

Abstract
Aims
In view of available targeted therapies, we investigated the presence of c-kit, PDGFR gene mutations and protein expression in cancer of unknown primary (CUP) in order to study their contribution in pathogenesis, their prognostic value and potential as therapeutic targets.
Methods
Mutations in hot spots c-kit exon 11 and PDGFR exons 12 and 18 were studied in paraffin-embedded tumour samples from 50 patients with CUP by means of PCR-based single-strand conformational polymorphism and protein expression by means of streptavidin-biotin immunoperoxidase assays. Molecular markers were screened for possible correlations with patient outcome.
Results
No shifted band was detected in any of the polyacrylamide gel electrophoreses, indicating absence of c-kit exon 11 and PDGFR exon 12, 18 mutations. Immunohistochemical analysis in 37 tumours revealed positive membranous CD117 expression in 30 samples (81%) of which five exhibited strong (+3), four moderate (+2) and 21 weak (+1) staining. PDGFRa protein staining was seen in 15 out of 30 (50%) cases, mostly weak (13) and rarely moderate (1) or strong (1). The expression of KIT or PDGFRa protein did not correlate with the clinical outcome of the patients in our cohort.
Conclusions
In a moderate-sized CUP patient cohort, KIT or PDGFRa protein overexpression is rare, does not have gross prognostic significance for survival and is not associated with presence of activating mutations.
Keywords: C-KIT, PDGFR, Mutations, Immunohistochemistry, Cancer of unknown primary
Introduction
Cancer of unknown primary (CUP) makes up to 3–5% of metastatic malignancies. It is characterised by failure to identify the primary site with appropriate clinical, imaging and laboratory work-up. The majority of CUP patients present with widespread metastases after a short symptomatic period, do not benefit from systemic therapy and succumb to malignancy within a few months. Although some progress has been made recently with the identification of favourable prognosis subgroups of CUP patients who occasionally enjoy long-term survival with chemotherapy, the molecular pathophysiology of the disease is still poorly understood (Pentheroudakis et al. 2007). No molecular markers of established predictive or prognostic significance have been found to date (Pentheroudakis and Pavlidis 2006).
The proto-oncogene c-kit encodes a type III transmembrane receptor (CD117) with cytoplasmic tyrosine kinase activity involved in inhibition of apoptosis, regulation of cell adhesion and induction of cellular proliferation. Constitutive ligand-independent KIT activation can occur by gain-of-function mutations mainly observed in gastrointestinal stromal tumours (GIST), an early oncogenic event that contributes to malignant transformation of the interstitial Cajal cells of the myenteric plexus. The most common such mutations are seen in exon 11 of the gene, the mutated exon encoding a juxtamembrane domain that facilitates spontaneous KIT receptor dimerisation and activation of tyrosine kinase (Hirota et al. 1998).
Two structurally related transmembrane protein tyrosine kinase receptor subunits, α- and β-, represent the PDGF receptor with activity involved in regulation of cell growth, actin reorganisation and chemotaxis, among others (Heldin and Westermark 1999). Constitutive ligand-independent PDGFRa activation can occur by gain-of-function mutations mainly observed in GISTs and most commonly found in exons 12 and 18 of the gene, the mutated exon encoding a juxtamembrane and a tyrosine kinase II domain, respectively (Hirota et al. 2003; Heinrich et al. 2003a).
The advent of receptor tyrosine kinase inhibitors provided impetus for the identification of target molecules driving neoplastic growth. Imatinib (STI 571, Gleevec®) inhibits KIT, BCR-ABL and PDGFR tyrosine kinases, producing remarkable clinical benefit in patients with GIST and chronic myelogenous leukemia (Buchdunger et al. 2000; Demetri et al. 2002). Sunitinib inhibits VEGFR, PDGFR, KIT and Fms-like tyrosine kinase 3 (Marx 2005), producing benefit in renal-cell carcinoma (Motzer et al. 2006a, b) and GIST patients, among others. The lack of insight into the molecular biology of CUP, lack of data on KIT and PDGFR status along with the prospect for beneficial tyrosine kinase inhibition with targeted drugs have prompted us to screen for exon 11 c-kit and exon 12 and 18 PDGFR gene mutations and respective protein expression in CUP.
