

Abstract
1. Alzheimer’s disease (AD) is pathologically defined by the deposition of amyloid peptide and neurofibrillary tangles and is characterized by a progressive loss of cognition and memory function, due to marked cortical cholinergic depletion.
2. Cholinergic cortical innervation is provided by basal forebrain cholinergic neurons. The neurotrophin Nerve Growth Factor (NGF) promotes survival and differentiation of basal forebrain cholinergic neurons.
3. This assertion has been at the basis of the hypothesis developed in the last 20 years, whereby NGF deprivation would be one of the factor involved in the etiology of sporadic forms of AD.
4. In this review, we shall summarize data that lead to the production and characterization of a mouse model for AD (AD11 anti-NGF mice), based on the expression of transgenic antibodies neutralizing NGF. The AD-like phenotype of AD11 mice will be discussed on the basis of recent studies that have posed NGF and its precursor pro-NGF back to the stage of AD-like neurodegeneration, showing the involvement of the precursor pro-NGF in one of the cascades leading to AD neurodegeneration.
KEY WORDS: Alzheimer’s disease, nerve growth factor, TrkA, P75NTR, sortilin, transgenic mice, neurodegeneration
Alzheimer’s disease (AD) is a progressive, incurable disease representing the major cause of dementia in the elderly. Two forms of AD exist, a familial one (multiple family members are affected) and a sporadic one, in which one or a few members of a family have the disease (Selkoe, 2001). From the neuropathological point of view, the presence of two characteristic hallmarks defines the disease: plaques of β-amyloid protein and neurofibrillary tangles, mainly constituted of paired helical filaments of abnormally phosphorylated tau proteins (Selkoe, 2001). As the disease progresses, neuronal death appears. In particular, cholinergic neurons of the basal forebrain are lost (Bartus et al., 1982; Whitehouse et al., 1982), accounting for the development of cognitive impairments (Perry et al., 1978; Collerton, 1986; DeKosky et al., 1992).
For over a decade, the predominant viewpoint among researchers and clinicians has been that the deposition of β-amyloid is the primary etiological event in AD, a theory known as the “amyloid cascade hypothesis” (Golde, 2005). According to this hypothesis, β-amyloid would originate from the proteolytic cleavage the amyloid precursor protein (APP), and it would deposit and aggregate in extracellular insoluble forms to constitute plaques. These processes would cause neurotoxic damage and the consequent neurodegeneration characterizing AD. However, doubts can be raised about the reliability of the amyloid cascade hypothesis. One of the major issues rests in the experiments performed in transgenic mice, where the over-expression of mutated forms of APP linked to the familial forms of AD did not trigger the amyloid cascade (Selkoe, 2002). In the brain of these mice β-amyloid plaques can be found, but not tangles, suggesting that tau pathology, which is an essential part of the dementing process, does not depend on the altered processing of APP. Such observations led to argue that some common factors should induce both plaque and tangle formation. Nerve growth factor (NGF) might be one of these.
MATURE NGF AND ITS RECEPTORS
NGF is a highly conserved protein that was first identified in two sarcoma tissues (Levi-Montalcini and Hamburger, 1951) and in certain snake venoms (Cohen, 1959). Murine and human NGF genes code for two transcripts to produce 34 and 27 kDa precursor (Scott et al., 1983; Ullrich et al., 1983). These NGF precursors are cleaved by convertases such as furin and convertases 1 and 2 to give rise to mature processed NGF of 13.2 kDa (Seidah et al., 1996). When fully processed, NGF exists as a non-covalently bound homodimer (Stach and Shooter, 1974) and it is released through constitutive secretory pathways (Mowla et al., 1999). NGF acts through binding to two classes of cell surface receptors, the tyrosine kinase receptor A (TrkA) and p75NTR.
TrkA is a type I transmembrane protein member of the receptor tyrosine kinase superfamily. It selectively binds to NGF, with a dissociation constant (kd) equal to 10−11 M (Cordon-Cardo et al., 1991; Kaplan et al., 1991; Klein et al., 1991). Binding of the NGF homodimer causes receptor dimerization (Jing et al., 1992) followed by autophosphorylation on tyrosine residues within the activation loop and by phosphorylation of the seven intracellular tyrosine residues (Cunningham et al., 1997). These phosphorylated residues represent docking sites for signaling molecules which regulate cell growth and survival through different signaling pathways (Kaplan and Miller, 2000; Huang and Reichardt, 2003).
The second NGF receptor is represented by p75NTR (Chao et al., 1986; Johnson et al., 1986). It non-specifically binds to all neurotrophins with similar affinity (kd=10−9M) (Rodriguez-Tebar et al., 1990; Squinto et al., 1991). P75NTR is a type I transmembrane protein with an extracellular domain (ECD) constituted by four cysteine-rich repeated domains (CRDs) (Yan and Chao, 1991; Baldwin et al., 1992). CRD3 is the domain responsible for the interaction with neurotrophins (Yan and Chao, 1991; Baldwin et al., 1992; Chapman and Kuntz, 1995; Shamovsky et al., 1999). The intracellular domain (ICD) contains several regions that mediate the interaction with signaling elements (Roux and Barker, 2002).
NGF AND ALZHEIMER’S DISEASE: THE CLASSICAL VIEW
During the last 20 years, several scientific supports have been given to the hypothesis that NGF might be one of the factors involved in the cascade of events leading to AD. NGF maintains and regulates the cholinergic phenotype of basal forebrain neurons (Mobley et al., 1986; Li et al., 1995) through the retrograde transport of the NGF/TrkA signaling complex from the cortex and hippocampus, where NGF is produced, to the basal forebrain (Mufson et al., 1999). NGF protects cholinergic neurons, following age-related atrophy (Holtzman et al., 1993; Smith et al., 1999) and experimental surgical lesions (Koliatsos et al., 1990, 1991a,b; Hefti et al., 1993) and contribute to enhance memory in aged rodents (Fischer et al., 1987; Markowska et al., 1994, 1996).
