

Abstract
Lactobacillus plantarum is a versatile and flexible species that is encountered in a variety of niches and can utilize a broad range of fermentable carbon sources. To assess if this versatility is linked to a variable gene pool, microarrays containing a subset of small genomic fragments of L. plantarum strain WCFS1 were used to perform stringent genotyping of 20 strains of L. plantarum from various sources. The gene categories with the most genes conserved in all strains were those involved in biosynthesis or degradation of structural compounds like proteins, lipids, and DNA. Conversely, genes involved in sugar transport and catabolism were highly variable between strains. Moreover, besides the obvious regions of variance, like prophages, other regions varied between the strains, including regions encoding plantaricin biosynthesis, nonribosomal peptide biosynthesis, and exopolysaccharide biosynthesis. In many cases, these variable regions colocalized with regions of unusual base composition. Two large regions of flexibility were identified between 2.70 and 2.85 and 3.10 and 3.29 Mb of the WCFS1 chromosome, the latter being close to the origin of replication. The majority of genes encoded in these variable regions are involved in sugar metabolism. This functional overrepresentation and the unusual base composition of these regions led to the hypothesis that they represented lifestyle adaptation regions in L. plantarum. The present study consolidates this hypothesis by showing that there is a high degree of gene content variation among L. plantarum strains in genes located in these regions of the WCFS1 genome. Interestingly, based on our genotyping data L. plantarum strains clustered into two clearly distinguishable groups, which coincided with an earlier proposed subdivision of this species based on conventional methods.
As an alternative to the complete sequencing of genomes, spotted DNA and oligonucleotide microarrays can be used to obtain a highly detailed view of the gene content of related organisms, especially of strains of closely related species or of the same species (6, 33, 48). In these studies, information about relative gene position and chromosomal organization is lost but may be obtained by PCR techniques (37). Nevertheless, large-scale mutation events (i.e., insertion and deletion events) can often, with a high degree of certainty, be reconstructed from a comparison of related strains. The results obtained with microarray-based genotyping support a model of high evolutionary plasticity of bacterial genomes (19, 36). It has become clear that horizontal gene transfer is an important mechanism for generating genotypic and phenotypic diversity in bacteria. The phenomenon has been studied in particular in relation to niche adaptations like the emergence of virulence, antibiotic resistance (20, 21, 45), and symbiosis or fitness (15). Although this plasticity may, in many cases, depend on the simple transfer of mobile genetic elements, it often appears to result in very complex mosaic-structured genomic rearrangements.
For a number of genes located in plasticity zones of other organisms, a function in niche adaptation has been shown or proposed, but in many cases, their function is unknown. In fact, as a result of an interspecies study it was conjectured that more than 70% of the genes that are obtained by horizontal transfer encode proteins of unknown function (13).
The genome of the lactic acid bacterium Lactobacillus plantarum strain WCFS1 has been sequenced (25). It has a size of 3.3 M, which is among the largest genome sizes known for lactic acid bacteria. The large size of its genome is thought to be related to the diversity of environmental niches in which L. plantarum is encountered. Its most prominent abundance is in the fermentation of plant-derived raw materials, which include several industrial and artisan food and feed fermentations, like must (7), olives (40), and a variety of vegetable fermentations (22). In addition to these environments, L. plantarum is also encountered in some dairy (16) and meat (4) fermentations and as a natural inhabitant of the gastrointestinal tract of humans and animals (2). Probably related to the diversity of niches is the fact that this bacterium is able to ferment a broad range of sugars (10). A large number of genes involved in these functions appear to be located in a region near the origin of replication of L. plantarum WCFS1. Moreover, many genes in this region have an unusual base composition compared to the rest of the genome, suggesting that they originate from recent horizontal transfers. Based on these findings, this region was designated a so-called “lifestyle adaptation island” (25). This island might represent a high-plasticity region in the L. plantarum genome and could be involved in niche adaptation of this species. A comparison of several strains of L. plantarum should provide evidence for or against this hypothesis.
Here we describe a comparison of the genomic contents of 20 L. plantarum strains by using microarrays. Sets of genes that are present in the reference strain, WCFS1, and not detected in other strains were analyzed with respect to their chromosomal location, base composition, and putative functions. Our observations support the high plasticity of the lifestyle adaptation island near the origin of replication in strain WCFS1. Assuming that most of the observed variability is due to nonneutral selection, our findings suggest an important place of sugar catabolism in niche adaptation of this microbe.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in this study are listed in Table 1. L. plantarum was grown in MRS (Difco, Molesey, Surrey, United Kingdom) at 30°C without aeration.
TABLE 1.
Lactobacillus plantarum strains used in this study
| Strain | Alternative designation | RAPD groupa | Origin | Source or referenceb |
|---|---|---|---|---|
| WCFS1 | NCIMB 8826 | GLp1 | Human saliva | NCIMB |
| ATCC 14917 | LMG 6907 | GLp1 | Pickled cabbage | ATCC |
| ATCC 8014 | LMG 1284 | GLp1 | Maize ensilage | ATCC |
| NCDO 1193 | LMG 9209 | GLp1 | Vegetables | NCIMB |
| 299 | DSM 6595 | Human colon | 32 | |
| 299v | DSM 9843 | GLp1c | Human intestine | 24 |
| LP80 | DSM 4229 | GLp1 | Silage | C. Platteeuw |
| LMG 18021 | GLp1c | Milk, Senegal | BCCM | |
| LM3 | Silage | 34 | ||
| NC8 | GLp1c | Grass silage | 3 | |
| CIP104440 | 61A | GLp1 | Human stool | CIP |
| CIP104441 | 61P | GLp1 | Human stool | CIP |
| SF2A35B | GLp2 | Sour cassava, South America | 18 | |
| NCIMB 12120 | GLp2 | Ogi, Nigeria | NCIMB | |
| LP85-2 | GLp2 | Silage, France | 9 | |
| CIP104452 | GLp1 | Human tooth abcess | CIP | |
| CIP104450 | 61BR | GLp1 | Human stool | CIP |
| CIP104451 | 61CA | GLp1 | Human urine | CIP |
| CIP104448 | 61BB | GLp1 | Human stool | CIP |
| CIP102359 | Human spinal fluid | CIP |
According to Bringel et al. (10).
NCIMB, National Collections of Industrial, Food and Marine Bacteria, Aberdeen, Scotland; ATCC, American Type Culture Collection, Manassas, Va.; BCCM, Belgian Co-ordinated Collections of Micro-organisms; CIP, Collection de l'Institut Pasteur, Paris, France.
