‐mediated antitumor immunity.( 11 )
In the present study, we found that IL‐2 induced antitumor immunity against mouse colon adenocarcinoma cells and that the population of tumor‐infiltrating CD4+CD25+Foxp3+ T cells was slightly expanded in tumors in mice treated with IL‐2. To clarify the effects of Treg‐cell depletion on IL‐2‐induced antitumor immunity, tumor growth and the proportion of tumor‐infiltrating lymphocytes (TIL) were examined. We found that depletion of Treg cells enhanced IL‐2‐induced antitumor immunity and that further expansion of tumor‐infiltrating CD4+, CD8+ and natural killer cells occurred in tumors in mice treated with IL‐2 plus anti‐CD25 mAb. Furthermore, systemic therapy with recombinant IL‐2 (rIL‐2) plus anti‐CD25 mAb reduced the occurrence of hepatic metastasis.
Materials and Methods
Mice and tumor cells. Male C57BL/6 (B6) mice (5–7 weeks old) were purchased from Japan SLC (Hamamatsu, Japan). All animal experiments were conducted according to the guidelines of the Committee on Animals of Gifu University Graduate School of Medicine. MC38, a mouse colon adenocarcinoma cell line, was kindly provided by Dr Alan B. Frey (New York University School of Medicine, New York, NY, USA). Tumor cells were cultured in RPMI‐1640 medium containing 10% fetal calf serum (FCS), l‐glutamate (Invitrogen, Carlsbad, CA, USA) and penicillin–streptomycin (Invitrogen) and maintained at 37°C in a humidified 5% CO2 atmosphere. Establishment of the IL‐2‐transfected MC38 cell line was reported previously.( 12 ) In brief, a mouse IL‐2 cDNA generated from splenocyte total RNA was amplified and inserted into pcDNA3.1/Hygro (Invitrogen). Purified plasmids and control vector (pcDNA3.1) were introduced into MC38 cells, and stable transfectants (MC38/IL‐2 and MC38/mock cells) were obtained. The rate of proliferation was similar between MC38/mock and MC38/IL‐2 cells in vitro. IL‐2 in the culture supernatant was assessed by enzyme‐linked immunosorbent assay; 7.8 ± 0.6 ng/mL IL‐2 was detected in the supernatant of MC38/IL‐2 cells (1 × 106 cells/well cultured in a 24‐well plate for 24 h), but none was found in that of MC38/mock cells.
Flow cytometric analysis. The following antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA, USA) and used for flow cytometric analysis: biotin‐labeled anti‐CD25 (7D4), allophycocyanin (APC) labeled anti‐CD3e (145‐2C11), anti‐CD25 (PC61), Fluorescein isothiocyanate (FITC)‐labeled anti‐CD4 (RM4‐5), anti‐CD8b.2 (53‐5.8), Phycoerythrin (PE) labeled anti‐CD4 (RM4‐5), anti‐CD8b.2 (53–5.8), anti‐NK‐1.1 (PK136), anti‐CD25 (PC61), anti‐CD122 (5H4), anti‐CD132 (4G3), streptavidin–PE, and streptavidin–APC. A PE anti‐Foxp3 staining set (eBioscience, San Diego, CA, USA) was used to stain intracellular Foxp3, and Via‐Probe (BD Biosciences Pharmingen) was used to discriminate viable from non‐viable cells. Immunofluorescence was analyzed with CellQuest software with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Immunohistochemical analysis. Tumors were embedded in Tissue‐Tek OCT compound (Miles, Elkhart, IN, USA) and frozen in liquid nitrogen. Sections were fixed with acetone (Wako Pure Chemical Industries, Osaka, Japan) at 4°C for 10 min followed by Fc‐receptor blocking with purified antimouse CD16/32 (eBioscience) at 4°C for 30 min. To examine tumor‐infiltrating CD8+ T cells, specimens were stained with FITC‐labeled anti‐CD8b.2 and PE‐labeled anti‐F4/80 (CI:A3‐1; Caltag Laboratories, Burlingame, CA, USA). Tumor‐infiltrating NK cells were stained with antiasialo GM1 primary antibody (Wako Pure Chemical Industries) and biotin‐labeled goat antirabbit secondary IgG (Wako Pure Chemical Industries). They were then stained with streptavidin–FITC (BD Biosciences Pharmingen) and PE‐labeled anti‐F4/80. After a wash in phosphate‐buffered saline (PBS), specimens were examined with a Leica DMRA microscope (Leica Microsystems, Wetzlar, Germany). Sections were then stained with hematoxylin and eosin and examined with the same microscope.
Subcutaneous and intrahepatic implantation of tumor cells, preparation of TIL, and depletion of Treg, CD8+ and NK cells. For subcutaneous implantation, MC38/mock and MC38/IL‐2 cells (5 × 106 cells) were injected subcutaneously, and the tumor area, calculated as the product of the longest and shortest diameters, was measured every other day with a micrometer caliper. For intrahepatic implantation, mice were anesthetized, and the liver was exposed through a small abdominal incision. Tumor cells (1 × 106 cells) were injected directly with a 10‐µL Hamilton syringe (Hamilton Company, Reno, NV, USA).
Cells were prepared as described elsewhere.( 12 ) Collected tumors were minced and digested, and total cells in tumor suspension were stained with Via‐Probe, APC‐labeled anti‐CD3e, and PE‐labeled anti‐CD4, anti‐CD8b.2 or anti‐NK‐1.1, and the percentage of CD4+CD3+, CD8+CD3+ or NK‐1.1+CD3− cells was analyzed by flow cytometry. To examine the characteristics of TIL, these cells were isolated from the tumor cell suspension using MACS separation columns (Miltenyi Biotec, Bergisch Gladbach, Germany) after incubation with CD4 or CD8a MicroBeads (Miltenyi Biotec).