Materials and methods
Tumour tissue from formalin-fixed paraffin embedded biopsy specimens was collected from 50 patients with CUP managed at the Department of Medical Oncology at the Ioannina University Hospital. All patients were treated with combination chemotherapy according to Hellenic Cooperative Oncology Group (HeCOG) protocols. Histological sections were used for histopathologic diagnosis with hematoxylin-eosin staining, for evaluation of CD117, PDGFRa protein expression and for mutational gene analysis. For all tumour samples included in the analysis, the number of malignant cells represented at least 75% of all nucleated cells, as judged by hematoxylin-eosin staining. The protocol was approved by the local research ethics committee of the Ioannina University Hospital and all patients provided written informed consent for research use of their biologic material.
Patient characteristics
Patient demographics are described in Table 1. Most patients were aged more than 60, had mild to moderate symptoms and were diagnosed with metastatic adenocarcinoma or undifferentiated carcinoma of unknown origin. The majority had metastases in two or more organ sites, mainly the liver, peritoneum, lymph nodes and skeleton. All received combination cytotoxic chemotherapy, platinum-based in 43.
Table 1.
Patient demographics
| N = 50 | |
|---|---|
| Median age (range) | 68 (45–85) |
| Sex (male/female) | 29/21 |
| Performance status | |
| 0 | 6 |
| 1 | 28 |
| 2 | 16 |
| Histology | |
| Adenocarcinoma | 28 |
| Undifferentiated carcinoma | 17 |
| Neuroendocrine carcinoma | 2 |
| Squamous carcinoma | 3 |
| Differentiation (grade) | |
| 1 | 5 |
| 2 | 12 |
| 3 | 25 |
| Not available | 8 |
| Metastatic sites | |
| Liver | 9 |
| Bone | 5 |
| Lung | 5 |
| Lymph nodes | 11 |
| Soft tissues | 4 |
| Brain | 2 |
| Peritoneum | 10 |
| Chemotherapy | |
| Platinum-based | 43 |
| Taxane-based | 30 |
| Non platinum, non taxane | 7 |
| Median survival (95% CI) | 9 months (7–11) |
Immunohistochemical assay
KIT and PDGFRa oncoproteins were evaluated immunohistochemically by means of streptavidin-biotin complex immunoperoxidase assay on 5 μm sections from formalin-fixed paraffin-embedded tissue in 37 and 30 cases of CUP, respectively. Histopathologic material from 13 and 20 cases could not be retrieved from referring hospitals and private practices for additional immunohistochemical studies; these cases were thus not available for CD117 and PDGFRa staining. The slides were immunostained with the polyclonal rabbit antibodies anti-CD117 (anti-human C-KIT A 4502, Dako, Glostrup, Denmark) and anti-PDGFRa (anti-human PDGFRa 3164, Cell Signaling Technologies, USA) diluted at 1:300, using streptavidin-biotin complex autostainer (Labvision Corp, Fremont, CA) with diaminobenzidine as chromogen. Staining was analysed with a detection kit (Envision Detection Kit, Peroxidase/DAB, Dako) designed for use with the automated immunostaining system.
Staining evaluation
The immunohistochemical CD117 and PDGFRa protein expression was evaluated semi-quantitatively using a four-tiered scale (score 0, +1, +2, +3) based on the intensity of membranous or membranous and cytoplasmic staining and the percentage of positive tumour cells (0–100%). Samples with less than 5% of weakly stained cells were considered to be negative (score 0), samples with 5–25% weak to moderate staining were weakly positive (+1), those with 25–50% moderate or strong staining were moderately positive (+2), whereas the ones with moderate or strong staining in more than 50% of cells were graded as strongly positive (+3).
