

Abstract
IL-17 is a potent proinflammatory cytokine that drives pathogenesis of multiple autoimmune diseases, including psoriasis. A major source of pathogenic IL-17 is a subset of γδ T cells (Tγδ17) that acquires the ability to produce IL-17 while developing in the thymus. The mechanisms that regulate homeostasis of Tγδ17 cells and their roles in psoriasis, however, are not fully understood. In this paper, we show that the heparan sulfate proteoglycan syndecan-1 (sdc1) plays a critical role in regulating homeostasis of Tγδ17 cells and modulating psoriasis-like skin inflammation in mice. sdc1 was predominantly expressed by Tγδ17 cells (but not IL-17− Tγδ cells) in the thymus, lymph nodes, and dermis. sdc1 deficiency significantly and selectively increased the frequency and absolute numbers of Tγδ17 cells by mechanisms that included increased proliferation and decreased apoptosis. Adoptive transfer experiments ruled out a significant role of sdc1 expressed on nonhematopoietic cells in halting expansion and proliferation of sdc1-deficient Tγδ17 cells. When subjected to imiquimod-induced psoriasiform dermatitis, Tγδ17 cells in sdc1KO mice displayed heightened responses accompanied by significantly increased skin inflammation than their wild-type counterparts. Furthermore, transferred sdc1-deficient γδ T cells caused more severe psoriasiform dermatitis than their sdc1-sufficient counterparts in TCR–βδ KO hosts. The results uncover a novel role for sdc1 in controlling homeostasis of Tγδ17 cells and moderating host responses to psoriasis-like inflammation.
Interleukin-17 is a potent proinflammatory cytokine that is required for host defense against bacterial and fungal infections (1, 2). Dysregulated IL-17 responses drive the pathogenesis of multiple autoimmune diseases, including psoriasis (3). The mechanisms that regulate IL-17 responses in host defense and autoimmune responses are incompletely understood. These mechanisms are particularly important for regulating innate-like T cells, which acquire their effector functions while developing in the thymus via agonist selection pathways and are prone to self-reactivity (4). After exiting the thymus, the IL-17–producing subset of γδ T cell (Tγδ17) localize not only in lymphoid tissues but also in epithelial barriers. In the dermis, Tγδ17 cells are involved in immunosurveillance and host defense against invading pathogens (5). Aberrant function of Tγδ17 cells contributes to autoimmunity. Indeed, Tγδ17 cells are considered a primary source of IL-17 involved in pathogenesis of psoriasis. Fruition of these observations are reflected in the approval of mAbs directed against IL-17A and IL-17 receptor A subunit (IL-17Ra) for clinical use to treat moderate to severe forms of psoriasis (6). In addition, mouse models of psoriasiform dermatitis have been developed to understand the underlying disease mechanisms. These include imiquimod (IMQ)-induced psoriasisform (7) in which Tγδ17 cells are important mediators of the disease pathologic condition. Thus, identifying the factors that control homeostasis of Tγδ17 cells is clinically relevant and could be useful for developing novel strategies to prevent aberrant production of IL-17 and alleviate its pathological consequences.
Syndecan-1 (sdc1, CD138) is a heparan sulfate proteoglycan (HSPG) that is predominantly expressed by epithelial cells and other nonhematopoietic cells and which regulates many of their properties, including proliferation and apoptosis (8, 9). Beyond being recognized as a marker for plasma cells and developing B cells, little is known about the role of sdc1 in immune cell functions (9, 10). Recently, our group and subsequently others have identified sdc1 as a specific marker of IL-17–producing subset of NKT cells (NKT17) (11–15). We have also shown that sdc1 deficiency significantly increases frequency of NKT17 cells both in the thymus and periphery (11) and hence act as a negative regulator of NKT17 cell homeostasis (16).
In this study, we show that sdc1 regulates homeostasis of Tγδ17 cells and pathogenesis of psoriasiform dermatitis in mice. sdc1 is expressed by most of the Tγδ17 cells in the thymus and lymph nodes and, to a lesser extent, on those localized in the dermis. Absence of sdc1 significantly increased the overall frequency and absolute numbers of Tγδ17 cells in different organs, including the dermis, by mechanisms that included increased proliferation and decreased apoptosis. Adoptive transfer experiments showed no major role of sdc1 expressed on non-hematopoietic cells in regulating homeostasis of Tγδ17 cells. Furthermore, sdc1 control of Tγδ17 cells are pathologically relevant as sdc1 deficiency significantly exacerbated IMQ-induced psoriasiform dermatitis even when expression is limited to the T cell compartment. Targeting increased expression or activation sdc1 could therefore represent a novel strategy to control IL-17 production by γδ T cells.