F. Bringel, unpublished data.
Chromosomal DNA isolation.
All L. plantarum strains were grown in 10 ml MRS to a turbidity at 600 nm of 1 (exponentially growing cells). Cells were harvested and suspended in 0.5 ml THMS (30 mM Tris [pH 8.0], 3 mM MgCl2 in 25% sucrose) containing 50 mg/ml lysozyme. This mixture was incubated at 37°C for 2 h, and cells were harvested by centrifugation. Cell pellets were resuspended in 0.5 ml TE (10 mM Tris, 1 mM EDTA, pH 7.4) containing 10 μg/ml RNase. Thirty microliters of a 10% (wt/vol) sodium dodecyl sulfate (SDS) solution was added, and the resulting mixture was incubated for 15 min at 37°C. Subsequently, 10 μl of a solution containing 20 mg/ml of proteinase K was added, followed by a 15-min incubation at 65°C. Phenol-chloroform extractions were repeated until the water phase was clear, and residual traces of phenol were removed from the water phase by chloroform extraction. Total DNA was precipitated from the water phase by addition of an equal volume of isopropanol (41). The DNA pellet was washed once with 70% ethanol, briefly vacuum dried, and dissolved overnight in 1 ml TE at 4°C. The concentration of DNA was estimated by measurement of the optical density at 260 nm (41).
Array design.
Clone-based DNA microarrays were used, which contain approximately 80% of all bases of the L. plantarum WCFS1 genome. Coverage of all bases was not possible due to technical limitations. Missing fragments are randomly distributed over the genome. A selected subset of genomic fragments was amplified by PCR from the genomic L. plantarum WCFS1 library in pBlueScript SK+ that was previously constructed for sequencing purposes (25). The primers used were universal forward and reverse primers with 5′-C6 amino linkers to facilitate cross-linking to the aldehyde-coated glass slides (Telechem). PCR was performed directly on cells from glycerol stocks, employing SuperTaq polymerase (SphaeroQ, Leiden, The Netherlands), PCR products were purified by ethanol precipitation, and the efficacy of PCR was confirmed by agarose gel electrophoresis. Purified PCR products were dissolved in 3× SSC (1× SSC is 150 mM sodium chloride plus 17 mM sodium citrate, pH 7.2) and subsequently arrayed in a controlled atmosphere on CSS-100 silylated aldehyde glass slides (Telechem) with quill pins (Telechem SMP3) in an SDDC 2 Eurogridder (ESI, Toronto, Canada). After drying, the slides were blocked with borohydride. The total number of clones spotted on the microarray was 3,692, having an average size of 1.2 kb. The coverage of the total genome was 80.8%, representing 2,683 genes. The overlap of clones in the covered part resulted in a 1.6-fold redundancy.
Fluorescent labeling and hybridization.
Differential DNA presence was determined by two-color fluorescent hybridizations of the corresponding genomic DNAs on the L. plantarum WCFS1 clone array. Genomic DNA of each strain was cohybridized once with the reference DNA from strain WCFS1. In half of the experiments, the WCFS1 reference DNA was labeled with Cy5 whereas in the other half it was labeled with Cy3. The samples were labeled by random primed labeling using the Bioprime labeling kit (Invitrogen) using Cy5- or Cy3-labeled dUTP (Amersham Biosciences). Unincorporated dyes were removed using AutoSeq G50 columns (Amersham Biosciences). DNA microarrays were prehybridized for 45 min at 42°C in prehybridization solution (1% bovine serum albumin, 5× SSC, and 0.1% SDS). Cohybridizations of the labeled genomic DNA samples were performed overnight at 42°C in Easyhyb buffer (Roche Applied Sciences) according to the manufacturer's protocol. Slides were washed twice in 1× SSC, 0.2% SDS, once in 0.5× SSC, and twice in 0.2× SSC at 37°C.
Scanning and primary data analysis.
After washing and drying, slides were scanned with a ScanArray Express 4000 scanner (Perkin-Elmer). Images were analyzed using ImaGene 4.2 software (BioDiscovery, Marina del Rey, CA). Criteria for flagging spots were as follows: (i) empty, spot threshold of 2.0; (ii) poor, spot threshold of 0.4; and (iii) negative spot detection. ImaGene output files were further analyzed in a spreadsheet. Spots flagged as either empty, poor, or negative by ImaGene software in either the Cy5 or the Cy3 channel were omitted, as were spots with signals less than twice the local background. Routinely over 80% of all spots passed these quality criteria.
Secondary data analysis.
Array measurements were normalized by local fitting of an M-A plot using the implementation of the LOWESS algorithm in R (http://www.r-project.org). Normalized data were analyzed using an adaptation of an error model (39). The random error present in the data was assessed in a self-hybridization of Cy3- and Cy5-labeled DNA from the reference strain WCFS1, using the statistic X = (a2 − a1)/[σ1 + σ2 + f · (a1 + a2)], where a1 and a2 are Cy3 and Cy5 signals, σ1 and σ2 are the standard deviations of the Cy3 and Cy5 background signals, and f is a proportional constant estimating the relative standard deviation in the signals. A histogram of this statistic was fitted to the logistic distribution with mean = 0 and scale parameter = √3/π by adapting f. In the experiments described below, f was equal to 5.6%. Independent duplicates of the self-hybridization experiment demonstrated that the observed deviations in normalized Cy3 and Cy5 signals (the errors) did not display clone-specific effects and may, therefore, be regarded as random. The one-sided probability of observing statistic X was calculated for each clone in each hybridization experiment and is referred to below as the P value. It indicates the probability of obtaining the observed Cy3 and Cy5 signals or more extreme values due to experimental error alone. The one-sided probability was calculated because the absence of a fragment in a test strain will be concluded from the observation that the signal of the test strain is much smaller than the signal of the control.
Since the DNA fragments (“clones”) on the array represent a minimal tiling coverage of the chromosome, a correction had to be made for overlapping parts. The complete set of clone-border coordinates on the chromosome was used to define a set of disjoint (nonoverlapping) “slices” of the chromosome. For each slice, a composite P value was calculated as the average of the weighted P values from the clones that covered this slice. A weight equal to the length of the slice divided by the length of the clone was used. To assess a P value for individual genes, the P values of all clones overlapping with a gene were taken. From these, a weighted average was calculated where the weights were equal to the square of the fraction of the clone that overlapped with the gene. In this way, clones that fully overlap with a gene and, consequently, do not contain signals from other genes have a more than proportional weight compared to clones that overlap with other genes too.