Molecular analysis
Genomic DNA isolation in five 10-μm thick cut sections from paraffin-embedded tissue blocks was performed using the QIAGEN DNEasy TM tissue kit (Westburg, Leusden, The Netherlands) according to the manufacturer’s instructions. PCR gene amplification was performed using a cycling thermal protocol with Taq DNA polymerase (Dia Tech, Padre V. Pellegrini, Italy) in 25 μl reaction volume containing 0.2 μΜ οf each primer, 1.5–2.5 mM MgCl2, 200 μM of each 5′-deoxytriphosphates (dNTPs) and 250 ng of sample DNA. The primer sequences used for amplification of the complete C-KIT exon 11 coding sequences (coding the juxtamembrane domain: codons 550–590) were 5′-CCA-GAG-TGC-TCT-AAT-GAC-TGA-GAC-3′ as forward primer and 5′-ACT-CAG-CCT-GTT-TCT-GGG-AAA-CTC-3′ as reverse primer. The primers used for PDGFR exon 12 (coding the juxtamembrane domain: codons 560–572) were 5′-TCC-AGT-CAC-TGT-GCT-GCT-TC-3′ as forward primer and 5′-GCA-AGG-GAA-AAG-GGA-GTC-TT-3′ as reverse primer and PDGFR exon 18 (coding the activating loop: codons 838–850) were 5′-ACC-ATG-GAT-CAG-CCA-GTC-TT-3′ as forward primer and 5′-TGA-AGG-AGG-ATG-AGC-CTG-ACC-3′ as reverse primer (Invitrogen Life Technologies, Merelbeke, Belgium). The PCR amplification process included denaturation at 95°C for 4 min followed by 40 cycles of 95°C denaturation for 30 s, 55–60°C annealing for 45 s, 72°C extension for 45 s. Amplified fragments were analysed on high resolution 2% w/v Metaphor agarose gel (BioWhittaker Molecular Applications, Rockland, ME, USA).
PCR amplified products were fully denatured with addition of loading buffer consisting of 95% v/v formamide, 10 mM NaOH, 0.03% w/v bromophenol blue, 0.03% w/v xylene cyanol and incubation at 95°C for 6 min. Subsequently, 4 μl aliquots were loaded on 10% w/v non-denaturing polyacrylamide gel containing 5% v/v glycerol. Single-strand conformational polymorphism (SSCP) analysis was carried out at a constant 12 W for 16–20 h at room temperature and visualisation of bands done by means of silver staining. In the absence of a supplementary strand, the single strand undergoes intrastrand base pairing that shapes a unique tri-dimensional (3D) structure. Subtle nucleotide changes (point mutations, insertions or deletions as small as less than 3 bp) alter the base pairing and 3D conformation of the strand, resulting in differential electrophoretic mobility through the gel.
Statistical analysis
Overall survival analyses (from diagnosis till date of death or last follow-up) were performed with the Kaplan-Meier product limit method. Cox regressional hazard analysis was applied to study prognostic significance of variables for survival and the χ2 Exact test for the predictive significance of variables for response to therapy. All analyses were performed with the use of the SPSS 13.0 statistical software package.
Results
Immunohistochemical protein expression
Immunostaining for the KIT protein (CD117) was available in 37 and for PDGFRa in 30 out of 50 cases. Seven of 37 analysed samples (19%) were graded as negative for CD117 expression. Τwenty-one samples were weakly positive (57%), four moderately positive (11%) while five tumours showed strong CD117 staining (13%), comparable to mast cells, commonly used as internal positive controls. PDGFRa staining was seen in 15 cases (50%), weak in 13 of them, moderate in one and strong in one. Stromal cells and pericytes exhibiting strong PDGFRa staining were used as internal positive controls.
The immunostaining in positive samples was either membranous only or membranous and cytoplasmic. Cytoplasmic only staining proved to be false-positive in preabsorption negative control experiments. No accentuation of perinuclear staining (Golgi apparatus) was seen in samples with membranous and cytoplasmic staining. In view of the high rate of CD117 positivity, we validated the immunohistochemical methodology and found positive CD117 immunostaining in 7/7 gastrointestinal stromal tumours, 4/12 seminomas and 3/15 small-cell lung carcinomas. Samples with representative negative and strongly positive CD117 and moderately positive PDGFRa staining are shown in Figs. 1, 2.
Fig. 1.