Anti‐CD25 (PC61, PC61 5.3), anti‐CD8a (53‐6.72) and anti‐NK‐1.1 mAb (PK136) were purified from the culture supernatant of a hybridoma cell line (American Type Culture Collection, Manassas, VA, USA). These antibodies (0.1 mg) were injected intraperitoneally every 3 days beginning 6 days before tumor cell implantation until the day of analysis, and depletion of specific subsets in the spleen was confirmed by flow cytometry (data not shown).
Cytotoxicity assay. Tumor‐infiltrating CD8+ T cells isolated on MACS separation columns were cultured in RPMI‐1640 medium containing 10% FCS overnight at 37°C with 5% CO2 and were used as effector cells. MC38 cells (1 × 106 cells) were labeled with 3.7 MBq Na2CrO4 for 1 h at 37°C with 5% CO2 and were used as target cells. These effector and target cells were combined at various ratios in 96‐well U‐bottom plates and incubated for 5 h at 37°C with 5% CO2. After incubation, supernatants were collected, and radioactivity was quantified with a γ counter. Spontaneous release was determined by incubation of target cells in the absence of effector cells, and maximum release was determined by incubation of target cells in 0.1% Triton X‐100. Specific cytotoxicity was calculated as follows:
| 51Cr release (%) = 100 × (cpm experimental − cpm spontaneous)/(cpm maximum − cpm spontaneous). |
Anti‐metastatic effects of IL‐2 and anti‐CD25 mAb. Human rIL‐2 was kindly provided by Shionogi and Co. (Osaka, Japan). After induction of anesthesia and exposure of the spleen through an abdominal incision, MC38/mock cells (1 × 106 cells) were injected intrasplenically on day 0. PC61 (0.1 mg) was administered intraperitoneally on days 7, 10, 13 and 16, and rIL‐2 (5 × 104 IU) was administered daily by intraperitoneal injection on days 8–16. PBS was administered instead of PC61 or rIL‐2 as a vehicle control. On day 17, the weights of spleens and livers were measured.
Statistical analysis. Differences were evaluated statistically with Student's t‐test, and P < 0.05 was considered significant.
Results
Numbers of tumor‐infiltrating CD4+CD25+ and CD8+ T cells are increased in MC38/IL‐2‐treated tumors. Subcutaneously implanted MC38/IL‐2 cells induced an antitumor response, and 40–60% of mice showed tumor rejection, whereas the remaining mice showed no rejection and tumors grew (data not shown). Mice implanted with MC38/mock cells showed no antitumor response. Similarly, about 50% of mice implanted intrahepatically with MC38/IL‐2 cells showed complete tumor rejection within 28 days.
To investigate immunological responses to IL‐2 in the tumor environment, we examined the proportions of tumor‐infiltrating CD4+ and CD8+ T cells in MC38/mock and MC38/IL‐2 tumors (Fig. 1a). Although the percentage of tumor‐infiltrating CD8+ T cells in MC38/IL‐2 tumors on day 11 after implantation was greater than that in MC38/mock tumors (P = 0.0196), that of tumor‐infiltrating CD4+ T cells was similar to that in MC38/mock tumors (P = 0.4994). Expansion of tumor‐infiltrating CD8+ T cells was confirmed by immunohistochemical analysis (Fig. 1b). The percentage of CD25+ cells among CD4+ T cells was greater in MC38/IL‐2 than in MC38/mock tumors (P = 0.0143), and these CD4+CD25+ T cells were predominantly positive for Foxp3 (Fig. 1c). To evaluate the advantageous antitumor effects of CD8+ T cells in combination with the disadvantageous effects of Treg cells, the ratio of CD8+ : CD4+Foxp3+ cells was examined. The average ratio of CD8+ : CD4+Foxp3+ cells was greater in MC38/IL‐2 than in MC38/mock tumors (7.77 ± 0.59 vs 4.48 ± 0.56, P < 0.0001). Although the percentage of CD25+Foxp3+ cells among CD4+ T cells was significantly greater in MC38/IL‐2 than in MC38/mock tumors (P = 0.0127), that of CD25+Foxp3+ cells among CD4+ T cells was not significantly greater in MC38/IL‐2 tumors (P = 0.0572), and the average ratio of CD4+CD25+Foxp3+ : CD4+CD25+Foxp3− cells in MC38/mock and MC38/IL‐2 tumors was 3.50 ± 1.11 and 2.26 ± 0.26, respectively (P = 0.2002, n = 3). The percentage of CD122+ cells among CD4+ T cells was greater in MC38/IL‐2 than in MC38/mock tumors (54.3 ± 5.3% vs 44.9 ± 4.8%, P = 0.0144). The percentage of CD132+ cells among CD4+ T cells was similar between MC38/mock and MC38/IL‐2 tumors (40.5 ± 4.3% vs 36.1 ± 7.6%, P = 0.2868). The percentage of CD25+ cells among tumor‐infiltrating CD8+ T cells was less than 10% in both MC38/mock and MC38/IL‐2 tumors (data not shown). These results suggest that IL‐2‐induced antitumor immunity is associated with tumor infiltration of CD8+ T cells. Although tumor‐infiltrating CD4+ T cells were predominantly Treg cells and these Treg cells were slightly increased in tumors in mice treated with IL‐2, it appears that IL‐2 has a more potent effect on non‐Treg cells than on Treg cells.
Figure 1.