The deviation of the local base composition from the average composition in the chromosome was assessed using the χ2 statistic calculated in a sliding window of 10 kb. This parameter is called “base frequency deviation index” or “base deviation index” below. It was calculated for both halves of the WCFS1 chromosome individually, since a clear GC-skew shift at approximately 180° of the circular chromosome indicated a difference in the average base composition of the halves. Regions were characterized as having unusual base composition when the base frequency deviation index was in the upper 5% quantile.
The distance metric δ used to construct a pairwise distance matrix for the strains was
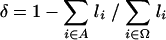 |
where li is the size in bases of the i-th slice, Α is the set of indices of slices that are present in both strains, and Ω is the set of indices of all slices that were tested in both strains. Since Α is a subset of Ω, δ will be between 0 and 1 and δ = 0 when Α = Ω.
RESULTS AND DISCUSSION
Genotyping results.
Strains of the species L. plantarum are encountered in a variety of ecological niches. It was anticipated that the diversity among strains is reflected in their genomes. Here we describe the application of L. plantarum WCFS1 DNA microarrays for the genotyping of 19 other L. plantarum strains isolated from different sources like food fermentations and human mucosa (Table 1). The probes on the DNA microarrays consisted of a subset of genomic fragments amplified by PCR from plasmids from the random insert library used for sequencing of the genome of strain WCFS1 (25). The microarray covered 80.8% of all bases of the WCFS1 genome (see the rectangle labeled “missing” in Fig. 1). The presence or absence of genomic fragments corresponding to the clones on the array was inferred from a statistical model that accounts for experimental error in the raw data. The statistical model was derived from observations on the experimental error in self-self hybridizations of WCFS1 DNA (see Materials and Methods). The criterion used to decide whether fragments were absent or present was set to a P value of 10−4, which corresponds on average to an erroneous conclusion only once every 10,000 clones that the corresponding fragment is absent in the tested genome. The overall genotyping results for the strains analyzed are displayed as “bar plots” with the chromosomal organization of strain WCFS1 as a template (Fig. 1, central panel), in which a black bar indicates the absence of a fragment in a specific strain. We wish to stress that the presentation of the results does not allow conclusions about the chromosomal localization of genes or genome fragments in the other strains. Neither the experimental setup nor the error model accounts for the possibility that hybridization may occur between similar but nonidentical DNA fragments. However, the hybridization conditions that were chosen are stringent, which is corroborated by the finding that in hybridizations with DNA of closely related species (Lactobacillus pentosus and Lactobacillus brevis) less than 35% of the clones gave a signal at all. Therefore, data sets obtained for these other Lactobacillus species could not be interpreted and limited the current study to strains of the species L. plantarum. To get an indication of the percentage of similarity necessary to obtain a hybridization signal in our experimental setup, sequence data of L. plantarum strains available in public database were compared. This showed that homologous sequences within the L. plantarum species are highly similar. One hundred sixty-seven homologous DNA sequences were found, which were on average 98% identical with a minimum of 86% identity.
FIG. 1.

A comparison of the gene contents of 20 strains of L. plantarum. The names of the strains tested are indicated to the left. For each strain, the gene content is indicated in a rectangle mapping onto the WCFS1 genome sequence. In these rectangles, the presence (no line) and absence (black line) of fragments is indicated. The upper rectangle, labeled “missing,” shows in black the fragments of the genome that were not spotted on the microarray (approximately 19% of the total chromosome). The panel labeled “sum of distances” shows the sum of distances over all strain combinations generated by the particular slice of the genome. It is a measure of the contribution of this slice to the total distance in the reconstructed phylogenetic tree and, thus, a measure of the genotypic variation mapping onto the slice. The lower panel shows the base deviation index along the chromosome of L. plantarum WCFS1. A high value of this index indicates an unusual base composition. The highest values are off scale in this panel. To the left, a tree is shown which represents a clustering of the strains that was based on a pairwise distance matrix derived from the results shown in the central panels. The distance metric used is described in Materials and Methods.
The only available sequence information that allowed us to test our method of detecting the presence or absence of genes in other strains concerned the plantaricin antibiotic gene cluster in strain NC8 (GenBank accession no. AF522077; 12.4 kb) (30). Our hybridization data agreed very well with the sequence information. The absence of plnN in the 1.6-kb deletion of NC8 and the complete 3.5-kb deletion between plnP and plnD were correctly predicted by our data. The only discrepancy is the absence in NC8 of one gene (plnO) which was predicted to be present in that strain by our data.
Taxonomy of strains.
A distance matrix representing the fractional similarity between strains was constructed by pairwise comparison, and a hierarchical tree was constructed from this matrix by tree clustering (Fig. 1). Insertions and deletions that have resulted in the variation among strains were probably random events in time. Therefore, the constructed tree should reflect historical branching events. However, the method does not take into account the fact that some singular events, like phage or transposon integration, may cause very large differences and, consequently, large distances. Nor does it account for the possibility that horizontal transfer within the L. plantarum species may take place frequently. Nevertheless, the method clearly distinguishes two groups of L. plantarum. This result is in accordance with earlier observations based on classical taxonomy data. These included distinct variations in randomly amplified polymorphic DNA (RAPD) patterns and sugar catabolism inventories, in particular catabolism of melizitose, dulcitol, α-methyl-d-mannoside, and α-methyl-d-glucoside (10). The upper three strains depicted in Fig. 1 (NCIMB 12120, SF2A35B, and LP85-2) were shown to belong to a distinct subgroup of L. plantarum strains, designated GLp2 (see Table 1 in reference 10), whereas the reference strain employed here, WCFS1, was shown to belong to another subgroup, designated GLp1 (Table 1). Based on the microarray analysis, it is estimated that the organisms of GLp2 lack approximately 20% of the genes present in WCFS1. In part, this may be due to a smaller genome size of these organisms (12). The absent genes are distributed over the functional catego-ries, in a similar manner to the genes found to be lacking in strains of the GLp1 subgroup (see below; Fig. 2). Nevertheless, gene homologues that encode some specific functions are predicted to be absent in GLp2 strains, including the genes coding for exodeoxyribonuclease III (gene lp_0812) and carbonic anhydrase (lp_2736), as well as several less-well-characterized genes, like those coding for transporters and extracellular proteins. Of course, it cannot be excluded that there are genes present in GLp2 strains with low or no similarity to the ones in WCFS1 but encoding proteins with similar functions. For example, a different class of carbonic anhydrase may be present in GLp2 strains (44).