Materials and Methods
Mice
sdc1 knockout (sdc1KO) mice of C57BL/6J background were provided by Dr. P. Park of Harvard University and Dr. M.A. Stepp (17) of George Washington University. C57BL/6J, B6 CD45.1, BALB/c, and C3H/HeJ mice were purchased from The Jackson Laboratory. All the mice were kept and bred under specific pathogen-free conditions at the animal facility of Johns Hopkins University. Mice between the ages of 8 and 10 wk were used in all experiments unless otherwise indicated. All experiments were performed using experimental protocols approved by the Animal Care and Use Committee of Johns Hopkins University.
Abs
All the fluorochrome-conjugated mAbs were from BD Biosciences, Bio-Legend, or eBioscience. These included APC-Cy7–anti-CD45 (30-F11), BV421–anti-αβTCR (H57–597), FITC–anti-γδTCR (UC7–13D5), APC-anti-CD138 (sdc1, 281–2), PerCP-Cy 5.5–anti-IL-17A (TC11–18H10), PerCP-Cy 5.5–anti–retinoid orphan receptor γ t (RORγt) (Q31–378), PE/Cy7–anti-CD27(LG.3A10), BV421–anti-CCR6 (140706), APC–anti-TCRVγ4 (UC3–10A6), BV421–anti-CD45.1 (A20), FITC–anti-Vγ3 TCR (536), and APC-Cy7–anti-CD45.2 (104) mAbs were used.
Single-cell preparation and counting of absolute number of γδ T cells in ear skin and lymphoid organs
Single-cell suspensions from dermal sheets of ear splits were isolated as described (18). In unmanipulated mice, dermal sheets of both ears of each mouse were pooled together, whereas in IMQ experiments the treated ear from each of two mice in the group were pooled together. Samples were treated with dispase (1 U/ml; STEMCELL Technologies, Canada) and Liberase TM Research Grade (85 μg/ml; Roche Diagnostics) digestion in DMEM, followed by single-cell preparation as described (18). Single-cell suspensions from thymus and draining lymph nodes of each mouse were prepared as previously described (19). Absolute number of γδ T cells in each preparation was determined using total number of viable cells and percentage of γδ T cells in the sample (20). Numbers of viable cells in single-cell suspensions were determined by using a hemocytometer and trypan blue exclusion (21), whereas frequencies of γδ T cells were determined by FACS (Supplemental Fig. 1). The absolute cell number of γδ T cells was determined by multiplying their frequency by the total number of cells in the sample. Results were normalized to show average number of γδ T cells per mouse.
Flow cytometry
Flow cytometric analysis was performed using standard methods. Acquisition of sample was performed using an LSR II (BD Biosciences, equipped with 405-, 488-, 561-, and 640-nm laser lines). The acquired data were analyzed using FlowJo v10 software (Treestar). Dermal cell analysis was limited to gated TCRmedγδ T cells as illustrated in the gating strategy (Supplemental Fig. 1A). To focus on dermal γδ T cells and exclude epidermal contamination, we excluded TCRhigh Vγ3+ epidermal γδ T cells (22) from our analysis by specific gating on TCRmedγ3− T cells as illustrated in the gating strategy (Supplemental Fig. 1B).
Intracellular analysis of IL-17 and RORγt
For intracellular cytokine staining, single-cell suspensions were stimulated with PMA (10 ng/ml) and Ionomycin (500 ng/ml; Sigma-Aldrich) and GolgiPlug (BD Biosciences) at 37°C in a 5% CO2 incubator for 4 h in RPMI 1640 medium containing 10% FCS. Lymphocytes were surface stained followed by intracellular staining according to manufacturer’s instructions (BD Biosciences). Briefly, surface-stained cells were washed, permeabilized with Cytofix/Cytoperm buffer, and subjected to intracellular staining using PerCP-Cy 5.5–conjugated anti–IL-17A (TC11–18H10 clone) and PerCP-Cy 5.5–conjugated anti-RORγt (Q31–378 clone) mAbs. Data were acquired with LSR II using FACSDiva software (BD Biosciences) and analyzed using FlowJo software. RORγt was detected by intracellular staining as described (11).
Immunofluorescence microscopy
Immunofluorescence microscopy for mouse CD3 was performed on deparaffinized histologic sections after heat-mediated Ag retrieval in Trilogy buffer (Cell Marque). Sections were blocked for 1 h at room temperature in PBS with 10% goat serum (blocking buffer) and then incubated at 4°C overnight with 10 μg/ml mouse anti-CD3 Ab (ab5690; Abcam) diluted in blocking buffer. The next day, sections were incubated for 1 h at room temperature with 2 μg/ml Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) diluted in blocking buffer and subsequently mounted in VECTASHIELD with DAPI (Vector Labs). Fluorescent images were taken at 400× magnification (DFC365FX; Leica).