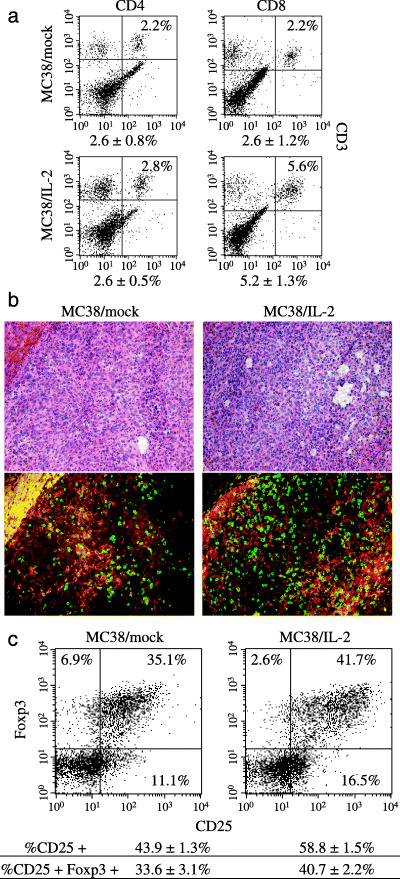
Infiltration of CD4+ and CD8+ T cells into MC38/mock and MC38/interleukin (IL)‐2 tumors in our intrahepatic implantation model. (a) On day 11 after intrahepatic implantation of 1 × 106 cells, the percentages of tumor‐infiltrating CD4+ (CD4+CD3+) and CD8+ (CD8+CD3+) T cells were examined by flow cytometry. One representative and the mean percentages ± SD for five different tumors are shown. (b) Immunohistochemical analysis of tumor‐infiltrating CD8+ T cells in MC38/mock and MC38/IL‐2 tumors. Tumor specimens on day 11 after implantation were stained with anti‐CD8b.2 antibody (green) and anti‐F4/80 antibody (red) (magnification, ×200). The peripheral area of each tumor section was evaluated for tumor‐infiltrating CD8+ T cells, and a hematoxylin and eosin‐stained image was also analyzed. (c) On day 11 after intrahepatic implantation of 1 × 106 cells, CD4+ T cells were separated from the tumor suspension of five mice. CD25 and Foxp3 expression in these CD4+ T cells was analyzed by flow cytometry. One representative and the mean percentages ± SD of CD25+ and CD25+Foxp3+ cells for three different experiments are shown.
Treg‐cell depletion enhances the IL‐2‐induced antitumor effect. We hypothesized that tumor‐infiltrating Treg cells would inhibit the effective induction of antitumor immunity and the therapeutic effect of IL‐2, and that depletion of Treg cells would enhance the antitumor effect of IL‐2. When MC38/IL‐2 cells were implanted subcutaneously, 40–60% of mice treated with PBS showed complete rejection of tumors (Fig. 2a). However, tumors in mice that did not show rejection grew. When PC61 was administered to mice implanted with MC38/mock cells, tumor growth was suppressed slightly from day 7 to day 11, whereas all tumors continued to grow after day 11. When PC61 was administered to mice implanted with MC38/IL‐2 cells, all mice showed complete tumor rejection. Results for intrahepatic implantation were similar to those for subcutaneous implantation; all mice implanted with MC38/IL‐2 cells and treated with PC61 showed complete tumor rejection on day 28 after implantation (Fig. 2b). These results suggest that depletion of Treg cells contributes to the suppression of tumor growth and that this depletion enhances the IL‐2‐induced antitumor effect.
Figure 2.

Tumor growth after subcutaneous or intrahepatic implantation of MC38/mock and MC38/interleukin (IL)‐2 cells in mice treated with anti‐CD25 monoclonal antibody. (a) MC38/mock or MC38/IL‐2 cells (5 × 106 cells) were implanted subcutaneously, and the area of each tumor was calculated every other day. PC61 and phosphate‐buffered saline (PBS) were administered every 3 days beginning 6 days before implantation. Values for individual mice are shown (n = 5 in each group). Similar results were obtained in two independent experiments. (b) MC38/IL‐2 cells (1 × 106 cells) were implanted intrahepatically, and PC61 and PBS were administered as in (a). On days 7, 11, 14, 21 and 28 after implantation, the area of each tumor was calculated. The mean ± SD is shown (n = 10 in each experiment). *P < 0.05.
Numbers of tumor‐infiltrating CD4+ and CD8+ T cells are further increased after Treg‐cell depletion in MC38/IL‐2 tumors. To evaluate the effect of Treg‐cell depletion on IL‐2‐induced antitumor immunity in the tumor environment, the proportions of tumor‐infiltrating CD4+ and CD8+ T cells were examined. The percentage of tumor‐infiltrating CD8+ T cells on day 11 in MC38/IL‐2 tumors treated with PC61 was greater than that in MC38/IL‐2 tumors treated with PBS (P = 0.0454; Fig. 3a); this expansion of tumor‐infiltrating CD8+ T cells was confirmed by immunohistochemical analysis (Fig. 3b). The cytotoxic effect of these tumor‐infiltrating CD8+ T cells was similar, regardless of the type of tumor cell or the presence of Treg‐cell depletion (Fig. 3c). In addition, these tumor‐infiltrating CD8+ T cells did not have cytotoxic effects on B16 melanoma cells, and splenic CD8+ T cells from normal mice did not have cytotoxic effect on MC38 cells (data not shown). The percentage of tumor‐infiltrating CD4+ T cells in MC38/IL‐2 tumors in mice treated with PC61 was also greater than that in MC38/IL‐2 tumors in mice treated with PBS (P = 0.0007; Fig. 3a). Tumor‐infiltrating CD4+CD25+ T cells were predominantly depleted in MC38/IL‐2 tumors treated with PC61, whereas some Foxp3+ T cells, which were predominantly negative for CD25, were not depleted (Fig. 3d). The average ratio of CD8+ : CD4+Foxp3+ cells was greater in MC38/IL‐2 tumors in mice treated with PC61 than in mice treated with PBS (34.95 ± 8.14 vs 9.58 ± 2.18, P = 0.0003). These results suggest that depletion of Treg cells enhances IL‐2‐induced antitumor immunity mediated by further expansion of tumor‐infiltrating CD4+ and CD8+ T cells, and that the CD4+ T cells are predominantly CD4+CD25− T cells.