In AD brains, the levels of NGF protein may be increased in the cortex and hippocampus (Crutcher et al., 1993; Scott et al., 1995; Fahnestock et al., 1996; Hellweg et al., 1998; Hock et al., 2000). By contrast, NGF protein levels in the basal forebrain are reduced (Mufson et al., 1994, 1995; Scott et al., 1995). The increased levels of NGF in the cortex and hippocampus have been correlated to a decreased expression of TrkA receptors (Mufson et al., 1996, 1997, 2000; Boissiere et al., 1997; Hock et al., 1998; Counts et al., 2004) which are necessary to transport NGF to the basal forebrain.
Thus, according to this classical view, the atrophy of cholinergic neurons in AD patients would be the consequence of a lack of trophic support due to the impairment of NGF retrograde transport system and the consequent accumulation of NGF protein in target areas (Mufson et al., 1999; Salehi et al., 2003).
A confirmation to this hypothesis could have been obtained from animal models. However, in no animal model a direct link to AD was found. A difficulty was also due to the fact that mice, in which NGF synthesis was deleted by gene homologous recombination (ngf −/− mice), die during the early postnatal period and do not allow to study effects of long term NGF deprivation (Crowley et al., 1994). Heterozygous NGF knockout mice can survive until adulthood but no signs of AD-like neurodegeneration can be observed, with the exception of a 20% loss of basal forebrain cholinergic neurons (Chen et al., 1997).
To overcome the problems encountered with ngf −/− mice, a new approach was used to neutralize NGF activity. The activity of proteins can be blocked using antibodies that can not only recognize a given protein but neutralize its activity (neuroantibody approach) (Cattaneo, 1998).
In the initial study, the efficiency of antibody secretion by different type of cells was analyzed and it was found that the secretion by neuronal and glial cells was very high, with efficiency comparable to that of lymphoid cells transfected with the same antibody genes (Cattaneo and Neuberger, 1987). Thus, after achieving the proof of principle by producing transgenic mice expressing recombinant antibodies neutralizing substance P (Piccioli et al., 1995), anti-NGF transgenic mice were generated (Ruberti et al., 2000).
AD11 ANTI-NGF MICE: THE αD11 ANTIBODY
AD11 anti-NGF mice express the recombinant version of the monoclonal antibody (mAb) αD11. This antibody was selected after immunization of rats with mouse NGF and neutralizes NGF activity by a direct competition between NGF itself and the TrkA receptor (Cattaneo et al., 1988). The epitope of mAb αD11 on NGF includes the loop region from residues 41–49, which contributes to the interaction surface between NGF and its high affinity receptor TrkA and distinguishes NGF from other members of the neurotrophin family (Ibanez et al., 1991). The neutralizing properties of mAb αD11 and the specificity of this inhibition were extensively proven both in vitro and in vivo. In the first set of analysis, αD11 prevented the NGF-induced elongation of processes in PC12 cells and inhibited the survival of neurons from dorsal root ganglia (Molnar et al., 1998). In the same experiment, mAb αD11 was not able to inhibit the pro-survival action of BDNF, NT-3 or NT-4 (Molnar et al., 1998). In vivo, the implantation of hybridoma cells secreting mAb αD11 induces the atrophy of NGF-dependent BFCNs in young rats (Molnar et al., 1998).
AD11 ANTI-NGF MICE: A COMPREHENSIVE MODEL FOR AD-LIKE NEURODEGENERATION
The starting point to obtain AD11 anti-NGF mice was to make it detectable against the mouse IgGs. For this purpose, the variable regions of the light and heavy chains of mAb αD11 were cloned and reassembled with the constant regions of the K and γ1 chains of human immunoglobulins (Ruberti et al., 1993). The chimeric αD11 antibody was placed under the transcriptional control of the early promoter region of human cytomegalovirus in two separate plasmids. The linearized DNA was then individually injected in mouse eggs (Ruberti et al., 2000).
Mice expressing the neutralizing antibody αD11 were obtained by crossing the line of mice expressing the light chain of the recombinant version of the αD11 antibody (AD11-VK mice) with mice expressing the heavy chain of the same recombinant antibody (AD11-VH mice) (Ruberti et al., 2000).
The two-tier approach used to obtain AD11 transgenic mice, allowed us to circumvent the effect of an early exposure to functional antibodies during the embryonic stage of development. Indeed, at embryonic day 13, transgenic antibodies are detectable but then their levels become undetectable in the prenatal and postnatal period during which NGF influences mouse development (Capsoni et al., 2000a). Only after postnatal day 45 the antibody levels reach values above the detection threshold (Capsoni et al., 2000a). The expression of the recombinant αD11 antibody allows neutralizing up to 50% of unbound NGF (Ruberti et al., 2000).
AD11 mice are characterized by a progressive neurodegeneration which resembles many features of AD. The characterization of the AD-like neurodegeneration in the brain of AD11 mice was performed using immunohistochemical and biochemical techniques. A detailed time course was performed to assess the presence of behavioral deficits, using different paradigms such as the object recognition test, the radial and the Morris water maze tests. As expected from previous studies on the effects of NGF deprivation in the basal forebrain (Molnar et al., 1997, 1998), AD11 mice are characterized by an atrophy and loss of cholinergic neurons in this brain region, that starts from 2 months of age remaining stable thereafter (Capsoni et al., 2000, 2002b) (Fig. 1(A) and (B)) and progresses until 6 months of age, remaining stable thereafter (Capsoni et al., 2002b) (Fig. 1(C)).
Fig. 1.

Progressive atrophy and loss of choline acetyltransferase-positive neurons in the basal forebrain of AD11 mice. (A) WT mice (B) 2-month-old and (C) 15-month-old AD11 mice. Scale bar = 50 μm.