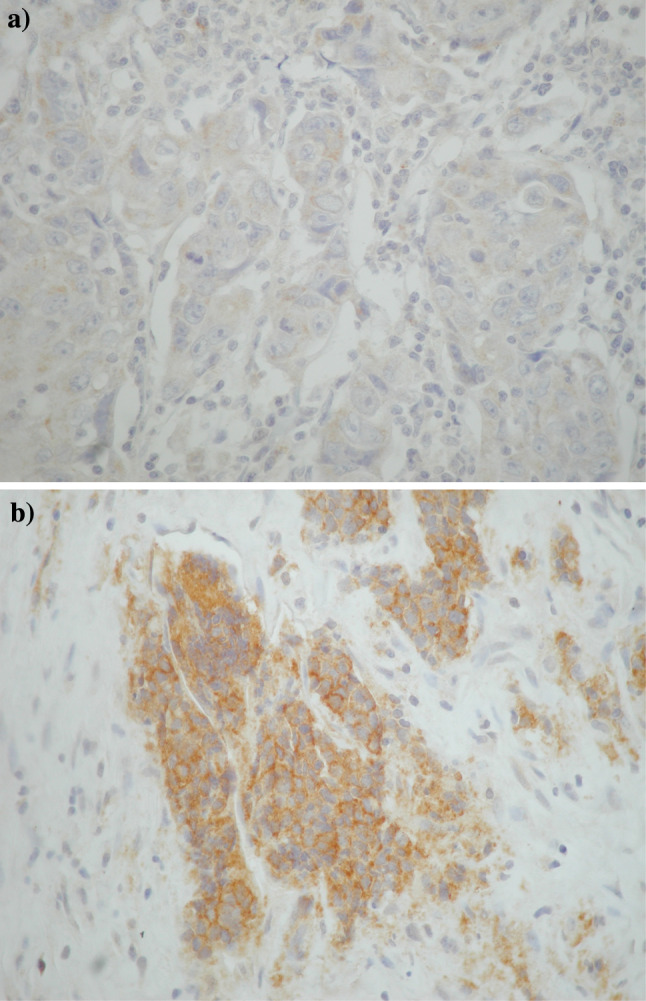
Case with (a) negative and (b) strongly positive C-KIT/CD117 immunohistochemical expression
Fig. 2.
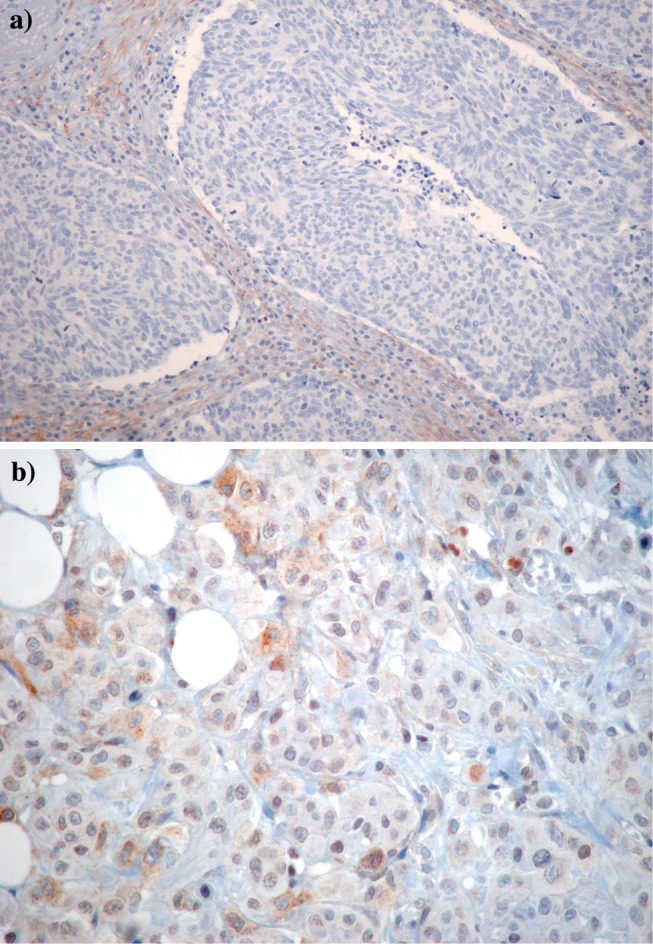
Case with (a) negative and (b) moderately positive PDGFRa immunohistochemical expression
Molecular analysis for C-KIT/PDGFR polymorphism
The complete exon 11 coding sequences (codons 550–590) of the juxtamembrane domain of c-kit, the coding sequences of exon 12 (codons 560–572, juxtamembrane domain), and of exon 18 (codons 838–850, activating loop) of the PDGFRa of 50 CUP tumours, as well as of two gastrointestinal stromal tumours (positive controls representing the c-kit exon 11 WK 557–558 deletion) and of peripheral blood lymphocytes from four healthy donors (negative controls) were PCR-amplified. The quality of the amplified fragments was tested by agarose gel electrophoresis. The samples were subsequently screened for mutational polymorphism by SSCP and non-denaturing gel electrophoresis. We were unable to observe c-kit exon 11 or PDGFR exon 12- or 18-point mutations, insertions or deletions in any of the 50 samples, including the four negative controls. No shifted band (aberrant pattern) was detected in any of the CUP tumours, in contrast to the two gastrointestinal stromal tumours serving as positive controls that harboured shifted bands characteristic for exon 11 c-kit mutations. In order to increase the method sensitivity and the likelihood of detecting subtle differences in DNA strand migration, polyacrylamide gels were run at room temperature as well as at 4°C, without observation of any aberrant bands. These results indicate the absence of c-kit exon 11 and PDGFR exon 12 or 18 mutations in our CUP patients who apparently carry tumours with wild-type c-kit and PDGFR genotype, at least as far as the specific “hot-spot” exons are concerned. A characteristic example of lack of c-kit polymorphism in amplified DNA bands on polyacrylamide gel electrophoresis is given in Fig. 3.
Fig. 3.

SSCP polyacrylamide gel electrophoresis in 7 CUP samples for the detection of C-KIT aberrant bands. The aberrant pattern of the GIST mutated exon 11 is represented on lanes 1 and 11 (positive controls). Wild type exon 11 is represented on lanes 2 and 10 (negative control). Lanes 3–9 are analysed CUP tumour samples