Figure 3.

Tumor infiltration of CD4+ and CD8+ T cells into MC38/interleukin (IL)‐2 tumors in mice treated with anti‐CD25 monoclonal antibody. (a) On day 11 after intrahepatic implantation of 1 × 106 MC38/IL‐2 cells, the percentages of tumor‐infiltrating CD4+ (CD4+CD3+) and CD8+ (CD8+CD3+) T cells were examined by flow cytometry. PC61 (0.1 mg) and phosphate‐buffered saline (PBS) were injected intraperitoneally every 3 days beginning 6 days before tumor cell implantation until the day of analysis. One representative and the mean percentages ± SD for five different tumors are shown. (b) Immunohistochemical analysis of tumor‐infiltrating CD8+ T cells in MC38/IL‐2 tumors in mice treated with PBS or PC61. Tumor specimens on day 11 after implantation were stained with anti‐CD8b.2 antibody (green) and anti‐F4/80 antibody (red) (magnification, ×200). The peripheral area of each tumor section was evaluated for tumor‐infiltrating CD8+ T cells, and a hematoxylin and eosin‐stained image was also analyzed. (c) 51Cr‐release assay. MC38 cells labeled with Na2CrO4 and tumor‐infiltrating CD8+ T cells isolated from tumor suspension were coincubated for 5 h at 37°C with 5% CO2. Radioactivity was quantified with a γ counter, and specific cytotoxicity was calculated. One representative of three different experiments is shown. (d) On day 11 after intrahepatic implantation, CD4+ T cells were separated from the tumor suspension of MC38/IL‐2 tumors collected from five mice treated with PC61. CD25 (7D4) and Foxp3 expression by these CD4+ T cells was analyzed by flow cytometry. One representative of three different experiments is shown.
NK cells are not important for the antitumor effect of IL‐2 plus anti‐CD25 mAb. The mean size of MC38/IL‐2 tumors in mice treated with PC61 was significantly larger on day 7 than in mice treated with PBS (26.1 ± 11.3 mm2 vs 14.8 ± 9.4 mm2, P = 0.0332; Fig. 2b). To determine the cause of this difference in tumor size, we investigated the histopathological characteristics of MC38/IL‐2 tumors and the proportion of tumor‐infiltrating NK cells on day 7 after implantation. Severe intratumoral edema was observed in most MC38/IL‐2 tumors in mice treated with PC61 (Fig. 4a). MC38/IL‐2 tumors in untreated mice also showed various degrees of intratumoral edema, which was generally mild compared to that in mice treated with PC61. The percentage of tumor‐infiltrating NK cells in MC38/IL‐2 tumors was greater than that in MC38/mock tumors (P = 0.0180; Fig. 4b), and that in MC38/IL‐2 tumors in mice treated with PC61 was greater than that in MC38/mock and MC38/IL‐2 tumors (P = 0.0015 and 0.0237, respectively). This expansion was confirmed by immunohistochemical analysis (Fig. 4c). To investigate the role of NK cells in antitumor immunity induced by IL‐2 plus Treg‐cell depletion, sizes of MC38/IL‐2 tumors after NK‐cell depletion were measured (Fig. 5). In MC38/IL‐2 tumors in mice treated with PBS or PC61, tumor size in NK‐cell‐depleted mice was significantly larger on day 7 after implantation than in non‐depleted mice (64.2 ± 22.4 mm2 vs 36.9 ± 5.7 mm2 and 65.9 ± 6.0 mm2 vs 31.4 ± 5.5 mm2; P = 0.0460 and P < 0.0001, respectively). In mice treated with PC61, all MC38/IL‐2 tumors were rejected after NK‐cell depletion. In contrast, in mice treated with PBS, only 20–40% of MC38/IL‐2 tumors were rejected after NK‐cell depletion. Interestingly, in NK‐cell‐depleted mice, the mean size of MC38/IL‐2 tumors on day 7 was similar between mice treated with PBS and mice treated with PC61 (65.9 ± 6.0 mm2 vs 64.2 ± 22.4 mm2, P = 0.8830). However, after CD8+ cell depletion, all MC38/IL‐2 tumors in mice treated with or without PC61 grew. These results suggest that CD8+ T cells play a critical role in the antitumor effect induced by IL‐2 plus anti‐CD25 mAb and that NK cells are relatively unimportant. We believe that it is inadequate to compare antitumor immunity on day 7 after implantation by means of MC38/IL‐2 tumor size because the degree of intratumoral edema may affect tumor size.
Figure 4.