Quantitative real-time PCR
Total RNA from thymus, lymph node, or dermis from wild type (WT) and sdc1KO was isolated using RNeasy mini kit (Qiagen, Valencia, CA) and reverse transcribed using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher, Waltham, MA). Gene-specific Taqman primer and probe sets (Applied Biosystems) were used to assess transcriptional status of TCR γ-chain in CFX96 real-time PCR (Bio-Rad, Hercules, CA). The specific primers used for the TCR γ-chain are as follows: forward-5′-CCATCCACTGCAGAAGTCCC-3′, reverse-5′-GGTTGTCTAGAGTGGGGGTC-3′. The gene expression was normalized with GAPDH, and the relative fold expression values were calculated using ΔΔCT method.
Apoptosis assay
Apoptosis was assessed by flow cytometry using FITC–Annexin V Apoptosis Kit according to manufacturer’s instruction (BD Pharmingen). Approximately 2 × 106 cells/ml were washed in PBS, surface stained, resuspended in binding buffer, and incubated with FITC-conjugated annexin V and propidium iodide (PI) for 15 min in dark at room temperature, washed, and resuspended in the binding buffer. Samples were acquired by LSR II and analyzed using FlowJo v10 software, and gated γδ T cells were analyzed for annexin Vand PI binding.
Proliferation assay
Proliferation was examined by analysis of intracellular expression of the Ki67 molecule using PerCP–eFluor 710–anti-Ki67 (clone, SclA1S). Lymphocytes from different organs were isolated and surface stained followed by intracellular staining for Ki67 with Foxp3 staining buffer (catalog no. 00-5523-00; eBioscience). Data were acquired and analyzed as described above.
Analysis of the role of sdc1 expressed on nonhemopoietic cells on homeostasis of Tγδ17 cells
We used adoptive transfer to determine whether sdc1 expressed on nonhemopoietic cells regulates homeostasis of Tγδ17 cells. Briefly, we purified T cells from WT or sdc1KO donors and used frequency of γδ T cells (determined by FACS) in each sample to adjust dose and inject 1.5 × 105 γδ T cells into sublethally irradiated (6 Gy or 600 rad) CD45.1+ congenic sdc1-sufficient recipients. On day 6, lymphocytes were isolated from the lymph nodes of each recipient, and viable cells were counted using trypan blue exclusion. Samples were stained, and donor cells (CD45.2+) were gated and analyzed for intracellular expression of IL-17 or Ki67 and apoptosis. Absolute numbers of donor γδ T cells recovered from respective recipients were determined by multiplying frequency by total cell count.
Induction of psoriasiform dermatitis using IMQ
We examined the effect of sdc1 on psoriasis using the IMQ-induced psoriasiform dermatitis mouse model (7). Briefly, age-matched sdc1KO and WT mice were anesthetized using isoflurane, and both sides of the right ear were treated topically with 84 mg of 5% IMQ cream (Taro Pharmaceuticals) for five consecutive days. The left ear was treated with vehicle and used as a placebo control. Skin thickness was measured daily using Dial Thickness Gauge (Ozaki Mfg., Japan). On day 6, mice in both groups were euthanized, and effects of sdc1 absence on their responses were analyzed. In one set of experiments, we analyzed the effects on γδ T cells in draining lymph nodes. Single-cell suspension from the lymph node draining the IMQ- or placebo-treated ear of each mouse was prepared, and γδ T cells were isolated and analyzed for various parameters described in the results section. In a second set of experiments, we used histological changes as readouts. IMQ- or placebo-treated ears were excised from each mouse and fixed in 10% formaldehyde. Tissue slices (5 μm) were prepared from paraffin sections, stained with H&E, and analyzed for pathological changes in a blind fashion. Epidermal thickness was precisely measured using ImageJ analysis software.
Analysis of the ability of sdc1-deficient Tγδ17 cells to cause IMQ-induced psoriasiform dermatitis in sdc1-sufficient TCR–βδ KO hosts
T cells were purified from lymph nodes of WTor sdc1KO mice, and 1 × 107 cells were injected into 8-wk-old TCR–βδ KO recipients. Mice in each group were treated topically with 84 mg of IMQ cream (5%) on both sides of the right ear for five consecutive days, whereas the left ear was vehicle treated and used as a placebo control. Ear thickness was measured daily using Dial Thickness Gauge as described above. On day 6, mice in each group were euthanized, and lymphocytes in respective draining lymph nodes of IMQ-treated or placebo-treated ears were isolated and analyzed separately as described above.
Statistical analysis
Data were expressed as means ± SEM using (Prism 6; GraphPad Software). Comparisons were made using a two-tailed Student t test. Comparisons between multiple groups were performed by a one-way ANOVA test followed by the Tukey multiple comparison test when appropriate. Statistical significance was determined as p < 0.05.