Clinical outcome and prognostic significance of protein expression
At a median follow-up time of 60 months, 87% of patients have died. The median overall survival of 50 patients was 9 months (95% CI 7–11 months). In the 37 patients with available immunohistochemical studies of KIT protein expression, the latter did not have any prognostic significance for survival when either positive versus negative or strong-moderate versus weak-negative staining patterns were analysed (Cox hazard ratio for death 1.02, 95% CI 0.4–2.7, P = 0.9 and 1.01, 95% CI 0.37–2.8, P = 0.98 respectively). Positive, strong/moderate or strong KIT staining also failed to predict for tumour response to antineoplastic chemotherapy (Fisher’s Exact test P > 0.05). In the 30 patients with available immunohistochemical studies of PDGFRa protein expression, the presence of any degree of positive staining did not have prognostic significance for survival (Cox hazard ratio for death 0.67, 95% CI 0.2–1.9, P = 0.47). Positive PDGFRa protein staining was not a predictive factor for tumour regression from chemotherapy (Fisher’s Exact test P > 0.05). The survival of patients with negative-weak or moderate-strong CD117 staining is shown in Fig. 4 and those with positive versus negative PDGFRa staining in Fig. 5.
Fig. 4.

Overall survival of patients with negative-weak and moderate-strong CD117 tumour staining
Fig. 5.

Overall survival of patients with positive versus negative PDGFRa protein tumour staining
Discussion
The c-kit and PDGFRa genes, located both on the long arm of chromosome 4, encode for the KIT and PDGFRa receptor tyrosine kinases consisting of an extracellular domain, the transmembrane segment and the juxtamembrane and cytoplasmic domains (Yarden et al. 1987; Claesson-Welsh et al. 1989). The juxtamembrane domain is responsible for the dimerisation of two KIT receptors upon binding to the extracellular segment of its ligand, Stem Cell Factor (SCF). The extracellular Ig domain 4 is responsible for the dimerisation of two PDGF receptors, in addition to the bridging effect of its ligand, PDGF. After ligand-induced activation and dimerisation, the cytoplasmic segment of both receptors undergoes autophosphorylation of tyrosine residues and activation of signal transduction pathways favouring cell survival and proliferation (Kitamura and Hirotab 2004; Kazlauskas and Cooper 1989).