FIG. 2.

Conservation and variability of genes in L. plantarum GLp1, grouped according to functional classes. For each gene, the fraction of strains in which it was absent was determined and it was accordingly classified in an absence category. Subsequently, for each gene category the fraction of genes per absence category was plotted. Note that the “0% absence” category represents those genes that are conserved in all strains. Panel A shows the results for all main functional classes, whereas in panel B selected subcategories are shown that deviate significantly from the distribution of other subclasses in their main class. Also in panel B, the absence score for genes with a high CAI is shown.
Because of the large differences in genotype between GLp1 and GLp2, unless stated otherwise, most analyses concerning conservation and variation of gene content were performed only within GLp1.
Distribution of genotypic variation among the functional categories of gene products.
All genes of L. plantarum WCFS1 have previously been assigned to one functional subcategory (25), which is grouped into the main functional categories. The variation and/or conservation of genes among the different L. plantarum GLp1 strains was investigated with respect to these functional categories. The results for all main functional categories were plotted as the fraction of genes per category that is missing in a certain fraction (interval) of the strains (Fig. 2A). Exceptional conservation is observed in the main categories that contain genes involved in biosynthesis or degradation of structural components, like proteins, nucleotides, and lipid biosynthesis. Since many of the genes in these categories are essential for growth, conservation of these genes might have been anticipated. The only main category that contains many genes that are absent in many strains is the category which contains phage- and transposon-related genes (termed “other categories”). In contrast to the majority of main functional categories, some subcategories contain genes that appeared to be absent in significantly more strains than average (Fig. 2B). Among these subcategories are those involved in sugar catabolism (i.e., energy metabolism of sugars and phosphotransferase sugar uptake systems) and genes encoding regulators of the DeoR and LacI families. Many members of these families are known to be involved in regulation of carbohydrate metabolism (5, 47) (see entry PF00455 in the Pfam database).
Genes grouped among the functional subcategory “glycolysis” (part of the main category “energy metabolism”) appeared to be 100% conserved. However, some genes of the pentose-phosphate pathway—in particular all three transaldolase genes, two out of three transketolase genes, and one of two phosphoketolase genes—were not conserved. Given the fact that glycolytic genes have a high codon adaptation index (CAI; calculated using the ribosomal proteins as a reference set) (25, 42), it was investigated whether genes with a high CAI are in general well conserved. The rationale to anticipate a high level of conservation for genes with a high CAI is provided by the fact that their codon bias is thought to be optimized for high expression, which would lead to the assumption that an extensive evolutionary history of these genes within a species exists (43). However, in L. plantarum this class of genes appears not much better conserved than average (Fig. 2B). The genes with a high CAI in strain WCFS1 that were not detected in at least one strain were not allocated to one or a small number of functional categories. It seems that in L. plantarum a high CAI is not correlated with conservation.
Assuming that most adaptations are nonneutral, genes putatively involved in very recent niche adaptations by strain WCFS1 are likely to be among those that are unique for this strain (Table 2). These genes are mainly concentrated in two clusters involved in non-ribosomal peptide (NRPS) biosynthesis and in exopolysaccharide biosynthesis. Both clusters also have an unusual base composition relative to the genome's average (see also below), which in case of the NRPS cluster is exemplified by an extremely high base deviation index. Both observations support the hypothesis that these clusters are recent acquisitions of strain WCFS1. NRPS gene clusters have not been found in any lactobacillus before and hence presumably have been acquired from another genus.
TABLE 2.
Genes that are only found in strain WCFS1
| Gene(s) | Product(s) or function(s) |
|---|---|
| lp_0483 | Hypothetical protein |
| lp_0488 | Bifunctional protein; acetyl-glutamate kinase; acetyl-ornithine deacetylase |
| lp_0491 | Transketolase, C-terminal fragment |
| lp_0578 to lp_0584 | Most genes from a non-ribosomal protein biosynthesis cluster |
| lp_1180, lp_1186, lp_1190, lp_1202 | Some genes from an exopolysaccharide biosynthesis cluster (glycosyltransferases, glucose-1-phosphate thymidylyltransferase, and dTDP-4-dehydrorhamnose reductase) |
| lp_1351 | Hypothetical protein |
| lp_2401 | Uncharacterized prophage protein |
| lp_2880 | Hypothetical protein |
| lp_3379, lp_3385, lp_3387 | Uncharacterized prophage proteins |
The function of uncharacterized genes can be revealed in a comparative study among related organisms provided that these genes lead to measurable phenotypes and that their presence in the genomes varies among these organisms. A statistically significant correlation between the occurrence of a certain gene in an organism and a trait that the organism displays may indicate the involvement of this gene in the trait. This approach for generating hypotheses about gene function has been proposed for interspecies comparison by correlating orthologs and phenotypes (23, 28). Using a similar approach, we could not find significant associations of genes with the sources from which the different strains were isolated (Table 1). Associations with isolation from human material (n = 10) or plant fermentation (n = 9) were tested. The absence of such associations may be due to the versatility of individual L. plantarum strains, which survive or even grow in different environments. Therefore, the source of isolation may not be a good indicator of the niche to which a strain is adapted. Nevertheless, niche specialization is likely to exist in the L. plantarum species, and, therefore, niche-specific genes are probably present. An example is the presence of proteins that promote adherence to gut cells in strain 299v (1, 38a). The identification of such genes using genotype-phenotype correlations will require more detailed phenotypic analysis of the individual strains or enlargement of the category sample sizes.
Localization of genotypic variation.
Much of the genotypic variation between the strains maps onto specific regions of the chromosome of strain WCFS1. The panel in Fig. 1 showing the sum of distances gives an indication of the genotypic variation between the strains as mapped onto a particular location of the WCFS1 genome. The panel showing the base deviation index indicates how the local base composition deviates from the overall base composition of the chromosome (25). Many regions that have an unusual base composition also have a high value of the sum of distances. Therefore, the current study supports the hypothesis (27) that unusual base composition is a good indicator for horizontal transfer events, at least in L. plantarum, as far as the variability of genes between related strains is an indicator of recent horizontal transfers. Other authors concluded that this relationship does not hold for Escherichia coli, using positional orthology between E. coli and Salmonella enterica serovar Typhi as an indicator for a common ancestral origin of genes (26). A selection of regions in WCFS1 with both high sum of distances and base deviation index values is listed in Table 3. Many of the variable regions have a distinct physiological function and are likely to have been obtained or deleted as functional cassettes. Clearly, this is the case for prophages and regions with transposon remnants, since we know the mechanisms that cause contiguous regions of variability in these cases. For other regions, it is less clear how they can be transferred as functional cassettes. Some of the functions encoded within these variable regions are also known to have an unusual base composition in other species or are known to vary among strains. These include the exopolysaccharide biosynthesis regions (11, 14, 17) and the restriction modification systems (38). Some variable regions have a base composition that is similar to the genome average. The absence of these regions may result from deletions relative to the parent organism or may be generated by horizontal transfer from closely related organisms or even the same species. An example is the region that lies between 1.350 and 1.376 Mb and that contains genes encoding subunits of a nitrate reductase and all genes involved in molybdopterin biosynthesis. This cluster is both present and absent in strains from distant branches of the L. plantarum tree (Fig. 1), including the three strains of the GLp2 group. This observation supports the hypothesis that this region was present in a common ancestor and was lost on several occasions during evolution of the progeny.