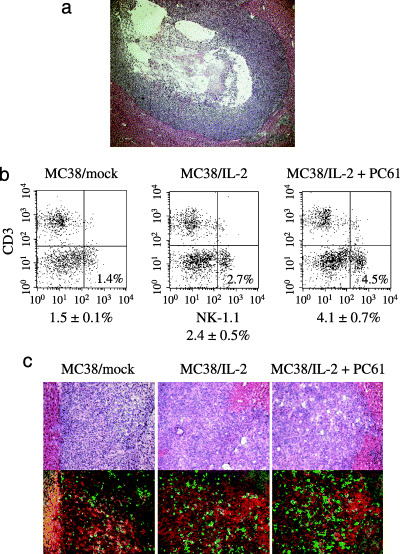
Analysis of tumor‐infiltrating natural killer (NK) cells. (a) Histopathology of a typical MC38/interleukin (IL)‐2 tumor in a mouse treated with PC61 on day 7 after intrahepatic implantation. Intratumoral edema is observed in the surrounding tumor tissue (hematoxylin and eosin; magnification, ×100). (b) MC38/IL‐2 cells (1 × 106 cells) were implanted into normal mice. On day 7 after intrahepatic implantation, the percentage of tumor‐infiltrating NK cells (NK‐1.1+CD3−) was examined by flow cytometry. One representative and the mean percentages ± SD for five different tumors are shown. (c) Immunohistochemical analysis. Tumor specimens were stained with antiasialo GM1 antibody (green) and anti‐F4/80 antibody (red) (magnification, ×200). The peripheral area of each tumor section was evaluated for tumor‐infiltrating NK cells, and a hematoxylin and eosin‐stained image was also analyzed.
Figure 5.

Effect of natural killer (NK) or CD8+ cell depletion on the growth of subcutaneous MC38/interleukin (IL)‐2 tumors in mice treated with anti‐CD25 monoclonal antibody (mAb). Anti‐NK‐1.1 mAb (PK136) or anti‐CD8a mAb (53–6.72) were injected intraperitoneally every 3 days beginning 6 days before subcutaneous implantation of 5 × 106 MC38/IL‐2 cells in mice treated with phosphate‐buffered saline (PBS) or PC61. PBS was administered instead of each monoclonal antibody as a vehicle control. The area of each tumor was calculated every other day, and the values for individual mice are shown (n = 5 in each group). Similar results were obtained in two independent experiments.
Proliferation of rechallenged MC38/mock cells is inhibited in mice that rejected MC38 tumors when treated with IL‐2 plus anti‐CD25 mAb. To examine immunological memory in Treg‐cell‐depleted mice, mice with or without PC61 treatment that had rejected MC38/IL‐2 tumors were rechallenged with MC38/mock cells. MC38/mock cells were implanted at least 28 days after the last PBS or PC61 injection in the previous experiment; cells were also implanted in normal mice as a control. All mice that had rejected MC38/IL‐2 tumors and received PC61 treatment showed complete rejection of newly implanted MC38/mock cells, whereas rejection was not observed in mice that had rejected MC38/IL‐2 tumors and received PBS treatment, or in control mice (Fig. 6). These results suggest that potent antitumor immunity is retained when mice reject tumor cells in response to IL‐2 and anti‐CD25 mAb treatment.
Figure 6.

Tumor growth after rechallenge of mice that had rejected subcutaneous MC38/interleukin (IL)‐2 tumors in mice treated with PC61. Normal mice and mice with or without PC61 treatment that had rejected MC38/IL‐2 tumors were rechallenged subcutaneously with 5 × 106 MC38/mock cells at a site distant from the initial implantation site at least 28 days after the last administration of phosphate‐buffered saline (PBS) or PC61. The area of each tumor was determined every other day, and the mean values for five mice are shown.
Systemic administration of rIL‐2 plus anti‐CD25 mAb inhibits hepatic metastasis of colon adenocarcinoma. To mimic a clinical scenario, the therapeutic efficacy of systemic administration of rIL‐2 plus anti‐CD25 mAb was evaluated in an intrasplenic implantation model. On day 17 after implantation, hepatic metastasis was clearly suppressed in mice treated with rIL‐2 plus PC61, and the average liver weight of these mice was less than that of mice treated with rIL‐2 alone (P = 0.0484) or PC61 alone (P = 0.0460) (Fig. 7). However, the average weight of primary tumors in the spleen was similar between groups (data not shown). These results suggest that rIL‐2 treatment in combination with Treg‐cell depletion effectively enhances the host immune response against hepatic metastasis.
Figure 7.

Systemic administration of recombinant interleukin (rIL)‐2 and anti‐CD25 monoclonal antibody after intrasplenic implantation of MC38/mock cells. (a) Average liver weight in each treatment group. On day 17 after implantation, liver weights were measured. The mean weight ± SD of livers from three different mice is shown. *P < 0.05. (b) Photographs of livers collected on day 17. Three livers from untreated mice (upper left) and mice treated with rIL‐2 alone (upper right), PC61 alone (lower left) and rIL‐2 plus PC61 (lower right) are shown.
Discussion
Recent studies have provided substantial evidence that elimination of Treg cells enhances antitumor immunity and improves the antitumor efficacy of cancer vaccines.( 9 , 10 ) How to manipulate Treg cells in cancer patients has been investigated. One way is to deplete Treg cells, and another way is to suppress Treg‐cell activation. Anti‐CD25 mAb is suitable for depletion of CD4+CD25+ Treg cells in mice; most CD4+CD25+ T cells were positive for Foxp3 and were considered to be Treg cells in the present study. This mAb is also useful in that it has no effect on CD25− cells, such as most CD8+ T and NK cells, and it does not directly suppress the effect of other IL‐2 cytokine family members such as IL‐21.( 13 ) Clinically, ONTAK (IL‐2 conjugated to diphtheria toxin) has been approved for depleting CD25+ T cells and its success in treating cancer patients has been documented.( 11 ) However, Attia et al. reported an inability of ONTAK to eliminate Treg cells in melanoma patients and suggested that repeated exposure to ONTAK generates antibodies directed against the diphtheria toxin.( 14 ) LMB‐2 (anti‐CD25 mAb conjugated to bacterial Pseudomonas toxin A) has been reported to specifically decrease human CD25+ Treg cells in vitro and is likely to enter clinical trials.( 15 ) We believe that it is difficult to selectively deplete human Treg cells with anti‐CD25 mAb because CD25+ Treg cells in humans comprise a small population of CD4+CD25+ T cells, unlike in mice.( 16 , 17 ) Recent studies targeting the immunoregulatory molecules expressed on the surface of Treg cells to suppress their activation have been reported, and clinical trials on human malignancies have been carried out. Cytotoxic T lymphocyte‐associated antigen (CTLA)‐4 is integral to the regulatory function of Treg cells, and anti‐CTLA‐4 blocking antibody has been used to effect tumor elimination in humans, although it also induces autoimmune disease.( 18 ) Glucocorticoid‐induced tumor necrosis factor receptor family related protein (GITR) is also expressed constitutively at high levels on Treg cells, and agonistic anti‐GITR mAb abrogates the suppressive effects of Treg cells.( 19 ) However, a recent study showed that this effect on vaccine‐induced CD8+ T‐cell responses was partially independent of Treg cells, consistent with a direct costimulatory effect on effector CD8+ cells themselves.( 20 ) Development of a mAb that can selectively deplete or inactivate Treg cells and further investigation of surface molecules that control Treg‐cell function are necessary.