In the cortex and hippocampus, NGF deprivation provokes a re-distribution of the phosphorylated form of the microtubule associated protein tau. In AD, phosphorylated tau is the main component of intracellular neurofibrillary tangles found in cell bodies, neuropil threads and dystrophic neurites (Binder et al., 2005; Iqbal et al., 2005). The phosphorylated state of tau influences the solubility of this protein which becomes insoluble and accumulates in form of fibrils (the so-called paired helical filaments (PHFs) (Iqbal et al., 2005). The regional progression of neurofibrillary accumulation is a characteristic of AD. Phosphorylated tau can be found first in transentorhinal and entorhinal regions of human brain (Braak and Braak, 1991). Interestingly, in 2-month-old AD11 mice the first accumulation of phosphorylated tau can also be found in the entorhinal region, spreading with age to other cortical and hippocampal areas (Capsoni et al., 2002b). At subcellular level, tau accumulates in neuronal perykarion and then in dystrophic neurites (Capsoni et al., 2000b, 2002b). From the biochemical point of view, aged AD11 mice show an accumulation of insoluble tau (Capsoni et al., 2000b, 2002b). Electron microscopy studies performed on insoluble brain extracts revealed that insoluble tau assembles in aggregates that morphologically resemble those found in human AD PHFs (Capsoni et al., 2002b) (Fig. 2).
Fig. 2.

Immuno electron microscopy demonstrating the presence of PHF-like insoluble aggregates of the microtubule associated protein tau in brains from (A) AD11 mice and (B) human AD. Black dots correspond gold particles.
In AD, abnormally phosphorylated tau provokes the disassembly of neuronal microtubules (Mandelkow et al., 1996; Alonso et al., 1997). Interestingly, in concomitance with appearance the progressive accumulation of phosphorylated tau, also in AD11 mice a re-distribution of neuronal microtubules can be observed in cortical areas (Capsoni et al., 2002b).
In AD11 mice, β-amyloid, which represents another principal endpoint of AD, is found in intracellular compartments, localized in MAP-2 positive dystrophic neurites of the hippocampus (Fig. 3), starting from 6 months of age (Capsoni et al., 2002b). In aged AD11 mice, β-amyloid is also found in extracellular, plaque-like deposits (Capsoni et al., 2002a) (Fig. 4). Interestingly, in AD11 mice, the deposition of β-amyloid occurs as a consequence of an altered processing of the endogenous β-amyloid precursor protein APP. This observation allows defining AD11 mice as a model for the sporadic form of AD, in opposition to other transgenic mice over-expressing mutated forms of human APP.
Fig. 3.
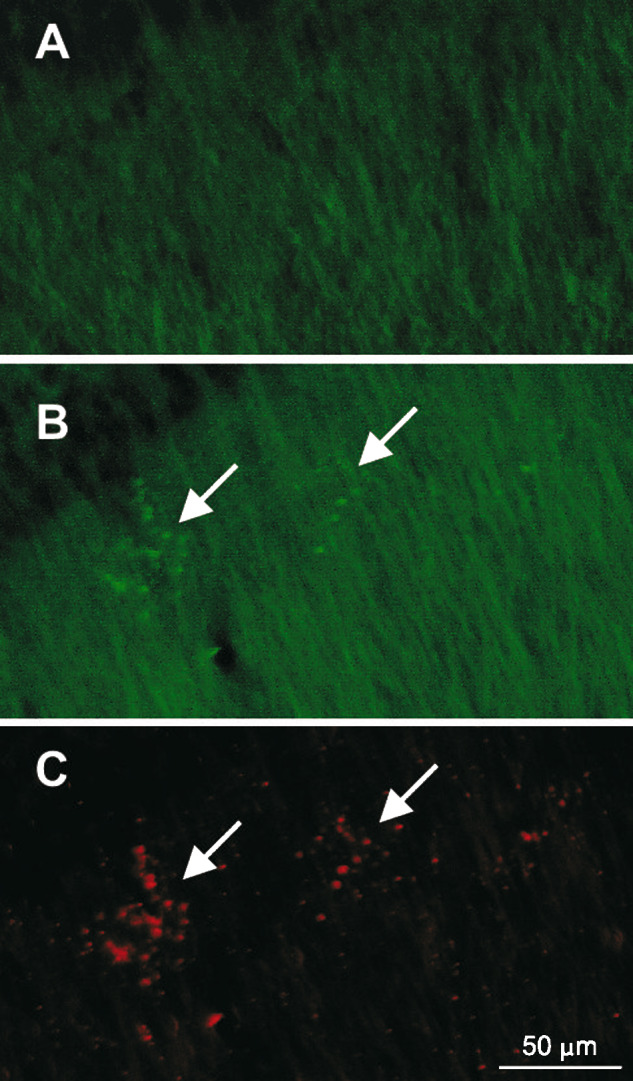
Microtubule associated protein 2 (MAP2) immunohistochemistry in (A) WT and (B) AD11 mouse demonstrating the presence of dystrophic neurites (green) in the radial layer of the AD11 mouse hippocampus. (C) These dystrophic neurites are positive for β-amyloid (red). Arrows point to clusters of dystrophic neurites. Scale bar = 50 μm.
Fig. 4.