TABLE 3.
Regions of high variability between strains as mapped on the chromosome of L. plantarum strain WCFS1 and main functions of the genes they containa
| Range on chromosome (Mb) | Size (kb) | Range of genes | Main function(s) |
|---|---|---|---|
| 0.142-0.156 | 14.6 | lp_0164-lp_0179 | Sugar catabolism, sugar ABC transporter |
| 0.220-0.229 | 9.1 | lp_0244-lp_0254 | Possibly sugar catabolism |
| 0.344-0.381 | 37.8 | lp_0373-lp_0426 | Plantaricin production and immunity |
| 0.432-0.447 | 15.2 | lp_0483-lp_0499 | Various |
| 0.533-0.552 | 18.5 | lp_0576-lp_0584 | Mannose PTS and non-ribosomal peptide biosynthesis cluster |
| 0.591-0.628 | 37.4 | lp_0625-lp_0683 | Prophage |
| 0.833-0.837 | 4.2 | lp_0904-lp_0906 | β-Glucoside PTS and hydrolysis |
| 0.869-0.878 | 9.0 | lp_0.939-lp_0946 | Restriction modification system |
| 1.060-1.068 | 8.4 | lp_1162-lp_1171 | Various |
| 1.072-1.087 | 14.9 | lp_1176-lp_1189 | Exopolysaccharide biosynthesis |
| 1.089-1.126 | 37.0 | lp_1193-lp_1238 | Exopolysaccharide biosynthesis |
| 1.235-1.247 | 12.1 | lp_1343-lp_1358 | Putative pheromone production, extracellular protein production |
| 1.275-1.283 | 7.8 | lp_1393-lp_1400 | β-glucosides PTS |
| 1.350-1.376 | 25.3 | lp_1473-lp_1503 | Nitrate reductase and molybdopterin biosynthesis cluster |
| 1.738-1.753 | 15.3 | lp_1920-lp_1933 | Various |
| 1.767-1.773 | 6.3 | lp_1953-lp_1958 | ABC transporter |
| 1.804-1.816 | 12.2 | lp_1994-lp_2015 | Transposon fragments |
| 1.869-1.876 | 7.1 | lp_2074-lp_2078 | Nitrate transporter |
| 2.061-2.069 | 7.9 | lp_2282-lp_2290 | Various |
| 2.116-2.120 | 4.2 | lp_2340-lp_2344 | Various |
| 2.160-2.202 | 41.9 | lp_2396-lp_2454 | Prophage |
| 2.205-2.217 | 11.8 | lp_2461-lp_2479 | Prophage |
| 2.284-2.288 | 4.2 | lp_2564-lp_2568 | Putative Mg2+ and Co2+ transport cluster |
| 2.310-2.323 | 12.8 | lp_2593-lp_2603 | Sugar metabolism |
| 2.563-2.577 | 14.3 | lp_2880-lp_2900 | Various |
| 2.658-2.663 | 4.9 | lp_2987-lp_2992 | Various |
| 2.691-2.698 | 7.3 | lp_3024-lp_3034 | Various |
| 2.70-2.85 | 150 | lp_3040-lp_3196 | Various; among others, extracellular proteins and sugar metabolism |
| 2.876-2.883 | 6.5 | lp_3228-lp_3234 | PTS system |
| 2.998-3.007 | 8.6 | lp_3377-lp_3390 | Prophage |
| 3.023-3.032 | 9.0 | lp_3411-lp_3416 | Extracellular protein biosynthesis |
| 3.10-3.29 | 190 | lp_3488-lp_3669 | Various, but mainly sugar metabolism |
Regions with a size of at least 4 kb that were absent in at least 30% of the strains.
Hybridization techniques such as the one used here do not give positional information about the genes. The preservation of gene linkage can be investigated using PCR techniques. This was attempted for the regions where the two prophages of WCFS1 are inserted (46). The surrounding genes of the Lp1 prophage (bases 589962 to 631801) were shown to be joined in six L. plantarum strains (including ATCC 8014), showing, at least for this region, positional conservation in the absence of Lp1 prophage.
Evidence for lifestyle adaptation regions in strain WCFS1.
The unusual base composition of two regions next to the origin of replication between 2.70 and 3.29 Mb of the chromosome of strain WCFS1 and the strong overrepresentation of genes involved in sugar metabolism in this region suggested that many of these genes were evolutionarily recent acquisitions of strain WCFS1 (25). Based on these findings, it was proposed that this chromosomal region represents a lifestyle adaptation island within the L. plantarum genome that displays a high degree of genomic plasticity. The present comparison of L. plantarum WCFS1 with other L. plantarum strains supports the hypothesis that many genes in this region were recently acquired. The genotypic variation in these regions is very high and is mainly concentrated in two large subregions (Fig. 1) which run from 2.70 to 2.85 Mb and from 3.10 to 3.29 Mb, amounting to 10% of the total chromosome. A total of 293 genes are present in these regions, and some functional categories are clearly overrepresented among these genes (Table 4). For example, almost half of the total number of genes in the category “energy metabolism; sugars” and more than one-third of the PTS-related genes are present in these regions. Other overrepresented classes are regulatory proteins, in particular two-component regulators and regulators of the LacI family, which are likely to be involved in the regulation of genes involved in sugar metabolism. Remarkably, these functional (sub)categories belong to the most variable identified in the L. plantarum genome (see above).
TABLE 4.