In the present study, we showed that Treg‐cell depletion enhances IL‐2‐induced antitumor immunity and that this results in further expansion of cytotoxic CD8+ T cells. The concept for this combination therapy resulted from our data showing that the number of tumor‐infiltrating Treg cells slightly increased in response to IL‐2 treatment. It has also been shown that the number of Treg cells was increased in cancer patients who received IL‐2 treatment.( 21 ) IL‐2 plays a critical role in the development of tolerance rather than immunity and is essential for the survival of mature Treg cells; however, it is not essential for the intrathymic development of Treg cells.( 8 , 22 ) Thus, it is generally believed that selective inhibition of IL‐2‐mediated expansion of Treg cells may improve the therapeutic effectiveness of IL‐2.( 21 ) It was recently reported that high‐dose IL‐2 treatment resulted in a significant decrease of Treg cells in those patients achieving an objective clinical response to IL‐2 therapy, and it was suggested that high‐dose IL‐2 may induce qualitatively different responses from those of low‐dose IL‐2, or that polymorphisms in immune response genes may mediate diverse responses to IL‐2 in individual patients.( 23 ) These mechanisms are not mutually exclusive and future studies are necessary. IL‐2 also potently activates CD4+ non‐Treg cells, and CD4+CD25+Foxp3+ T cells are increased in MC38/IL‐2 tumors. Despite the depletion of these activated non‐Treg cells as well as Treg cells with anti‐CD25 mAb, anti‐CD25 mAb enhanced IL‐2‐induced antitumor immunity, indicating that Treg cells are detrimental to the induction of an effective antitumor response. There has been only one study investigating the antitumor effect of IL‐2 treatment in combination with Treg‐cell depletion in mice. That study concluded that antitumor activity was eliminated by coadministration of IL‐2 and anti‐CD25 mAb, although anti‐CD25 mAb or IL‐2 administration suppressed tumor growth in a mouse model of renal cell carcinoma.( 24 ) It is true that anti‐CD25 mAb may have a detrimental effect on CD8+CD25+ activated T cells, but these effects should be minimal because the percentage of CD25+ cells among tumor‐infiltrating CD8+ T cells was low in our experiments. Differences in the tumor cells used, the IL‐2 treatment method, or the susceptibility of tumor cells to IL‐2 may also be involved. In humans, treatment metastatic melanoma patients with IL‐2 plus anti‐CTLA‐4 mAb has been investigated.( 25 ) Although there is no evidence to support a synergistic effect, long‐term cancer regression was observed in patients undergoing combined treatment. In our hepatic metastasis model, IL‐2 plus anti‐CD25 mAb treatment did not completely inhibit hepatic metastasis and had no effect on primary tumors. For patients with metastasis, it will be necessary to combine additional treatments with IL‐2 plus Treg‐cell depletion.
Potent antitumor immunity is retained by mice that reject tumor cells in response to IL‐2 and anti‐CD25 mAb treatment. How Treg‐cell depletion increases the memory response is an active area of exploration and was not examined in the present study. It has been suggested that IL‐2 inhibits proliferation of CD8+ memory T cells at least in part by stimulating the survival and perhaps function of Treg cells.( 26 ) Therefore, Treg‐cell depletion may also ameliorate the unfavorable effect of IL‐2 in terms of immunological memory.
We showed that CD4+CD25+Foxp3+ T cells accounted for 30–40% of tumor‐infiltrating CD4+ T cells. In patients with ovarian cancer, high Foxp3 expression or a low CD8+ : CD25+Foxp3+ cell ratio in the tumor is suggested to be associated with poor prognosis.( 27 , 28 ) Our results showed a similar tendency; the average ratio of CD8+ : CD4+Foxp3+ cells in MC38/IL‐2 tumors was greater than that in MC38/mock tumors, and that in MC38/IL‐2 tumors in Treg‐cell depleted mice was greater than that in MC38/IL‐2 tumors in non‐depleted mice. These findings indicate that the CD8+ : Treg ratio of tumor‐infiltrating T cells is an important parameter that reflects the potency and efficacy of antitumor immune response. Recently, the mechanisms underlying the increase in tumor‐infiltrating Treg cells have been investigated. Transforming growth factor‐β (TGF‐β) released by tumor cells or tumor‐recruited immature myeloid dendritic cells has been reported to induce Treg‐cell proliferation.( 29 ) In addition, the main mechanism of Treg‐cell expansion in tumor hosts has been reported to be conversion of CD4+CD25− T cells to CD4+CD25+ T cells.( 30 ) Elucidation of the mechanisms whereby tumors induce Treg‐cell expansion will allow for the specific inhibition of tumor‐induced Treg‐cell expansion without adverse systemic events, such as autoimmune disease, which are associated with Treg‐cell depletion.