The pivotal oncogenic effect of KIT activation has been demonstrated primarily in malignant gastrointestinal stromal tumours (GISTs). Gain-of-function mutations of the c-kit gene that result in ligand-independent constitutive KIT receptor protein dimerisation and tyrosine kinase activation has been found in 85–90% of these tumours (Rubin et al. 2001). These small in-frame deletions, insertions or point mutations were most frequently found (80% of mutated cases) at exon 11 encoding for the juxtamembrane domain (mostly in codons 550–562) and occasionally at exons 9, 13, 14 and 17, encoding for the extracellular and tyrosine kinase domains. Preclinical experiments along with clinical observations proved that C-KIT constitutive activation is an early, necessary and sufficient oncogenic stimulus for malignant transformation and that GISTs remain dependent on it for continuing growth (Hirota et al. 1998; Kitamura and Hirotab 2004; de Silva and Reid 2003).
Expression of PDGF receptor has been linked to the development of several tumours, including gliomas and GISTs (Shih and Holland 2006; Corless et al. 2004). Activating mutations were found in exons 12 and 18 of the PDGF receptor, which encode parts of the juxtamembrane and the tyrosine kinase regions. The signal transduction profiles of the PDGFR-mutant GISTs were indistinguishable from KIT-mutant tumours, suggesting that PDGFRa can substitute for KIT in GIST oncogenesis (Heinrich et al. 2003).
The advent of imatinib mesylate (Gleevec®, Novartis) and sunitinib (Sutent®, Pfizer) has inaugurated new therapeutic avenues with drugs targeting tumour-specific molecular abnormalities. Dramatic responses are seen in metastatic GIST patients treated with imatinib (Buchdunger et al. 2000; Demetri et al. 2002) and sunitinib (Demetri et al. 2006). Interestingly, GIST patients with c-kit mutations in exon 11 respond better to imatinib (tumour regression rate 72%) and live longer than patients with mutations in other exons or wild type c-kit (tumour regression rate 32 and 12%, respectively) (Heinrich et al. 2003b; Debiec-Rychter et al. 2004). Implications have also been made concerning a possible correlation between PDGF receptor mutations and response to imatinib treatment (Corless et al. 2005).
The results of our study show that in patients with unknown primary carcinoma neither KIT oncoprotein expression, present in 81% of our cases, nor PDGFRa protein expression, present in 50% of our cases, are associated with exon 11 of c-kit or exons 12 or 18 of PDGFR gene activating mutations. PCR-based SSCP is a fast, economic and highly sensitive molecular technique that allows reliable detection of nucleotide changes in samples containing as little as 10% of mutated DNA. Immunohistochemical KIT protein expression has been reported by other investigators in as many as 70% of small cell lung carcinomas, 84% of seminomas, 65% of adenoid cystic carcinomas, 35% of melanomas and occasionally in 47 additional tumour types (Went et al. 2004). Immunohistochemical PDGFRa expression has been reported in head-and-neck (Ongkeko et al. 2005), melanoma (Ugurel et al. 2005), renal cell carcinoma (Sulzbacher et al. 2003a), osteosarcoma (Sulzbacher et al. 2003b), mesothelioma (Langerak et al. 1996) and ovarian carcinoma (Wilczynski et al. 2005), among others. Despite the relatively frequent oncoprotein expression, activating c-kit gene mutations were only observed in GISTs and never reported in other tumour types (Went et al. 2004; Burger et al. 2003; Sihto et al. 2005). Similarly, none or few exon 12 or 18 PDGFR gene mutations have been so far reported in the various series analysed (Liegl et al. 2006; Gomes et al. 2007; Peterson et al. 2006; Carvalho et al. 2005). In contrast to our findings, Rashid et al. recently reported KIT immunohistochemical expression in only 9 out of 76 patients with CUP (Rashid et al. 2005). This highlights the inherent heterogeneity that probably characterises every CUP patient cohort. KIT protein expression did not have prognostic significance for survival in our cohort, in keeping with previous experience published (Debiec-Rychter et al. 2004). We failed to find prognostic significance for PDGFRa protein expression either, at a time when no other published studies, to our current knowledge, have examined immunohistochemical expression of PDGF receptor in CUP patient cohorts. The moderate sample size, a restriction common in all CUP studies published, makes it impossible to rule out a weaker prognostic utility of these molecular markers.