Results
Expression of sdc1 by Tγδ17 cells
We began this study by determining whether sdc1 is expressed on γδT cells. We isolated lymphocytes from the thymus, lymph nodes, and dermis of C57BL/6 mice and stained and analyzed gated γδT cells for sdc1 expression (for gating strategy see Supplemental Fig. 1). Sizable fractions of γδT cells in the thymus, lymph nodes, and dermis expressed sdc1 (Fig. 1A). To focus on dermal γδ T cells and exclude epidermal contamination, we excluded TCRhigh γδ cells from our analysis by specific gating on TCRmedγδ T cells as illustrated in the gating strategy (Supplemental Fig. 1). Expression of sdc1 by a subset of γδ T cells was also observed in BALB/c and C3H/HeJ mice (Supplemental Fig. 2), indicating that it is not limited to a single mouse strain. Recently, we and others (11–14) have shown that among NKT cells, sdc1 is selectively expressed on the NKT17 subset. We therefore determined whether this is the case for γδ T cells. IL-17–producing Tγδ17 cells have been described to use certain TCRVγ (Vγ4 and Vγ6) chains and to express CCR6 and lack CD27 on their surface (22–25). Tγδ17 cells, however, are definitively defined by intracellular expression of IL-17. Expression of sdc1 by γδ T cells isolated from the thymus or the dermis did not correlate with usage of Vγ4, surface expression CCR6, or lack of CD27, but the majority of sdc1+ γδ T cells in the lymph nodes used Vγ4 and expressed CCR6 (Fig. 1B–D). However, we observed significant correlation between expression of IL-17 and sdc1 in the thymus and lymph nodes and, to a lesser extent, in the dermis (Fig. 2A, upper panel and Fig. 2B). The majority of Tγδ17 cells (unlike IL-17− γδ) in thymus (62 ± 10%) and lymph nodes (58 ± 8%) but not dermis (31 ± 5%) expressed sdc1 (Fig. 2A, 2B). In contrast, only a minority of IL-17− γδ T cells in thymus (5 ± 3% of), lymph nodes (13 ± 7%), and dermis (4 ± 2%) expressed sdc1. As specificity control, we show that no significant sdc1 signal was detectable in samples from sdc1KO mice (Fig. 2A, middle panel), which were stained in a similar manner as their WT counterparts. Likewise, no significant sdc1 signal was detected in samples stained with isotype-matched Ab (Fig. 2A, lower panel). Taken together, these results show that sdc1 expression is found more often in Tγδ17 cells than IL-17− γδ T cells.
FIGURE 1.

Expression of sdc1 by γδ T cells. Lymphocytes from thymus, lymph nodes, and dermis of WT C57BL/6 mice were isolated and stained, and TCRγδ+ cells were gated and analyzed for the expression of sdc1 (A) as described in Materials and Methods. Graph shows cumulative data (mean ± SEM) pooled from nine mice from three independent experiments. (B–D) Representative dot plots and cumulative graphs compare expression of sdc1 with that of Vγ4 (B), CCR6 (C), or CD27 (D) by gated γδ T cells in the three organs. Results (mean ± SEM) are from three independent experiments and six mice per genotype.
FIGURE 2.

sdc1 is predominantly expressed by IL-17–producing subset of γδ T cells. (A) Dot plots show representative expression of sdc1 by gated γδ T cells in each organ. Top representative dot plots show expression of sdc1 versus that of IL-17 by gated γδ T cells in WT mice. Middle dot plots show no nonspecific sdc1 signals in sdc1KO stained with sdc1-specific mAb. Bottom dot plots show background staining of WT mice with isotype-matched Ab in different organs. Numbers indicate percentages in quadrants. (B) Graph shows cumulative data of percentages of IL-17+ or IL-17− that expressed sdc1 in each organ. Percentage of IL-17+ cells in each organ that expressed sdc1 was determined using data in representative dot plots (right upper quadrant/upper left and upper right quadrants × 100). Percentage of IL-17− cells that expressed sdc1 in each organ was determined using the same formula (lower right quadrant/lower left and right quadrants × 100). Results from two independent experiments and six individually analyzed mice.