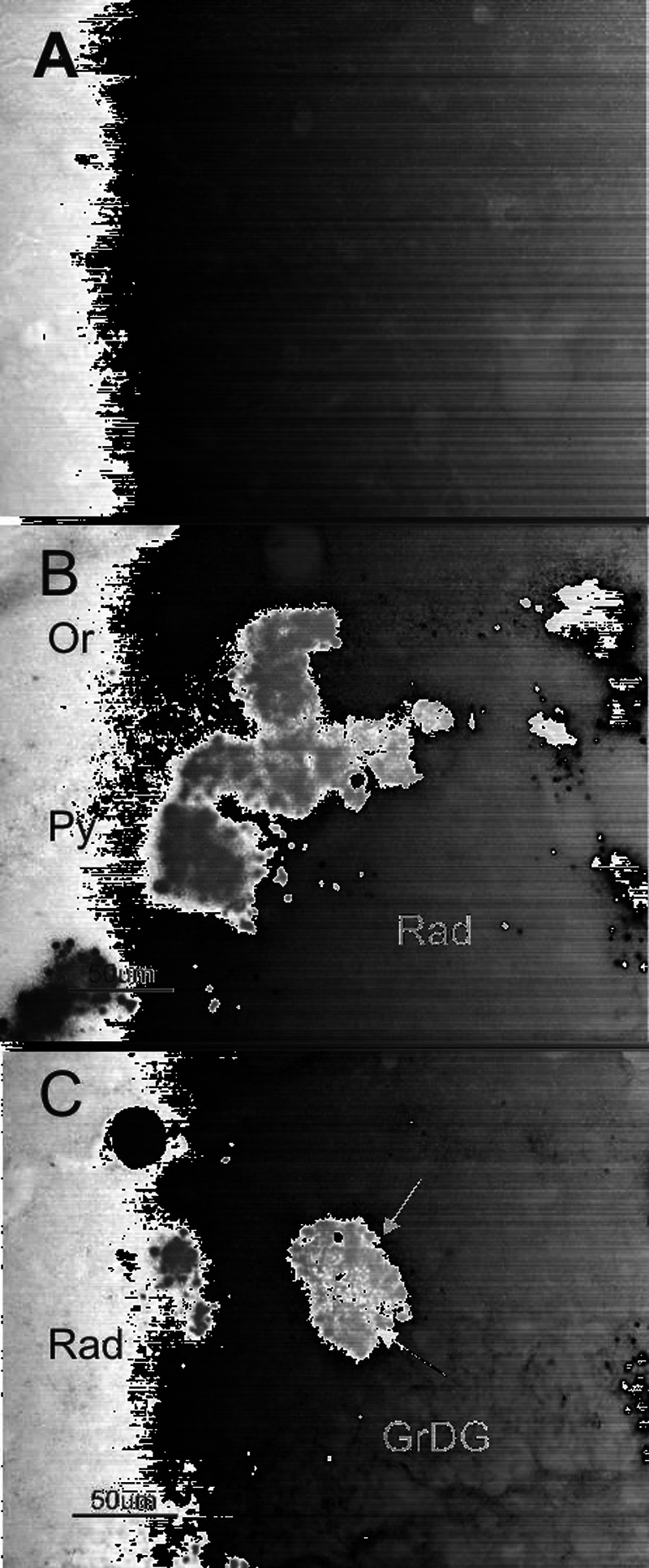
β-amyloid immunohistochemistry in 15-month-old (A) WT and (B, C) AD11 mouse brain demonstrating the absence of plaques in WT mice and the accumulation of extracellular deposits in AD11 mice. In panel (B), extracellular deposition is intermingled to dystrophic neurites while in (C) plaques are constituted by a core of extracellular material (red arrow) surrounded by dystrophic neurites (black arrow). GrDG = granule layer of dentate gyrus; Py = pyramidal layer; Or = oriens layer; rad = stratum radiatum of the hippocampus. Scale bar = 50 μm.
In AD11 mice, the neurodegeneration is accompanied by functional alterations that match with the appearance of the endpoints characterizing the neurodegeneration. Object recognition deficits appear early (at 4 months of age) soon after the onset of cholinergic deficits and tau accumulation (De Rosa et al., 2005). At 6 months of age, when β-amyloid intracellular deposits start to appear, AD11 mice show impairment of synaptic plasticity in the cortex (Pesavento et al., 2002) and hippocampus (E. Sola, S. Capsoni, A. Cattaneo, and E. Cherubini, unpublished data) and a progression of object recognition deficits (De Rosa et al., 2005). In aged AD11 mice, synaptic plasticity deficits are severe and can no more be reverted by cholinesterase inhibitors (N. Origlia, S. Capsoni, L. Domenici, and A. Cattaneo, unpublished data). Behavioral deficits are severe, with impairment in working and spatial memory (Capsoni et al., 2000b; Ruberti et al., 2000; De Rosa et al., 2005).
The complexity of the phenotype raised the doubt that only the effects of the local, decreased NGF signaling to basal forebrain neurons might not explain it.
Administering this neurotrophin to AD11 mice proved the fact that this neurodegeneration was specifically due to NGF deprivation. Since NGF, when injected peripherally, cannot cross the blood brain barrier, the classical approach used to deliver NGF to the brain is to directly inject NGF in brain parenchyma (Blesch and Tuszynski, 2004). In the case of AD11 mice, it was decided to apply an approach first discovered by Frey et al. whereby NGF can be delivered to the brain in pharmacologically relevant concentrations through non-invasive intranasal injections (Frey et al., 1997; Chen et al., 1998). In AD11 mice, the intranasal injections were performed at early and at moderate stages of neurodegeneration, characterized by a loss of cholinergic neurons, intracellular accumulation of hyperphosphorylated tau and β-amyloid and behavioral deficits (Capsoni et al., 2002c). In all cases, NGF was able to rescue the cholinergic deficit, the over-expression of hyperphosphorylated tau and beta amyloid (Capsoni et al., 2002c). In these experiments, testing the ability of AD11 mice to recognize new objects from the familiar ones assessed behavioral deficits. AD11 mice do not recognize the two types of objects, while wild-type mice explore more the new object. The intranasal administration of NGF rescued the object recognition deficits in AD11 mice (De Rosa et al., 2005). Thus, these experiments demonstrate that the AD-like neurodegeneration is due to alterations in NGF signaling. However, these experiments were inconclusive to clarify whether the rescue of the cholinergic phenotype was sufficient and essential to determine the rescue of the other relevant AD endpoints, such as phosphorylated tau and β-amyloid phenotype. Indeed, intranasal NGF might act not only on cholinergic neurons but also on other neuronal populations. The hint came from experiments during which the acetylcholinesterase inhibitors galantamine and donepezil were administered to AD11 mice (Capsoni et al., 2002c, 2004). Both drugs were able to rescue basal forebrain cholinergic neurons but neither of them determined the amelioration of the tau-related phenotype (Capsoni et al., 2000c, 2004). Thus, it might be concluded that the complex AD-like phenotype in AD11 mice was not a mere consequence of atrophy of basal forebrain cholinergic neurons, and a new hypothesis, linked to the new complexity of NGF signaling, had to be formulated.
NGF AND ALZHEIMER’S DISEASE: THE NEW PERSPECTIVE
Two recent findings changed the perspective of the involvement of NGF and p75NTR in AD and helped in formulate a new theory.