In the present study, systemic administration of IL‐2 did not inhibit the formation of hepatic metastasis. This is likely due to an inadequate dose of IL‐2. Although doses of IL‐2 of 1 × 104−105 IU/day are sufficient to induce antitumor effects in mouse models of renal cell carcinoma and melanoma, higher doses of IL‐2 (3 × 104−105 IU/day) are required in mouse models of colon adenocarcinoma.( 31 , 32 , 33 , 34 ) In humans, increased doses of IL‐2 may induce potent antitumor effects, but toxicity remains a problem. It has been shown that localized treatment of cancer with IL‐2, particularly intratumoral administration, is superior to systemic administration with respect to the occurrence of severe adverse side‐effects.( 35 , 36 ) Thus, how to deliver IL‐2 or how to maintain IL‐2 concentrations around the tumor site should be considered.
In addition to tumor‐infiltrating CD4+ and CD8+ T cells, NK cells were also increased in tumors in mice treated with IL‐2 plus Treg‐cell depletion compared with mice treated with IL‐2 alone. Although NK cells do not play a major role in IL‐2 plus Treg‐cell depletion, they may play an essential role in the early antitumor response. Previous studies have shown that Treg cells inhibit NK‐cell cytotoxicity and interferon‐γ production in a TGF‐β‐dependent manner and that Treg cells directly inhibit NKG2D‐mediated NK‐cell cytotoxicity.( 37 , 38 ) Despite the expansion of NK cells in MC38/IL‐2 tumors in Treg‐cell depleted mice, the larger size of MC38/IL‐2 tumors in Treg‐cell depleted mice compared with non‐depleted mice appears inconsistent with this concept. However, our histopathological observations suggested that intratumoral edema is an important factor with regard to tumor size in the early phase after implantation. NK cells are thought to be involved in IL‐2‐induced vascular leak syndrome, which is characterized by increased vascular permeability accompanied by extravasation of fluids and proteins from capillary vessels into tissues, resulting in interstitial edema, decreased microcirculatory perfusion, and organ damage.( 39 , 40 ) Thus, the severe intratumoral edema observed in MC38/IL‐2 tumors in Treg‐cell depleted mice may be attributable to the expansion of tumor‐infiltrating NK cells. The similar size of MC38/IL‐2 tumors on day 7 in Treg‐cell depleted mice and non‐depleted mice after NK‐cell depletion supports this concept. We speculate that the expansion of tumor‐infiltrating NK cells may be associated with both enlargement and reduction of tumors in the early phase of tumor implantation. Elucidation of the effects of Treg‐cell depletion on IL‐2‐induced adverse events is necessary to evaluate the safety and efficacy of this therapy with regard to vascular leak syndrome.
In conclusion, we showed that Treg‐cell depletion enhances IL‐2‐induced antitumor immunity against mouse colon adenocarcinoma and that IL‐2 treatment in combination with Treg‐cell depletion inhibits hepatic metastasis. However, the antitumor immunity induced by this combination therapy may be insufficient to eliminate tumors completely. Future studies will combine IL‐2 plus Treg‐cell depletion and chemotherapy, vaccines, or modalities that enhance the cytotoxicity of CD8+ T cells and improve methods of IL‐2 administration.
References
- 1. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976; 193: 1007–8. [DOI] [PubMed] [Google Scholar]
- 2. Taniguchi T, Matsui H, Fujita T et al. Structure and expression of a cloned cDNA for human interleukin‐2. Nature 1983; 302: 305–10. [DOI] [PubMed] [Google Scholar]
- 3. Rosenberg SA, Mule JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high‐dose recombinant interleukin 2. J Exp Med 1985; 161: 1169–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atkins MB, Lotze MT, Dutcher JP et al. High‐dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999; 17: 2105–16. [DOI] [PubMed] [Google Scholar]
- 5. Yang JC, Sherry RM, Steinberg SM et al. Randomized study of high‐dose and low‐dose interleukin‐2 in patients with metastatic renal cancer. J Clin Oncol 2003; 21: 3127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDermott DF, Regan MM, Clark JI et al. Randomized phase III trial of high‐dose interleukin‐2 versus subcutaneous interleukin‐2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 2005; 23: 133–41. [DOI] [PubMed] [Google Scholar]
- 7. Correale P, Cusi MG, Tsang KY et al. Chemo‐immunotherapy of metastatic colorectal carcinoma with gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte macrophage colony‐stimulating factor and interleukin‐2 induces strong immunologic and antitumor activity in metastatic colon cancer patients. J Clin Oncol 2005; 23: 8950–8. [DOI] [PubMed] [Google Scholar]
- 8. Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin‐2. J Immunother 2005; 28: 120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti‐CD25 (interleukin‐2 receptor alpha) monoclonal antibody. Cancer Res 1999; 59: 3128–33. [PubMed] [Google Scholar]
- 10. Kudo‐Saito C, Schlom J, Camphausen K, Coleman CN, Hodge JW. The requirement of multimodal therapy (vaccine, local tumor radiation, and reduction of suppressor cells) to eliminate established tumors. Clin Cancer Res 2005; 11: 4533–44. [DOI] [PubMed] [Google Scholar]
- 11. Dannull J, Su Z, Rizzieri D et al. Enhancement of vaccine‐mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest 2005; 115: 3623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tamakawa N, Saio M, Suwa T et al. Interleukin‐2 activated microglia engulf tumor infiltrating T cells in the central nervous system. Int J Mol Med 2004; 13: 497–503. [PubMed] [Google Scholar]