sdc1 deficiency selectively increases Tγδ17 cells in various organs
The role of Tγδ17 cells in driving skin inflammation was previously examined using the C57BL/6 strain (26); hence, we limited our analysis to the C57BL/6 strain to examine mechanisms and assess effect of sdc1 deficiency on the pathogenesis of psoriasiform dermatitis. To investigate the effect of sdc1 deficiency on homeostasis of γδ T cells, we compared frequency and absolute numbers of γδ T cells in sdc1KO and age-matched WT C57BL/6 mice. There was a significant increase in the absolute number of γδ T cells in thymus, lymph nodes, and dermis of sdc1KO as compared with age-matched WT mice (Fig. 3A, 3B). The increase of γδ T cells in sdc1KO mice was confirmed by quantitative real-time PCR (Fig. 3C). Given the biased sdc1 expression by Tγδ17 cells, we determined whether sdc1 deficiency cause generalized increase of γδ T cells or whether it selectively affects the Tγδ17 subset. The nuclear hormone receptor RORγt is a transcription factor that regulates IL-17 production, and its expression is used to identify Tγδ17 cells (27). Therefore, first, we compared the frequencies and total numbers of RORγt+ or RORγt−γδ T cells in sdc1KO and age-matched WT mice. There were significantly higher frequencies of RORγt+ γδ T cells in the examined organs (thymus, lymph node, and dermis) of sdc1KO mice as compared with their counterparts in WT mice (Fig. 4A). The changes were caused by increases of absolute numbers of RORγt+γδ T cells, which were significantly higher in sdc1KO than WT mice (Fig. 4B). In contrast, absolute numbers of RORγt−γδ T cells were comparable in the aforementioned organs of sdc1KO and WT mice (Fig. 4B). Thus, sdc1 deficiency selectively increased percentages and absolute numbers of RORγt+ but not RORγt− subset of γδ T cells. Second, we examined the direct effect of sdc1 deficiency on Tγδ17 cells. The results confirmed the above finding, as sdc1 deficiency significantly increased the percentages and absolute numbers of Tγδ17 cells, with no measurable effect on the absolute numbers of IL-17− γδ T cells in any of the examined organs (Fig. 4C, 4D). Thus, using either RORγt or IL-17 expression as readouts, our results show that sdc1 deficiency significantly and specifically increases the frequency and absolute numbers of Tγδ17 cells in the thymus, lymph nodes, and dermis with minimal or no effect on RORγt−/IL-17− γδ T cells.
FIGURE 3.
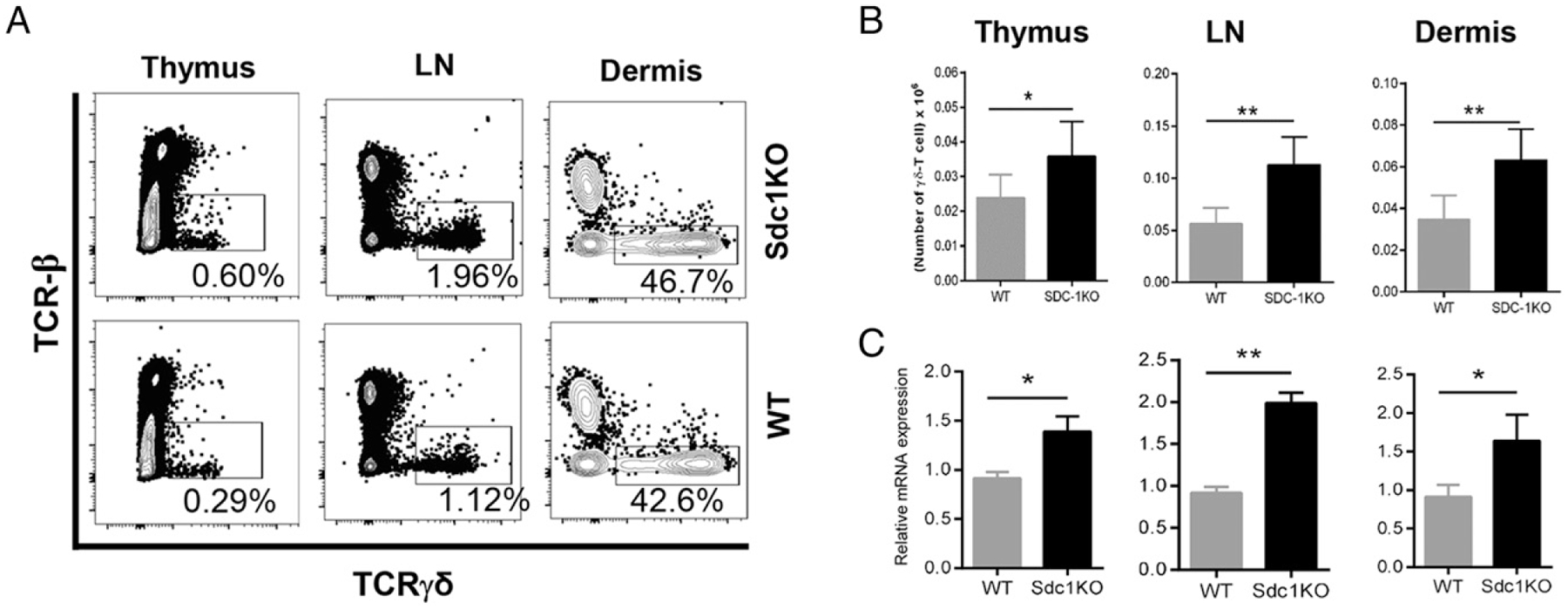
Absence of sdc1 increases γδ T cells in different organs. (A) Single-cell suspensions from indicated organs of sdc1KO and WT C57BL/6 mice were analyzed by FACS for the presence of γδ T cells. Representative dot plots show frequencies of γδ T cells in thymus, draining lymph nodes, or dermal sheets of the ear skins of sdc1KO or WT mice. (B) Graphs show the absolute numbers of γδ T cells in different organs (n = 6 mice per genotype). The absolute numbers were determined as described in Materials and Methods. (C) Graphs show the relative mRNA expression of TCR γ-chain in different organs as determined by quantitative real-time PCR, n = 3 per genotype. Results are expressed as mean ± SEM. *p < 0.05, **p < 0.01.