Functional classes of genes that are significantly overrepresented in the lifestyle regions (2.70 to 2.85 Mb and 3.10 to 3.29 Mb)
| Functional category | No. of genes in:
|
% in lifestyle regions | Overrepresentation (fold)a | P valueb | |
|---|---|---|---|---|---|
| Lifestyle regions | Total chromosome | ||||
| Energy metabolism; sugars | 28 | 58 | 48 | 5.0 | 7.9E-13 |
| Transport and binding proteins; PTS systems | 25 | 67 | 37 | 3.9 | 2.6E-09 |
| Regulatory functions; two-component regulators | 11 | 31 | 35 | 3.7 | 5.6E-05 |
| Regulatory functions; LacI family regulators | 7 | 15 | 47 | 4.9 | 1.2E-04 |
| Transport and binding proteins; carbohydrates, organic alcohols, and acids | 12 | 44 | 27 | 2.9 | 3.9E-04 |
| Cell envelope; cell surface proteins, other | 14 | 57 | 25 | 2.6 | 4.7E-04 |
| Regulatory functions; other | 15 | 69 | 22 | 2.3 | 1.2E-03 |
| Regulatory functions; BglB family regulators | 3 | 5 | 60 | 6.3 | 1.5E-03 |
| Energy metabolism; aerobic | 9 | 36 | 25 | 2.6 | 2.8E-03 |
| Regulatory functions; AraC family regulators | 3 | 7 | 43 | 4.5 | 4.9E-03 |
| Energy metabolism; pentose phosphate pathway | 5 | 17 | 29 | 3.1 | 6.3E-03 |
| Energy metabolism; phenolic acids | 3 | 8 | 38 | 3.9 | 7.7E-03 |
| Regulatory functions; TetR/AcrR family regulators | 6 | 25 | 24 | 2.5 | 0.01 |
Overrepresentation in lifestyle regions was calculated using the null hypothesis that a proportional part of the total number of genes in a functional category may be expected in the lifestyle regions. The proportionality constant used was equal to the number of genes in the lifestyle region, 293, divided by the total number of genes in the chromosome, 3,064.
Significance of overrepresentation was calculated using the cumulative binomial distribution with a probability of success equal to the proportion of genes present genomewide in the particular category.
These findings support the proposed genomic plasticity of this specific region of the L. plantarum WCFS1 genome and are in agreement with the lifestyle adaptation function proposed for this region. Lifestyle adaptation within this region would encompass the frequent acquisition and loss of genes involved in adaptation to niche functions, in particular to the catabolism of different sugars in decaying organic material. We hypothesize that similar lifestyle adaptation islands will exist in other strains of L. plantarum. This would also imply that these strains contain a high number of genes with related functions accumulated within their lifestyle adaptation region that are absent from the WCFS1 genome. Using microarrays, it is not possible to confirm these hypotheses. Also, the expected variation in gene content is too large to allow topology analysis by means of PCR approaches (unpublished observations).
Can the evolutionary history of the lifestyle regions be reconstructed?
The complexity of the pattern of gene content mapping onto the L. plantarum WCFS1 lifestyle region suggests that extensive gain and/or loss of genes represented in this region of the WCFS1 genome has taken place in L. plantarum. Our question was whether a clonal evolutionary tree of the strains (i.e., one in which no horizontal transfer of genetic material between strains takes place) could be reconstructed based on the content of genes that map onto this region in the WCFS1 genome. In such a tree, branching points indicate “gain” or “loss” events of genes or gene clusters and edges represent putative ancestors. It was concluded that a simple clonal tree, where genes or gene clusters are only gained or lost once, could not be reconstructed. This is exemplified for a few gene clusters and a small selection of strains in Fig. 3A and B. Either the same genes or gene clusters were gained or lost in multiple independent events, as shown in the examples, or, alternatively, extensive horizontal transfer (nonclonal inheritance) of genes within the species took place. As was already argued above, it is likely that a large proportion of the genes of the lifestyle adaptation island were acquired by “gain” events through horizontal transfer of genes from species with a different base composition. Nevertheless, subsequent loss of these genes should not be excluded. Alternative explanations for the complex gene content maps are that certain gene clusters have been independently gained (or lost) by previously diverged ancestors of the current strains or that extensive horizontal exchange of clusters (nonclonal inheritance) takes place among L. plantarum strains. The same phenomenon can be observed when comparing the WCFS1 lifestyle adaptation gene content maps of a GLp1 strain like CIP104441 and GLp2 strain Lp85-2. The latter contains many more genes represented in the WCFS1 lifestyle region than the former, suggesting that these genes were present before the GLp1-GLp2 divergence and were lost independently afterwards. Alternatively, they may have been independently gained by or horizontally transferred between both groups. The biological mechanisms underlying this high rate of exchange or gain of foreign genes is highly intriguing. Natural competence has hitherto not been observed in L. plantarum but should not be excluded as a mechanism, since several genes involved in natural competence are present in the genome of strain WCFS1 (25). Moreover, unraveling of the mechanism underlying the concentration of sugar catabolism genes in the 3.10- to 3.29-kb region of the WCFS1 chromosome presents another important challenge. This concentration might be linked to the mechanism of horizontal transfer of genes between L. plantarum and other species or between different strains of L. plantarum. Transposases (lp_3496 at 3.10 kb, lp_3569 and lp_3570 at 3.19 kb) or other mobile elements could be involved in the efficient integration of genes in these regions. A mobile element belonging to a different family was shown to be active in other strains of L. plantarum (35). However, we did not find putative remnants of events induced by mobile elements, like an overrepresentation of transposon fragments, putative integration sites, integration near tRNA genes, or the presence of typical plasmid remnants in the 3.10- to 3.29-kb region. The genomic islands that have been described before also generally have a much smaller size than the lifestyle adaptation island described here, although some large islands of similar size have been found in Salmonella enterica serovar Typhi among others (31). Another reason for the presence of horizontally transferred genes in this region could be that there is an advantage of placing them near the origin of replication. A possible advantage is the relatively high copy number of regions near the origin of replication in fast-growing cells (8), which leads to increased levels of transcription of the genes by a gene dosage effect (29). This effect could be especially beneficial for expression of genes with nonoptimal codon usage, as horizontally transferred genes may be expected to have. Therefore, strains that, possibly coincidentally, integrate such genes near the origin of replication would obtain a selective advantage compared to strains that integrate these genes elsewhere in the genome.
FIG. 3.