The absence of activating mutations of c-kit as well as the lack of firm evidence that the oncoprotein activity is important for cell transformation and survival in tumour types other than GIST may explain the failure of imatinib therapy in patients with soft tissue sarcomas, small cell lung cancer and germ cell tumours (Kitamura and Hirotab 2004; Heinrich et al. 2003b; Johnson et al. 2003). The prognostic implications of PDGFR gene mutations, if any, is not clarified from the evidence collected so far. Our data highlight absence of established molecular predictors of benefit from tyrosine kinase inhibitors in CUP, adding to accumulating evidence that its molecular pathogenesis is complex and heterogeneous. We recently failed to find epidermal growth factor receptor (EGFR) activating mutations or amplification in 50 CUP tumours (Dova et al. 2007). Although mechanisms other than gain-of-function mutations such as wild-type gene amplification, SCF overproduction, cross-activation by other kinases, gene promoter demethylation may result in KIT/PDGFR activation in solid tumours, our data pinpoint lack of known molecular markers of benefit from imatinib or sunitinib (Sihto et al. 2005). Consequently, further preclinical studies exploring alternative routes of activation of C-KIT and PDGFR should be carried out before embarking on CUP clinical trials that involve administration of TKI compounds.
References
- Buchdunger E, Cioffi CL, Law N et al (2000) Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther 295(1):139–145 [PubMed] [Google Scholar]
- Burger H, den Bakker MA, Stoter G, Verweij J, Nooter K (2003) Lack of c-kit exon 11 activating mutations in c-KIT/CD117-positive SCLC tumour specimens. Eur J Cancer 39(6):793–799 [DOI] [PubMed] [Google Scholar]
- Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F (2005) Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res 7(5):R788–R795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson-Welsh L, Eriksson A, Westermark B, Heldin CH (1989) cDNA cloning and expression of the human A-type platelet-derived growth factor (PDGF) receptor establishes structural similarity to the B-type PDGF receptor. Proc Natl Acad Sci USA 86(13):4917–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corless CL, Fletcher JA, Heinrich MC (2004) Biology of gastrointestinal stromal tumors. J Clin Oncol 22(18):3813–3825 [DOI] [PubMed] [Google Scholar]
- Corless CL, Schroeder A, Griffith D et al (2005) PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 23(23):5357–5364 [DOI] [PubMed] [Google Scholar]
- Debiec-Rychter M, Dumez H, Judson I et al (2004) EORTC Soft Tissue and Bone Sarcoma Group. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 40(5):689–695 [DOI] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD et al (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347(7):472–480 [DOI] [PubMed] [Google Scholar]
- Demetri GD, van Oosterom AT, Garrett CR et al (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368(9544):1329–1338 [DOI] [PubMed] [Google Scholar]
- de Silva CM, Reid R (2003) Gastrointestinal stromal tumors (GIST): C-kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib. Pathol Oncol Res 9(1):13–19 [DOI] [PubMed] [Google Scholar]
- Dova L, Pentheroudakis G, Georgiou I et al (2007) Global profiling of EGFR gene mutation, amplification, regulation and tissue protein expression in unknown primary carcinomas: to target or not to target? Clin Exp Metastases 24(2):79–86 [DOI] [PubMed] [Google Scholar]
- Gomes AL, Bardales RH, Milanezi F, Reis RM, Schmitt F (2007) Molecular analysis of c-Kit and PDGFRA in GISTs diagnosed by EUS. Am J Clin Pathol 127(1):1–8 [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Duensing A et al (2003a) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299(5607):708–710 [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Demetri GD et al (2003b) Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21(23):4342–4349 [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79(4):1283–1316 [DOI] [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y et al (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279(5350):577–580 [DOI] [PubMed] [Google Scholar]