FIGURE 4.

Deletion of sdc1 increases percentage and absolute numbers of RORγt+ Tγδ17 cells in different organs. Lymphocytes isolated from indicated organs of sdc1KO and WT C57BL/6 mice were analyzed for the expression of RORγt and IL-17 as described in Materials and Methods. (A) Representative dot plots show expression of RORγt by gated γδ T cells in indicated organs of sdc1KO (top panel) and WT (bottom panel) mice. Numbers indicate percentages of cells positive for RORγt expression. (B) Graphs show cumulative data of the frequency and absolute number of RORγt+ γδ T cells or RORγt− γδ T cells in each organ of sdc1KO or WT mice. (C) Representative dot plots show IL-17 expression by gated γδ T cells in indicated organs of sdc1KO (top panel) and WT (bottom panel) C57BL/6 mice. (D) Graphs show cumulative frequencies and absolute numbers of IL-17+ γδ T cells in different organs. Data are pooled from three independent experiments and 12 mice per genotype. Results are expressed as mean ± SEM. Unpaired t test (two-tailed). ***p < 0.001, **p < 0.01, *p < 0.05.
Absence of sdc1 increases proliferation of RORγt+ subset of γδ T cells in lymph nodes and dermis
Cell expansion could be cause by increased proliferation, decreased apoptosis, or both. To address this question and gain insights into mechanisms underlying RORγt+γδ T cell increase in sdc1KO mice, we compared their proliferation and apoptosis to those of their counterparts in WT mice. We first assessed expression of Ki67 and analyzed the effects of sdc1 deficiency on proliferation of RORγt+γδ T cells by assessing expression of a nuclear element associated with cell cycle (28) on the RORγt+ and RORγt− subpopulations of γδ T cells to determine the effect of sdc1 deficiency on proliferation. We did not observe a statistical difference in Ki67 expression by γδ T cells in thymi of sdc1KO mice as compared with WT mice (Fig. 5A). However, there were significantly higher frequencies of Ki67 expression by γδ T cells in lymph nodes and dermis of sdc1KO than in WT mice (Fig. 5A, 5B). When we focused the analysis on RORγt+ γδ T cells, the percentages of those expressing Ki67 in the thymus were not significantly different between sdc1KO and WT mice (Fig. 5C, 5D). RORγt+ γδ T cells, however, proliferated at significantly higher rates in lymph nodes and dermis of sdc1KO than WT mice as indicated by Ki67 expression (Fig. 5C, 5D). In contrast, there were no significant differences in expression of Ki67 by RORγt− γδ T cells in sdc1KO and WT controls (Fig. 5E). These results indicate that sdc1 deficiency causes significant increases in proliferation of RORγt+ subset of γδ T cells in the lymph nodes and dermis, with minimal or no effect on the RORγt− subset.