Fahnestock et al. performed the first observation. They found that the major form of NGF accumulating in cerebral cortex and hippocampus of AD brains is indeed the precursor pro-NGF (Fahnestock et al., 2001). The consequences of this accumulation might be explained by the second finding. In the past, it was a dogma thinking that the precursor pro-NGF was just a molecule devoid of signaling properties, with the mere function of being the precursor of mature NGF (Shooter, 2001). The only signaling properties taken into consideration for NGF-induced cell survival/death were those mediated by the TrkA receptors and p75NTR. While the first receptor is by definition the cell survival mediator (Fig. 5(A)), p75NTR has a twofold role. P75NTR increases the affinity and specificity of TrkA to NGF (Hempstead et al., 1991; He and Garcia, 2004) but it also mediates cell apoptosis, in absence of TrkA signaling (Fig. 5(A)) (Kaplan and Miller, 2000). Recently, it was reported that pro-NGF preferentially binds, in presence of a co-receptor belonging to the Vpss10p-domain receptors, sortilin, to p75NTR receptor (Nykjaer et al., 2004). The binding of pro-NGF to the p75NTR/sortilin complex determines cell death and apoptosis in neuronal cells (Fig. 5(B)) (Lee et al., 2001; Harrington et al., 2004).
Fig. 5.

Schematic representation of TrKA and p75NTR receptors. (A) According to the classical concept, P75NTR interacts with the TrkA receptor and increases its affinity for NGF, determining cell growth and survival. (B) The p75NTR/sortilin complex binds pro-NGF, determining cell death and neurodegeneration.
These findings prompted us to explore the possibility that the recombinant antibody αD11 preferentially binds to one of the two forms of NGF. Indeed, using plasmon resonance assays, it was found that mAb αD11 binds with picomolar affinity to mature NGF, while it has a very fast dissociation constant (and thus a very low affinity) for pro-NGF (F. Paoletti and A. Cattaneo, unpublished data). As a consequence, we hypothesized that the AD-like phenotype in AD11 mice could be due to the fact that, while mature NGF activity is neutralized by the recombinant anti-NGF antibody (Fig. 6(A)), pro-NGF is left free to interact with the p75NTR/sortilin complex (Fig. 6(B)). A first demonstration of the goodness of this hypothesis comes from the results obtained by crossing a line of mice expressing the recombinant anti-NGF antibody to p75 knockout mice, in which the ectodomain of the receptors has been ablated (Lee et al., 1992). Thus, we obtained a line of mice (AD12 mice) in which mature NGF is neutralized and pro-NGF cannot signal through p75NTR. The result obtained so far showed that, in AD12 mice, no β-amyloid intracellular deposits or extracellular plaques could be found (Fig. 7). Thus, the study of the interactions between pro-NGF/NGF and their receptors holds great promise as one of the keys to clarifying how they are involved in the signaling cascade leading to AD. As a consequence, new diagnostic and therapeutic perspective will be designed and developed in the attempt to cure AD.
Fig. 6.

Schematic diagram showing the effects of the αD11 antibody on pro-NGF/NGF signaling. (A) The αD11 antibody prevents binding of mature NGF to the TrKA/p75NTR complex determining a decrease in cell survival. (B) MAb αD11 does not bind pro-NGF that is free to interact with the p75NTR/sortilin complex, triggering neurodegeneration.
Fig. 7.

The graph shows that β-amyloid plaques are not detectable in AD12 mice in which the neutralizing antibody anti-NGF is still expressed, but the p75NTR receptor was knocked out (#P < 0.05 AD11 mice vs. WT and p75NTR knockout mice; * P < 0.05 AD12 mice vs. AD11 mice).
ACKNOWLEDGMENTS
The authors are grateful to Dr. Piero Giulio Giulianini (University of Trieste) for performing electron microscopy analysis of AD11 brain extracts and to Dr. Gabriele Ugolini for helpful discussions and Mrs. Lucia de Caprio for reading the manuscript.
REFERENCES
- Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U. S. A.94:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, A. N., Bitler, C. M., Welcher, A. A., and Shooter, E. M. (1992). Studies on the structure and binding properties of the cysteine-rich domain of rat low affinity nerve growth factor receptor (p75NGFR). J. Biol. Chem.267:8352–8359. [PubMed] [Google Scholar]
- Bartus, R. T., Dean, R. L., 3rd, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science217:408–414. [DOI] [PubMed] [Google Scholar]
- Binder, L. I., Guillozet-Bongaarts, A. L., Garcia-Sierra, F., and Berry, R. W. (2005). Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta1739:216–223. [DOI] [PubMed] [Google Scholar]
- Blesch, A., and Tuszynski, M. H. (2004). Gene therapy and cell transplantation for Alzheimer’s disease and spinal cord injury. Yonsei. Med. J.45(Suppl):28–31. [DOI] [PubMed] [Google Scholar]
- Boissiere, F., Hunot, S., Faucheux, B., Hersh, L. B., Agid, Y., and Hirsch, E. C. (1997). Trk neurotrophin receptors in cholinergic neurons of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord.8:1–8. [DOI] [PubMed] [Google Scholar]
- Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta. Neuropathol. (Berl.) 82:239–259. [DOI] [PubMed] [Google Scholar]
- Capsoni, S., Giannotta, S., and Cattaneo, A. (2002a). Beta-amyloid plaques in a model for sporadic Alzheimer’s disease based on transgenic anti-nerve growth factor antibodies. Mol. Cell. Neurosci.21:15–28. [DOI] [PubMed] [Google Scholar]
- Capsoni, S., Giannotta, S., and Cattaneo, A. (2002b). Early events of Alzheimer-like neurodegeneration in anti-nerve growth factor transgenic mice. Brain. Aging2:24–43. [Google Scholar]