- Hirota S, Ohashi A, Nishida T et al (2003) Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 125(3):660–667 [DOI] [PubMed] [Google Scholar]
- Johnson BE, Fischer T, Fischer B et al (2003) Phase II study of imatinib in patients with small cell lung cancer. Clin Cancer Res 9(16 Pt 1):5880–5887 [PubMed] [Google Scholar]
- Kazlauskas A, Cooper JA (1989) Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins. Cell 58(6):1121–1133 [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Hirotab S (2004) Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci 61(23):2924–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak AW, De Laat PA, Van Der Linden-Van Beurden CA et al (1996) Expression of platelet-derived growth factor (PDGF) and PDGF receptors in human malignant mesothelioma in vitro and in vivo. J Pathol 178(2):151–160 [DOI] [PubMed] [Google Scholar]
- Liegl B, Leithner A, Bauernhofer T et al (2006) Immunohistochemical and mutational analysis of PDGF and PDGFR in desmoid tumours: is there a role for tyrosine kinase inhibitors in c-kit-negative desmoid tumours? Histopathology 49(6):576–581 [DOI] [PubMed] [Google Scholar]
- Marx J. (2005) Cancer. Encouraging results for second-generation antiangiogenesis drugs. Science 308(5726):1248–1249 [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Rini BI, Bukowski RM et al (2006a) Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295(21):2516–2524 [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Michaelson MD, Redman BG et al (2006a) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24(1):16–24 [DOI] [PubMed] [Google Scholar]
- Ongkeko WM, Altuna X, Weisman RA, Wang-Rodriguez J (2005) Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. Am J Clin Pathol 124(1):71–76 [DOI] [PubMed] [Google Scholar]
- Pentheroudakis G, Pavlidis N (2006) Perspectives for targeted therapy in cancer of unknown primary site. Cancer Treat Rev 32:637–644 [DOI] [PubMed] [Google Scholar]
- Pentheroudakis G, Briasoulis E, Pavlidis N (2007) Cancer of unknown primary site: missing primary or missing biology? Oncologist 12(4):418–425 [DOI] [PubMed] [Google Scholar]
- Peterson MR, Piao Z, Weidner N, Yi ES (2006) Strong PDGFRA positivity is seen in GISTs but not in other intra-abdominal mesenchymal tumors: immunohistochemical and mutational analyses. Appl Immunohistochem Mol Morphol 14(4):390–6 [DOI] [PubMed] [Google Scholar]
- Rashid A, Hess KR, Lenzi R, et al (2005) Overexpression and prevalence of molecular markers in patients with cancer of unknown primary (CUP). Journal of Clinical Oncology 2005, ASCO Annual Meeting Proceedings, Vol 23, No 16S, Part 1, June 1 Supplement, Abstract number 9683
- Rubin BP, Singer S, Tsao C et al (2001) KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 61(22):8118–8121 [PubMed] [Google Scholar]
- Shih AH, Holland EC (2006) Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett 232(2):139–147 [DOI] [PubMed] [Google Scholar]
- Sihto H, Sarlomo-Rikala M, Tynninen O et al (2005) KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin Oncol 23(1):49–57 [DOI] [PubMed] [Google Scholar]
- Sulzbacher I, Birner P, Träxler M, Marberger M, Haitel A (2003a) Expression of platelet-derived growth factor-alpha alpha receptor is associated with tumor progression in clear cell renal cell carcinoma. Am J Clin Pathol 120(1):107–112 [DOI] [PubMed] [Google Scholar]
- Sulzbacher I, Birner P, Trieb K, Träxler M, Lang S, Chott A (2003b) Expression of platelet-derived growth factor-AA is associated with tumor progression in osteosarcoma. Mod Pathol 16(1):66–71 [DOI] [PubMed] [Google Scholar]
- Ugurel S, Hildenbrand R, Zimpfer A et al (2005) Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer 92(8):1398–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Went PT, Dirnhofer S, Bundi M et al (2004) Prevalence of KIT expression in human tumors. J Clin Oncol 22(22):4514–4522 [DOI] [PubMed] [Google Scholar]
- Wilczynski SP, Chen YY, Chen W, Howell SB, Shively JE, Alberts DS (2005) Expression and mutational analysis of tyrosine kinase receptors c-kit, PDGFRalpha, and PDGFRbeta in ovarian cancers. Hum Pathol 36(3):242–249 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Kuang WJ, Yang-Feng T et al (1987) Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6(11):3341–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]