FIGURE 5.
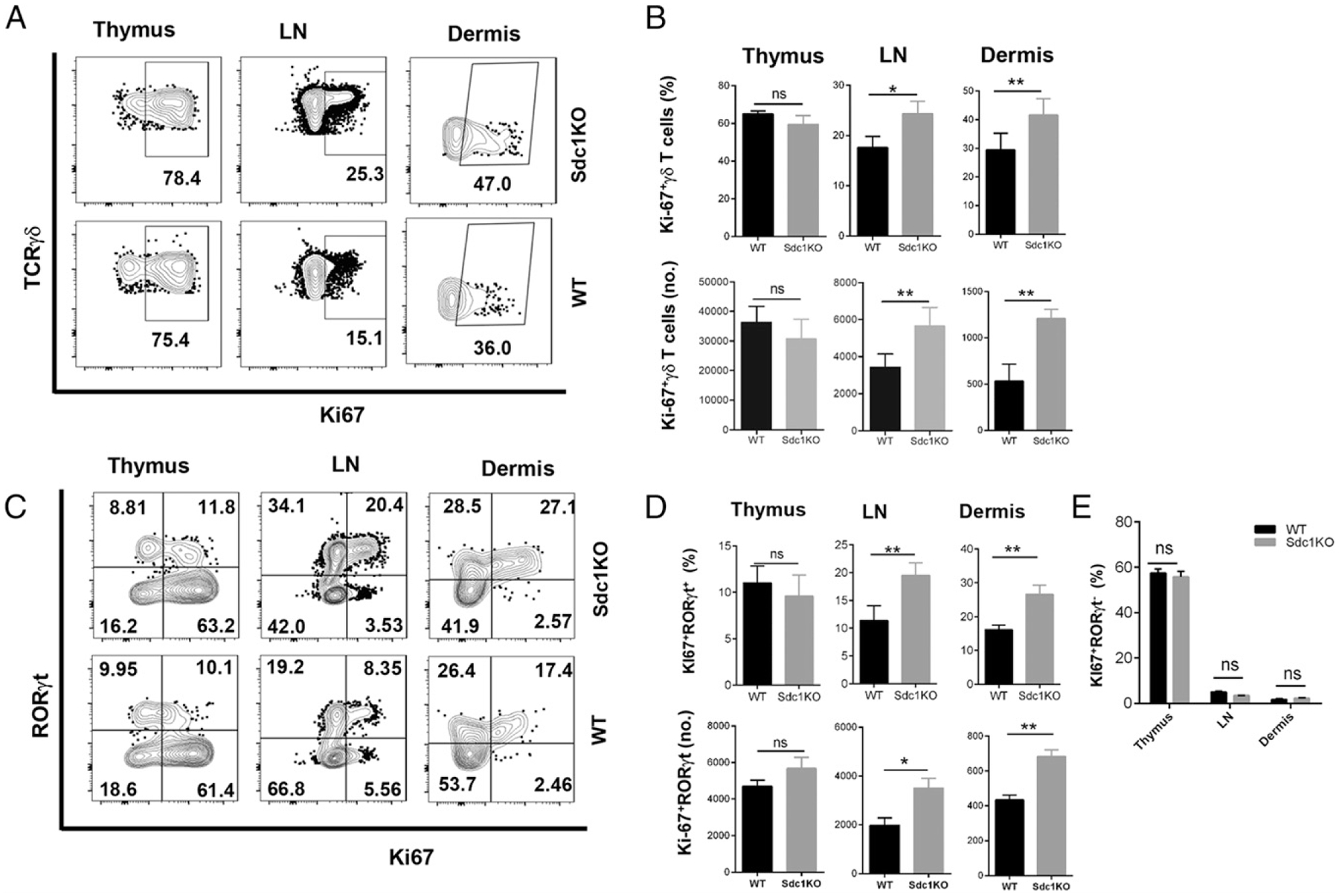
sdc1 deficiency increases proliferation of RORγt+ Tγδ17 cells in lymph nodes and dermis but not thymus. Single-cell suspensions from indicated organs of sdc1KO or WT C57BL/6 mice were stained and acquired, and gated γδ T cells were analyzed for intracellular expression of Ki67 and RORγt as described in the Materials and Methods. (A) Representative dot plots show expression of Ki67 by gated γδ T cells in sdc1KO (top panel) and WT (bottom panel) mice. (B) Graphs show cumulative frequencies and absolute numbers of Ki67+ γδ T cells in sdc1KO and WT mice. (C) Representative dot plots show expression of Ki67 versus that of RORγt by gated Tγδ17 cells in sdc1KO (top panel) and WT (bottom panel) mice. (D) Graphs show cumulative frequencies and absolute numbers of Ki67+RORγt+γδ T cells in different organs of sdc1KO or WT mice. (E) shows cumulative frequencies and absolute numbers of Ki67+RORγt−γδ T cells in different organs of sdc1KO or WT mice. Data are pooled from three independent experiments and nine mice per genotype. Unpaired t test (two-tailed); ns, not statistically significant. **p < 0.01, *p < 0.05.
Absence of sdc1 reduces apoptosis of RORγt+ subset of γδ T cells in different organs
Next, we analyzed the effect of sdc1 deficiency on apoptosis of γδ T cells. Because staining for either IL-17 or RORγt requires fixation of the cells, we limited our analysis to surface-stained γδ T cells. Our analysis showed that apoptosis of γδ T cells was significantly reduced in different organs, particularly in lymph node and dermis of sdc1KO mice as compared with WT controls (Fig. 6). Thus, sdc1 deficiency significantly reduces apoptosis of γδ T cells. Furthermore, given that sdc1 deficiency specifically affected Tγδ17 cells, we feel it is safe to extrapolate that changes in apoptosis affected mainly in Tγδ17 cells.
FIGURE 6.

sdc1 deficiency decreases late apoptosis of native γδ T cells in different organs. Isolated lymphocytes from sdc1KO and WT C57BL/6 mice were surface stained, and γδ T cells were gated and analyzed for late apoptosis as described in Materials and Methods. (A) Representative dot plots show annexin and PI binding by gated γδ T cells in sdc1KO (top panel) and WT (bottom panel) mice. (B) Graphs show cumulative frequencies and absolute numbers of γδ T cells undergoing late apoptosis (annexin V+, PI+) in different organs of sdc1KO and WT mice. Unpaired t test (two-tailed). **p < 0.01, *p < 0.05.