- 13. Comes A, Rosso O, Orengo AM et al. CD25+ regulatory T cell depletion augments immunotherapy of micrometastases by an IL‐21‐secreting cellular vaccine. J Immunol 2006; 176: 1750–8. [DOI] [PubMed] [Google Scholar]
- 14. Attia P, Maker AV, Haworth LR, Rogers‐Freezer L, Rosenberg SA. Inability of a fusion protein of IL‐2 and diphtheria toxin (Denileukin Diftitox, DAB389IL‐2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother 2005; 28: 582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Attia P, Powell DJ Jr, Maker AV, Kreitman RJ, Pastan I, Rosenberg SA. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB‐2. J Immunother 2006; 29: 208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baecher‐Allan C, Viglietta V, Hafler DA. Human CD4+CD25+ regulatory T cells. Semin Immunol 2004; 16: 89–98. [DOI] [PubMed] [Google Scholar]
- 17. Wing K, Suri‐Payer E, Rudin A. CD4+CD25+‐regulatory T cells from mouse to man. Scand J Immunol 2005; 62: 1–15. [DOI] [PubMed] [Google Scholar]
- 18. Phan GQ, Yang JC, Sherry RM et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte‐associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 2003; 100: 8372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological selftolerance. Nat Immunol 2002; 3: 135–42. [DOI] [PubMed] [Google Scholar]
- 20. Cohen AD, Diab A, Perales MA et al. Agonist anti‐GITR antibody enhances vaccine‐induced CD8(+) T‐cell responses and tumor immunity. Cancer Res 2006; 66: 4904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmadzadeh M, Rosenberg SA. IL‐2 administration increases CD4+CD25hiFoxp3+ regulatory T cells in cancer patients. Blood 2006; 107: 2409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. D’Cruz LM, Klein L. Development and function of agonist‐induced CD25+FoxP3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol 2005; 6: 1152–9. [DOI] [PubMed] [Google Scholar]
- 23. Cesana GC, DeRaffele G, Cohen S et al. Characterization of CD4+CD25+ regulatory T cells in patients treated with high‐dose interleukin‐2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol 2006; 24: 1169–77. [DOI] [PubMed] [Google Scholar]
- 24. Takeuchi T, Konno‐Takahashi N, Kasuya Y, Ogushi T, Nishimatsu H, Kitamura T. Interleukin‐2 blocks the antitumour activity caused by depletion of CD25 cells in a murine renal adenocarcinoma model. BJU Int 2004; 94: 171–6. [DOI] [PubMed] [Google Scholar]
- 25. Maker AV, Phan GQ, Attia P et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte‐associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol 2005; 12: 1005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murakami M, Sakamoto A, Bender J, Kappler J, Marrack P. CD25+CD4+ T cells contribute to the control of memory CD8+ T cells. Proc Natl Acad Sci USA 2002; 99: 8832–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolf D, Wolf AM, Rumpold H et al. The expression of the regulatory T cell‐specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res 2005; 11: 8326–31. [DOI] [PubMed] [Google Scholar]
- 28. Sato E, Olson SH, Ahn J et al. Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 2005; 102: 18 538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghiringhelli F, Puig PE, Roux S et al. Tumor cells convert immature myeloid dendritic cells into TGF‐β‐secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med 2005; 202: 919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor‐induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res 2006; 66: 4488–95. [DOI] [PubMed] [Google Scholar]
- 31. Masumori N, Tsukamoto T, Kumamoto Y. Interferon‐γ and interleukin‐2 suppress the experimental metastasis of mouse renal adenocarcinoma. Int J Urol 1995; 2: 6–11. [DOI] [PubMed] [Google Scholar]
- 32. Nakajima I, Chu TM. Synergistic antitumor activity of interleukin‐2 and cimetidine against syngeneic murine tumor. Cancer Immunol Immunother 1991; 33: 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodolfo M, Salvi C, Bassi C, Parmiani G. Adoptive immunotherapy of a mouse colon carcinoma with recombinant interleukin‐2 alone or combined with lymphokine‐activated killer cells or tumor‐immune lymphocytes. Survival benefit of adjuvant post‐surgical treatments and comparison with experimental metastases model. Cancer Immunol Immunother 1990; 31: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bernsen MR, Tang JW, Everse LA, Koten JW, Den Otter W. Interleukin 2 (IL‐2) therapy: potential advantages of locoregional versus systemic administration. Cancer Treat Rev 1999; 25: 73–82. [DOI] [PubMed] [Google Scholar]
- 35. Jacobs JJ, Sparendam D, Den Otter W. Local interleukin 2 therapy is most effective against cancer when injected intratumourally. Cancer Immunol Immunother 2005; 54: 647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barnard AL, Farzaneh F, Gaken J, Darling D. Local versus systemic interleukin‐2. tumor formation by wild‐type and B7‐1‐positive murine melanoma cells. Cancer Gene Ther 2000; 7: 207–14. [DOI] [PubMed] [Google Scholar]
- 37. Ghiringhelli F, Menard C, Terme M et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor‐β‐dependent manner. J Exp Med 2005; 202: 1075–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+CD25+ T regulatory cells suppress NK cell‐mediated immunotherapy of cancer. J Immunol 2006; 176: 1582–7. [DOI] [PubMed] [Google Scholar]
- 39. Baluna R, Vitetta ES. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology 1997; 37: 117–32. [DOI] [PubMed] [Google Scholar]
- 40. Assier E, Jullien V, Lefort J et al. NK cells and polymorphonuclear neutrophils are both critical for IL‐2‐induced pulmonary vascular leak syndrome. J Immunol 2004; 172: 7661–8. [DOI] [PubMed] [Google Scholar]