- Capsoni, S., Giannotta, S., and Cattaneo, A. (2002c). Nerve growth factor and galantamine ameliorate early signs of neurodegeneration in anti-nerve growth factor mice. Proc. Natl. Acad. Sci. U. S. A.99:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsoni, S., Giannotta, S., Stebel, M., Garcia, A. A., De Rosa, R., Villetti, G., Imbimbo, B. P., Pietra, C., and Cattaneo, A. (2004). Ganstigmine and donepezil improve neurodegeneration in AD11 antinerve growth factor transgenic mice. Am. J. Alzheimers Dis. Other Demen.19:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsoni, S., Ruberti, F., Di Daniel, E., and Cattaneo, A. (2000a). Muscular dystrophy in adult and aged anti-NGF transgenic mice resembles an inclusion body myopathy. J. Neurosci. Res.59:553–560. [DOI] [PubMed] [Google Scholar]
- Capsoni, S., Ugolini, G., Comparini, A., Ruberti, F., Berardi, N., and Cattaneo, A. (2000b). Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc. Natl. Acad. Sci. U. S. A.97:6826–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo, A. (1998). Selection of intracellular antibodies. Bratisl. Lek. Listy.99:413–418. [PubMed] [Google Scholar]
- Cattaneo, A., and Neuberger, M. S. (1987). Polymeric immunoglobulin M is secreted by transfectants of non-lymphoid cells in the absence of immunoglobulin J chain. EMBO J.6:2753–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo, A., Rapposelli, B., and Calissano, P. (1988). Three distinct types of monoclonal antibodies after long-term immunization of rats with mouse nerve growth factor. J. Neurochem.50:1003–1010. [DOI] [PubMed] [Google Scholar]
- Chao, M. V., Bothwell, M. A., Ross, A. H., Koprowski, H., Lanahan, A. A., Buck, C. R., and Sehgal, A. (1986). Gene transfer and molecular cloning of the human NGF receptor. Science232:518–521. [DOI] [PubMed] [Google Scholar]
- Chapman, B. S., and Kuntz, I. D. (1995). Modeled structure of the 75-kDa neurotrophin receptor. Protein Sci.4:1696–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. S., Nishimura, M. C., Armanini, M. P., Crowley, C., Spencer, S. D., and Phillips, H. S. (1997). Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J. Neurosci.17:7288–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. Q., Fawcett, J. R., Rahman, Y. E., Ala, T. A., and Frey, I. W. (1998). Delivery of nerve growth factor to the brain via the olfactory pathway. J. Alzheimers Dis.1:35–44. [DOI] [PubMed] [Google Scholar]
- Cohen, S. (1959). Purification and metabolic effects of a nerve growth-promoting protein from snake venom. J. Biol. Chem.234:1129–1137. [PubMed] [Google Scholar]
- Collerton, D. (1986). Cholinergic function and intellectual decline in Alzheimer’s disease. Neuroscience19:1–28. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo, C., Tapley, P., Jing, S. Q., Nanduri, V., O’Rourke, E., Lamballe, F., Kovary, K., Klein, R., Jones, K. R., Reichardt, L. F. et al. (1991). The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell66:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts, S. E., Nadeem, M., Wuu, J., Ginsberg, S. D., Saragovi, H. U., and Mufson, E. J. (2004). Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol.56:520–531. [DOI] [PubMed] [Google Scholar]
- Crowley, C., Spencer, S. D., Nishimura, M. C., Chen, K. S., Pitts-Meek, S., Armanini, M. P., Ling, L. H., MacMahon, S. B., Shelton, D. L., Levinson, A. D. et al. (1994). Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell76:1001–1011. [DOI] [PubMed] [Google Scholar]
- Crutcher, K. A., Scott, S. A., Liang, S., Everson, W. V., and Weingartner, J. (1993). Detection of NGF-like activity in human brain tissue: Increased levels in Alzheimer’s disease. J. Neurosci.13:2540–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, M. E., Stephens, R. M., Kaplan, D. R., and Greene, L. A. (1997). Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J. Biol. Chem.272:10957–10967. [DOI] [PubMed] [Google Scholar]
- De Rosa, R., Garcia, A., Braschi, C., Capsoni, S., Maffei, L., Berardi, N., and Cattaneo, A. (2005). Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. U. S. A.102:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky, S. T., Harbaugh, R. E., Schmitt, F. A., Bakay, R. A., Chui, H. C., Knopman, D. S., Reeder, T. M., Shetter, A. G., Senter, H. J., and Markesbery, W. R. (1992). Cortical biopsy in Alzheimer’s disease: Diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Intraventricular Bethanecol Study Group. Ann. Neurol.32:625–632. [DOI] [PubMed] [Google Scholar]
- Fahnestock, M., Michalski, B., Xu, B., and Coughlin, M. D. (2001). The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci.18:210–220. [DOI] [PubMed] [Google Scholar]
- Fahnestock, M., Scott, S. A., Jette, N., Weingartner, J. A., and Crutcher, K. A. (1996). Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Brain Res. Mol. Brain Res.42:175–178. [DOI] [PubMed] [Google Scholar]
- Fischer, W., Wictorin, K., Bjorklund, A., Williams, L. R., Varon, S., and Gage, F. H. (1987). Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature329:65–68. [DOI] [PubMed] [Google Scholar]
- Frey, I. W. H., Liu, J., Chen, X. Q., Thorne, R. G., Fawcett, J. R., Ala, T. A., and Rahman, Y. E. (1997). Delivery of 125I-NGF to the brain via the olfactory route. Drug Deliv.4:87–92. [Google Scholar]
- Golde, T. E. (2005). The Abeta hypothesis: Leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol.15:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington, A. W., Leiner, B., Blechschmitt, C., Arevalo, J. C., Lee, R., Morl, K., Meyer, M., Hempstead, B. L., Yoon, S. O., and Giehl, K. M. (2004). Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc. Natl. Acad. Sci. U. S. A.101:6226–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. L., and Garcia, K. C. (2004). Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science304:870–875. [DOI] [PubMed] [Google Scholar]
- Hefti, F., Knusel, B., and Lapchak, P. A. (1993). Protective effects of nerve growth factor and brain-derived neurotrophic factor on basal forebrain cholinergic neurons in adult rats with partial fimbrial transections. Prog. Brain Res.98:257–263. [DOI] [PubMed] [Google Scholar]