Demonstration of complex history of the 3.10- to 3.29-Mb lifestyle region. A region of the gene content maps, mapping on 3.10 to 3.29 Mb of the chromosome of WCFS1, is enlarged for a selection of strains (A). It is to be understood that the organization of genes in the maps does not necessarily represent the physical organization in the strains. Gene clusters (or combinations thereof) are labeled A to D, and the resulting “genotypes” are shown behind the maps, where a “−” stands for the absence of a cluster. Strains 299 and 299v or LM3 and NCDO 1193 have the same genotype and are grouped in panel B. Panel B shows putative clonal histories: one starting from a strain possessing all gene clusters (B1) and the other from a strain lacking all gene clusters (B2). These are only two examples of the many putative histories and are shown here to demonstrate that no clonal tree can be constructed without allowing for multiple independent gain or loss events for the same gene clusters in different ancestors. For example, in tree B1, A, B, and C are lost multiple times in different putative ancestors. The same is true for gain of C and D in tree B2.
Acknowledgments
We thank Peter Bron and Roger Bongers for technical assistance, Bernadet Renckens for in silico genome comparisons, and Ted van der Lende for analysis of microarrays.
REFERENCES
- 1.Adlerberth, I., S. Ahrné, M.-L. Johansson, G. Molin, L. Å. Hanson, and A. E. Wold. 1996. A mannose-specific adherence mechanism in Lactobacillus plantarum conferring binding to the human colonic cell line HT-29. Appl. Environ. Microbiol. 62:2244-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahrne, S., S. Nobaek, B. Jeppsson, I. Adlerberth, A. E. Wold, and G. Molin. 1998. The normal Lactobacillus flora of healthy human rectal and oral mucosa. J. Appl. Microbiol. 85:88-94. [DOI] [PubMed] [Google Scholar]
- 3.Aukrust, T., and H. Blom. 1992. Transformation of Lactobacillus strains used in meat and vegetable fermentations. Food Res. Int. 25:253-261. [Google Scholar]
- 4.Aymerich, T., B. Martin, M. Garriga, and M. Hugas. 2003. Microbial quality and direct PCR identification of lactic acid bacteria and nonpathogenic staphylococci from artisanal low-acid sausages. Appl. Environ. Microbiol. 69:4583-4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bateman, A., L. Coin, R. Durbin, R. D. Finn, V. Hollich, S. Griffiths-Jones, A. Khanna, M. Marshall, S. Moxon, E. L. Sonnhammer, D. J. Studholme, C. Yeats, and S. R. Eddy. 2004. The Pfam protein families database. Nucleic Acids Res. 32(Database issue):D138-D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behr, M. A., M. A. Wilson, W. P. Gill, H. Salamon, G. K. Schoolnik, S. Rane, and P. M. Small. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284:1520-1523. [DOI] [PubMed] [Google Scholar]
- 7.Beneduce, L., G. Spano, A. Vernile, D. Tarantino, and S. Massa. 2004. Molecular characterization of lactic acid populations associated with wine spoilage. J. Basic Microbiol. 44:10-16. [DOI] [PubMed] [Google Scholar]
- 8.Bipatnath, M., P. P. Dennis, and H. Bremer. 1998. Initiation and velocity of chromosome replication in Escherichia coli B/r and K-12. J. Bacteriol. 180:265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bringel, F., M.-C. Curk, and J.-C. Hubert. 1996. Characterization of lactobacilli by Southern-type hybridization with a Lactobacillus plantarum pyrDFE probe. Int. J. Syst. Bacteriol. 46:588-594. [DOI] [PubMed] [Google Scholar]
- 10.Bringel, F., P. Quenee, and P. Tailliez. 2001. Polyphasic investigation of the diversity within Lactobacillus plantarum related strains revealed two L. plantarum subgroups. Syst. Appl. Microbiol. 24:561-571. [DOI] [PubMed] [Google Scholar]
- 11.Broadbent, J. R., D. J. McMahon, D. L. Welker, C. J. Oberg, and S. Moineau. 2003. Biochemistry, genetics, and applications of exopolysaccharide production in Streptococcus thermophilus: a review. J. Dairy Sci. 86:407-423. [DOI] [PubMed] [Google Scholar]
- 12.Chevallier, B., J. C. Hubert, and B. Kammerer. 1994. Determination of chromosome size and number of rrn loci in Lactobacillus plantarum by pulsed-field gel electrophoresis. FEMS Microbiol. Lett. 120:51-56. [DOI] [PubMed] [Google Scholar]
- 13.Daubin, V., N. A. Moran, and H. Ochman. 2003. Phylogenetics and the cohesion of bacterial genomes. Science 301:829-832. [DOI] [PubMed] [Google Scholar]
- 14.Dharmapuri, S., J. Yashitola, M. R. Vishnupriya, and R. V. Sonti. 2001. Novel genomic locus with atypical G+C content that is required for extracellular polysaccharide production and virulence in Xanthomonas oryzae pv. oryzae. Mol. Plant-Microbe Interact. 14:1335-1339. [DOI] [PubMed] [Google Scholar]
- 15.Dobrindt, U., B. Hochhut, U. Hentschel, and J. Hacker. 2004. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2:414-424. [DOI] [PubMed] [Google Scholar]
- 16.Ercolini, D., P. J. Hill, and C. E. R. Dodd. 2003. Bacterial community structure and location in Stilton cheese. Appl. Environ. Microbiol. 69:3540-3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabret, C., Y. Quentin, N. Chapal, A. Guiseppi, J. Haiech, and F. Denizot. 1996. Integrated mapping and sequencing of a 115 kb DNA fragment from Bacillus subtilis: sequence analysis of a 21 kb segment containing the sigL locus. Microbiology 142:3089-3096. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa, C., A. Davila, and J. Pourquié. 1995. Lactic acid bacteria of the sour cassava starch fermentation. Lett. Appl. Microbiol. 21:126-130. [Google Scholar]
- 19.Fitzgerald, J. R., and J. M. Musser. 2001. Evolutionary genomics of pathogenic bacteria. Trends Microbiol. 9:547-553. [DOI] [PubMed] [Google Scholar]
- 20.Fitzgerald, J. R., S. D. Reid, E. Ruotsalainen, T. J. Tripp, M. Liu, R. Cole, P. Kuusela, P. M. Schlievert, A. Järvinen, and J. M. Musser. 2003. Genome diversification in Staphylococcus aureus: molecular evolution of a highly variable chromosomal region encoding the staphylococcal exotoxin-like family of proteins. Infect. Immun. 71:2827-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgerald, J. R., D. E. Sturdevant, S. M. Mackie, S. R. Gill, and J. M. Musser. 2001. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. USA 98:8821-8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner, N. J., T. Savard, P. Obermeier, G. Caldwell, and C. P. Champagne. 2001. Selection and characterization of mixed starter cultures for lactic acid fermentation of carrot, cabbage, beet and onion vegetable mixtures. Int. J. Food Microbiol. 64:261-275. [DOI] [PubMed] [Google Scholar]