- Hellweg, R., Gericke, C. A., Jendroska, K., Hartung, H. D., and Cervos-Navarro, J. (1998). NGF content in the cerebral cortex of non-demented patients with amyloid-plaques and in symptomatic Alzheimer’s disease. Int. J. Dev. Neurosci.16:787–794. [DOI] [PubMed] [Google Scholar]
- Hempstead, B. L., Martin-Zanca, D., Kaplan, D. R., Parada, L. F., and Chao, M. V. (1991). High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature350:678–683. [DOI] [PubMed] [Google Scholar]
- Hock, C., Heese, K., Hulette, C., Rosenberg, C., and Otten, U. (2000). Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol.57:846–851. [DOI] [PubMed] [Google Scholar]
- Hock, C., Heese, K., Muller-Spahn, F., Hulette, C., Rosenberg, C., and Otten, U. (1998). Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer’s disease. Neurosci. Lett.241:151–154. [DOI] [PubMed] [Google Scholar]
- Holtzman, D. M., Li, Y., Chen, K., Gage, F. H., Epstein, C. J., and Mobley, W. C. (1993). Nerve growth factor reverses neuronal atrophy in a Down syndrome model of age-related neurodegeneration. Neurology43:2668–2673. [DOI] [PubMed] [Google Scholar]
- Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem.72:609–642. [DOI] [PubMed] [Google Scholar]
- Ibanez, C. F., Ebendal, T., and Persson, H. (1991). Chimeric molecules with multiple neurotrophic activities reveal structural elements determining the specificities of NGF and BDNF. EMBO J.10:2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal, K., Alonso Adel, C., Chen, S., Chohan, M. O., El-Akkad, E., Gong, C. X., Khatoon, S., Li, B., Liu, F., Rahman, A., Tanimukai, H., and Grundke-Iqbal, I. (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta1739:198–210. [DOI] [PubMed] [Google Scholar]
- Jing, S., Tapley, P., and Barbacid, M. (1992). Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron9:1067–1079. [DOI] [PubMed] [Google Scholar]
- Johnson, D., Lanahan, A., Buck, C. R., Sehgal, A., Morgan, C., Mercer, E., Bothwell, M., and Chao, M. (1986). Expression and structure of the human NGF receptor. Cell47:545–554. [DOI] [PubMed] [Google Scholar]
- Kaplan, D. R., Martin-Zanca, D., and Parada, L. F. (1991). Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature350:158–160. [DOI] [PubMed] [Google Scholar]
- Kaplan, D. R., and Miller, F. D. (2000). Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol.10:381–391. [DOI] [PubMed] [Google Scholar]
- Klein, R., Jing, S. Q., Nanduri, V., O’Rourke, E., and Barbacid, M. (1991). The trk proto-oncogene encodes a receptor for nerve growth factor. Cell65:189–197. [DOI] [PubMed] [Google Scholar]
- Koliatsos, V. E., Applegate, M. D., Knusel, B., Junard, E. O., Burton, L. E., Mobley, W. C., Hefti, F. F., and Price, D. L. (1991a). Recombinant human nerve growth factor prevents retrograde degeneration of axotomized basal forebrain cholinergic neurons in the rat. Exp. Neurol.112:161–173. [DOI] [PubMed] [Google Scholar]
- Koliatsos, V. E., Clatterbuck, R. E., Nauta, H. J., Knusel, B., Burton, L. E., Hefti, F. F., Mobley, W. C., and Price, D. L. (1991b). Human nerve growth factor prevents degeneration of basal forebrain cholinergic neurons in primates. Ann. Neurol.30:831–840. [DOI] [PubMed] [Google Scholar]
- Koliatsos, V. E., Nauta, H. J., Clatterbuck, R. E., Holtzman, D. M., Mobley, W. C., and Price, D. L. (1990). Mouse nerve growth factor prevents degeneration of axotomized basal forebrain cholinergic neurons in the monkey. J. Neurosci.10:3801–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. F., Li, E., Huber, L. J., Landis, S. C., Sharpe, A. H., Chao, M. V., and Jaenisch, R. (1992). Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell69:737–749. [DOI] [PubMed] [Google Scholar]
- Lee, R., Kermani, P., Teng, K. K., and Hempstead, B. L. (2001). Regulation of cell survival by secreted proneurotrophins. Science294:1945–1948. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini, R., and Hamburger, V. (1951). Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool.116:321–361. [DOI] [PubMed] [Google Scholar]
- Li, Y., Holtzman, D. M., Kromer, L. F., Kaplan, D. R., Chua-Couzens, J., Clary, D. O., Knusel, B., and Mobley, W. C. (1995). Regulation of TrkA and ChAT expression in developing rat basal forebrain: Evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J. Neurosci.15:2888–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow, E. M., Schweers, O., Drewes, G., Biernat, J., Gustke, N., Trinczek, B., and Mandelkow, E. (1996). Structure, microtubule interactions, and phosphorylation of tau protein. Ann. N. Y. Acad. Sci.777:96–106. [DOI] [PubMed] [Google Scholar]
- Markowska, A. L., Koliatsos, V. E., Breckler, S. J., Price, D. L., and Olton, D. S. (1994). Human nerve growth factor improves spatial memory in aged but not in young rats. J. Neurosci.14:4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska, A. L., Price, D., and Koliatsos, V. E. (1996). Selective effects of nerve growth factor on spatial recent memory as assessed by a delayed nonmatching-to-position task in the water maze. J. Neurosci.16:3541–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley, W. C., Rutkowski, J. L., Tennekoon, G. I., Gemski, J., Buchanan, K., and Johnston, M. V. (1986). Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res.387:53–62. [DOI] [PubMed] [Google Scholar]
- Molnar, M., Ruberti, F., Cozzari, C., Domenici, L., and Cattaneo, A. (1997). A critical period in the sensitivity of basal forebrain cholinergic neurones to NGF deprivation. Neuroreport8:575–579. [DOI] [PubMed] [Google Scholar]
- Molnar, M., Tongiorgi, E., Avignone, E., Gonfloni, S., Ruberti, F., Domenici, L., and Cattaneo, A. (1998). The effects of anti-nerve growth factor monoclonal antibodies on developing basal forebrain neurons are transient and reversible. Eur. J. Neurosci.10:3127–3140. [DOI] [PubMed] [Google Scholar]
- Mowla, S. J., Pareek, S., Farhadi, H. F., Petrecca, K., Fawcett, J. P., Seidah, N. G., Morris, S. J., Sossin, W. S., and Murphy, R. A. (1999). Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J. Neurosci.19:2069–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson, E. J., Conner, J. M., and Kordower, J. H. (1995). Nerve growth factor in Alzheimer’s disease: Defective retrograde transport to nucleus basalis. Neuroreport6:1063–1066. [DOI] [PubMed] [Google Scholar]
- Mufson, E. J., Conner, J. M., Varon, S., and Kordower, J. H. (1994). Nerve growth factor-like immunoreactive profiles in the primate basal forebrain and hippocampal formation. J. Comp. Neurol.341:507–519. [DOI] [PubMed] [Google Scholar]
- Mufson, E. J., Kroin, J. S., Sendera, T. J., and Sobreviela, T. (1999). Distribution and retrograde transport of trophic factors in the central nervous system: Functional implications for the treatment of neurodegenerative diseases. Prog. Neurobiol.57:451–484. [DOI] [PubMed] [Google Scholar]
- Mufson, E. J., Lavine, N., Jaffar, S., Kordower, J. H., Quirion, R., and Saragovi, H. U. (1997). Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp. Neurol.146:91–103. [DOI] [PubMed] [Google Scholar]
- Mufson, E. J., Li, J. M., Sobreviela, T., and Kordower, J. H. (1996). Decreased trkA gene expression within basal forebrain neurons in Alzheimer’s disease. Neuroreport8:25–29. [DOI] [PubMed] [Google Scholar]
- Mufson, E. J., Ma, S. Y., Cochran, E. J., Bennett, D. A., Beckett, L. A., Jaffar, S., Saragovi, H. U., and Kordower, J. H. (2000). Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol.427:19–30. [DOI] [PubMed] [Google Scholar]
- Nykjaer, A., Lee, R., Teng, K. K., Jansen, P., Madsen, P., Nielsen, M. S., Jacobsen, C., Kliemannel, M., Schwarz, E., Willnow, T. E., Hempstead, B. L., and Petersen, C. M. (2004). Sortilin is essential for proNGF-induced neuronal cell death. Nature427:843–848. [DOI] [PubMed] [Google Scholar]
- Perry, E. K., Tomlinson, B. E., Blessed, G., Bergmann, K., Gibson, P. H., and Perry, R. H. (1978). Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J.2:1457–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento, E., Capsoni, S., Domenici, L., and Cattaneo, A. (2002). Acute cholinergic rescue of synaptic plasticity in the neurodegenerating cortex of anti-nerve-growth-factor mice. Eur. J. Neurosci.15:1030–1036. [DOI] [PubMed] [Google Scholar]
- Piccioli, P., Di Luzio, A., Amann, R., Schuligoi, R., Surani, M. A., Donnerer, J., and Cattaneo, A. (1995). Neuroantibodies: ectopic expression of a recombinant anti-substance P antibody in the central nervous system of transgenic mice. Neuron15:373–384. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Tebar, A., Dechant, G., and Barde, Y. A. (1990). Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron4:487–492. [DOI] [PubMed] [Google Scholar]
- Roux, P. P., and Barker, P. A. (2002). Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol.67:203–233. [DOI] [PubMed] [Google Scholar]
- Ruberti, F., Bradbury, A., and Cattaneo, A. (1993). Cloning and expression of an anti-nerve grwoth factor (NGF) antibody for studies using the neuroantibody approach. Cell. Mol. Neurobiol.13:559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruberti, F., Capsoni, S., Comparini, A., Di Daniel, E., Franzot, J., Gonfloni, S., Rossi, G., Berardi, N., and Cattaneo, A. (2000). Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J. Neurosci.20:2589–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi, A., Delcroix, J. D., and Mobley, W. C. (2003). Traffic at the intersection of neurotrophic factor signaling and neurodegeneration. Trends. Neurosci.26:73–80. [DOI] [PubMed] [Google Scholar]
- Scott, J., Selby, M., Urdea, M., Quiroga, M., Bell, G. I., and Rutter, W. J. (1983). Isolation and nucleotide sequence of a cDNA encoding the precursor of mouse nerve growth factor. Nature302:538–540. [DOI] [PubMed] [Google Scholar]
- Scott, S. A., Mufson, E. J., Weingartner, J. A., Skau, K. A., and Crutcher, K. A. (1995). Nerve growth factor in Alzheimer’s disease: Increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci.15:6213–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah, N. G., Benjannet, S., Pareek, S., Savaria, D., Hamelin, J., Goulet, B., Laliberte, J., Lazure, C., Chretien, M., and Murphy, R. A. (1996). Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem. J.314(Pt. 3):951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. (2001). Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev.81:741–766. [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science298:789–791. [DOI] [PubMed] [Google Scholar]
- Shamovsky, I. L., Ross, G. M., Riopelle, R. J., and Weaver, D. F. (1999). The interaction of neurotrophins with the p75NTR common neurotrophin receptor: A comprehensive molecular modeling study. Protein Sci.8:2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shooter, E. M. (2001). Early days of the nerve growth factor proteins. Annu. Rev. Neurosci.24:601–629. [DOI] [PubMed] [Google Scholar]
- Smith, D. E., Roberts, J., Gage, F. H., and Tuszynski, M. H. (1999). Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc. Natl. Acad. Sci. U. S. A.96:10893–10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squinto, S. P., Stitt, T. N., Aldrich, T. H., Davis, S., Bianco, S. M., Radziejewski, C., Glass, D. J., Masiakowski, P., Furth, M. E., Valenzuela, D. M. et al. (1991). trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell65:885–893. [DOI] [PubMed] [Google Scholar]
- Stach, R. W., and Shooter, E. M. (1974). The biological activity of cross-linked beta nerve growth factor protein. J. Biol. Chem.249:6668–6674. [PubMed] [Google Scholar]
- Ullrich, A., Gray, A., Berman, C., and Dull, T. J. (1983). Human beta-nerve growth factor gene sequence highly homologous to that of mouse. Nature303:821–825. [DOI] [PubMed] [Google Scholar]
- Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science215:1237–1239. [DOI] [PubMed] [Google Scholar]
- Yan, H., and Chao, M. V. (1991). Disruption of cysteine-rich repeats of the p75 nerve growth factor receptor leads to loss of ligand binding. J. Biol. Chem.266:12099–12104. [PubMed] [Google Scholar]