- 23.Jim, K., K. Parmar, M. Singh, and S. Tavazoie. 2004. A cross-genomic approach for systematic mapping of phenotypic traits to genes. Genome Res. 14:109-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson, M.-L., G. Molin, B. Jeppsson, S. Nobaek, S. Ahrné, and S. Bengmark. 1993. Administration of different Lactobacillus strains in fermented oatmeal soup: in vivo colonization of human intestinal mucosa and effect on the indigenous flora. Appl. Environ. Microbiol. 59:15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleerebezem, M., J. Boekhorst, R. van Kranenburg, D. Molenaar, O. P. Kuipers, R. Leer, R. Tarchini, S. A. Peters, H. M. Sandbrink, M. W. Fiers, W. Stiekema, R. M. Lankhorst, P. A. Bron, S. M. Hoffer, M. N. Groot, R. Kerkhoven, M. de Vries, B. Ursing, W. M. de Vos, and R. J. Siezen. 2003. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. USA 100:1990-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koski, L. B., R. A. Morton, and G. B. Golding. 2001. Codon bias and base composition are poor indicators of horizontally transferred genes. Mol. Biol. Evol. 18:404-412. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence, J. G., and H. Ochman. 1998. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. USA 95:9413-9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levesque, M., D. Shasha, W. Kim, M. G. Surette, and P. N. Benfey. 2003. Trait-to-gene: a computational method for predicting the function of uncharacterized genes. Curr. Biol. 13:129-133. [DOI] [PubMed] [Google Scholar]
- 29.Løbner-Olesen, A., M. G. Marinus, and F. G. Hansen. 2003. Role of SeqA and Dam in Escherichia coli gene expression: a global/microarray analysis. Proc. Natl. Acad. Sci. USA 100:4672-4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maldonado, A., R. Jimenez-Diaz, and J. L. Ruiz-Barba. 2004. Induction of plantaricin production in Lactobacillus plantarum NC8 after coculture with specific gram-positive bacteria is mediated by an autoinduction mechanism. J. Bacteriol. 186:1556-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantri, Y., and K. P. Williams. 2004. Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res. 32(Database issue):D55-D58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molin, G., B. Jeppsson, M. L. Johansson, S. Ahrne, S. Nobaek, M. Stahl, and S. Bengmark. 1993. Numerical taxonomy of Lactobacillus spp. associated with healthy and diseased mucosa of the human intestines. J. Appl. Bacteriol. 74:314-323. [DOI] [PubMed] [Google Scholar]
- 33.Murray, A. E., D. Lies, G. Li, K. Nealson, J. Zhou, and J. M. Tiedje. 2001. DNA/DNA hybridization to microarrays reveals gene-specific differences between closely related microbial genomes. Proc. Natl. Acad. Sci. USA 98:9853-9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muscariello, L., R. Marasco, M. De Felice, and M. Sacco. 2001. The functional ccpA gene is required for carbon catabolite repression in Lactobacillus plantarum. Appl. Environ. Microbiol. 67:2903-2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicoloff, H., and F. Bringel. 2003. ISLpl1 is a functional IS30-related insertion element in Lactobacillus plantarum that is also found in other lactic acid bacteria. Appl. Environ. Microbiol. 69:6032-6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299-304. [DOI] [PubMed] [Google Scholar]
- 37.Ohnishi, M., J. Terajima, K. Kurokawa, K. Nakayama, T. Murata, K. Tamura, Y. Ogura, H. Watanabe, and T. Hayashi. 2002. Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. Proc. Natl. Acad. Sci. USA 99:17043-17048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poly, F., D. Threadgill, and A. Stintzi. 2004. Identification of Campylobacter jejuni ATCC 43431-specific genes by whole microbial genome comparisons. J. Bacteriol. 186:4781-4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38a.Pretzer, G., J. Snel, D. Molenaar, A. Wiersma, P. A. Bron, J. Lambert, W. M. de Vos, R. van der Meer, M. A. Smits, and M. Kleerebezem. 2005. Biodiversity-based identification and functional characterization of the mannose-specific adhesin of Lactobacillus plantarum. J. Bacteriol. 187:6129-6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts, C. J., B. Nelson, M. J. Marton, R. Stoughton, M. R. Meyer, H. A. Bennett, Y. D. He, H. Dai, W. L. Walker, T. R. Hughes, M. Tyers, C. Boone, and S. H. Friend. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287:873-880. [DOI] [PubMed] [Google Scholar]
- 40.Ruiz-Barba, J. L., J. C. Piard, and R. Jimenez-Diaz. 1991. Plasmid profiles and curing of plasmids in Lactobacillus plantarum strains isolated from green olive fermentations. J. Appl. Bacteriol. 71:417-421. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 42.Sharp, P. M., and W. H. Li. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15:1281-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharp, P. M., and G. Matassi. 1994. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 4:851-860. [DOI] [PubMed] [Google Scholar]
- 44.Smith, K. S., and J. G. Ferry. 2000. Prokaryotic carbonic anhydrases. FEMS Microbiol. Rev. 24:335-366. [DOI] [PubMed] [Google Scholar]
- 45.Suerbaum, S., C. Josenhans, T. Sterzenbach, B. Drescher, P. Brandt, M. Bell, M. Droge, B. Fartmann, H. P. Fischer, Z. Ge, A. Horster, R. Holland, K. Klein, J. Konig, L. Macko, G. L. Mendz, G. Nyakatura, D. B. Schauer, Z. Shen, J. Weber, M. Frosch, and J. G. Fox. 2003. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA 100:7901-7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ventura, M., C. Canchaya, M. Kleerebezem, W. M. de Vos, R. J. Siezen, and H. Brussow. 2003. The prophage sequences of Lactobacillus plantarum strain WCFS1. Virology 316:245-255. [DOI] [PubMed] [Google Scholar]
- 47.Weickert, M. J., and S. Adhya. 1992. A family of bacterial regulators homologous to Gal and Lac repressors. J. Biol. Chem. 267:15869-15874. [PubMed] [Google Scholar]
- 48.Winzeler, E. A., B. Lee, J. H. McCusker, and R. W. Davis. 1999. Whole genome genetic-typing in yeast using high-density oligonucleotide arrays. Parasitology 118:S73-S80. [DOI] [PubMed] [Google Scholar]