sdc1 expressed on nonhematopoietic cells plays no major role in regulating homeostasis of Tγδ17 cells
Epithelia and other nonhematopoietic cells are the primary cell types that express sdc1 (8, 9, 16). We therefore determined whether sdc1 expressed on nonhematopoietic cells plays a role in regulating homeostasis of Tγδ17 cells. This notion was examined by comparing overall expansion, proliferation, and apoptosis of adoptively transferred CD45.2+ γδ T cells (1.5 × 105 per recipient) from sdc1KO or WT donors into congenic CD45.1+ sdc1-sufficient hosts as illustrated (Fig. 7A) and described in detail in Materials and Methods. Our results showed that Tγδ17 cells from sdc1KO donors significantly expanded in the WT environment as compared with their counterparts from WT donors (Fig. 7B). The frequency and absolute numbers of sdc1KO Tγδ17 cells were significantly increased as compared with their WT counterparts (Fig. 7B). In contrast, the absolute numbers of IL-17− γδ T cells from sdc1KO and WT donors remained comparable (Fig. 7B). This confirms that specific effect of sdc1 deficiency on Tγδ17 cells and affirmed that mice were reconstituted with similar numbers of γδ T cells. The frequency of donor sdc1KO Tγδ17 cells that expressed Ki67 was significantly higher than that of WT Tγδ17 cells, showing their increased proliferation is not driven by sdc1 expressed on nonhematopoietic cells (Fig. 7C). Proliferation of transferred cells was also directly visualized using the CFSE dilution assay (Supplemental Fig. 3). In contrast, we did not observe significant differences in apoptosis of the adoptively transferred sdc1KO and WT γδ T cells (Fig. 7D). As discussed in detail below, the absence of significant effect of sdc1 deficiency on apoptosis of adoptively transferred γδ T cells could be due to the high background apoptosis that likely emanated from in vitro manipulation and the harsh conditions in the irradiated lymphopenic hosts. Taken together, these results show that sdc1 expressed on nonhematopoietic cells plays no significant role in halting proliferation and expansion of sdc1-deficient Tγδ17 cells.
FIGURE 7.

sdc1 expressed on nonhematopoietic cells plays no major role in regulating homeostasis of Tγδ17 cells. Purified T cells containing (1.5 × 105 γδ T cells) from CD45.2+ WT or sdc1KO donors were injected i.p. into sublethally irradiated CD45.1+ sdc1-sufficient recipients. Five days later, single-cell suspensions were isolated from lymph nodes (LN) of recipients, stained, and acquired by FACS, and donor CD45.2+ γδ T cells were gated and analyzed. (A) Scheme of experimental strategy. (B) Representative dot plots show intracellular expression of IL-17 by gated WT or sdc1KO donor γδ T cells. Left and middle graphs show cumulative frequencies (left) and absolute numbers (middle) of WT or sdc1KO donor Tγδ17 cells. Right graph shows absolute numbers of WT or sdc1KO donor IL-17− γδ cells. (C) Representative dot plots show Ki67 expression by WT or sdc1KO γδ T cells. Graphs show cumulative data of frequencies and absolute numbers of Ki67-expressing γδ T cells of WT or sdc1KO origin. (D) Representative dot plots show annexin V binding by donors γδ T cells. Graphs show cumulative data of frequencies and absolute numbers of annexin V+ γδ T cells of WT or sdc1KO origin. Data are pooled from eight mice per group from four independent experiments. Results are shown as mean ± SEM. Unpaired t test (two-tailed); ns, not statistically significant. **p < 0.01, *p < 0.05.
Deficiency of sdc1 promotes IMQ-induced psoriasiform dermatitis
IL-17 is a major driver of skin inflammation in humans, and IL-17–neutralizing mAbs are in clinical use for treatment of psoriasis (22, 29). Tγδ17 cells are also major drivers of IMQ-induced skin inflammation (7, 29). Thus, we hypothesized that sdc1 deficiency could have significant pathologic implications in this model of skin inflammation. To this end, we determined whether sdc1 deficiency increases susceptibility to IMQ-induced psoriasiform. The first indication supporting this possibility was our findings that sdc1 deficiency significantly enhanced responses of Tγδ17 cells to IMQ treatment (Fig. 8). Percentages and absolute numbers of Tγδ17 cells were significantly higher in draining lymph nodes IMQ-treated ears than draining lymph nodes placebo-treated ears in both sdc1KO and WT mice. The placebo control was normal mice. Furthermore, frequencies and absolute numbers of Tγδ17 cells were significantly higher in IMQ lymph nodes of sdc1KO as compared with their counterparts in WT mice (Fig. 8A–C). The endpoints used to evaluate the effects of IMQ treatment are changes in ear thickness and epidermal thickness (22). sdc1 deficiency significantly increased both parameters (Fig. 8D–F). In contrast, there were no apparent changes in skin histology or epidermal thickness of sdc1KO mice as depicted in those in the placebo group (Fig. 8D–F). Thus, treatment with IMQ led to significant increases in Tγδ17 cells and resulted in exacerbated skin inflammation in sdc1KO as compared with WT controls. We concluded that sdc1 expression is important for controlling IMQ-induced skin inflammation.
FIGURE 8.
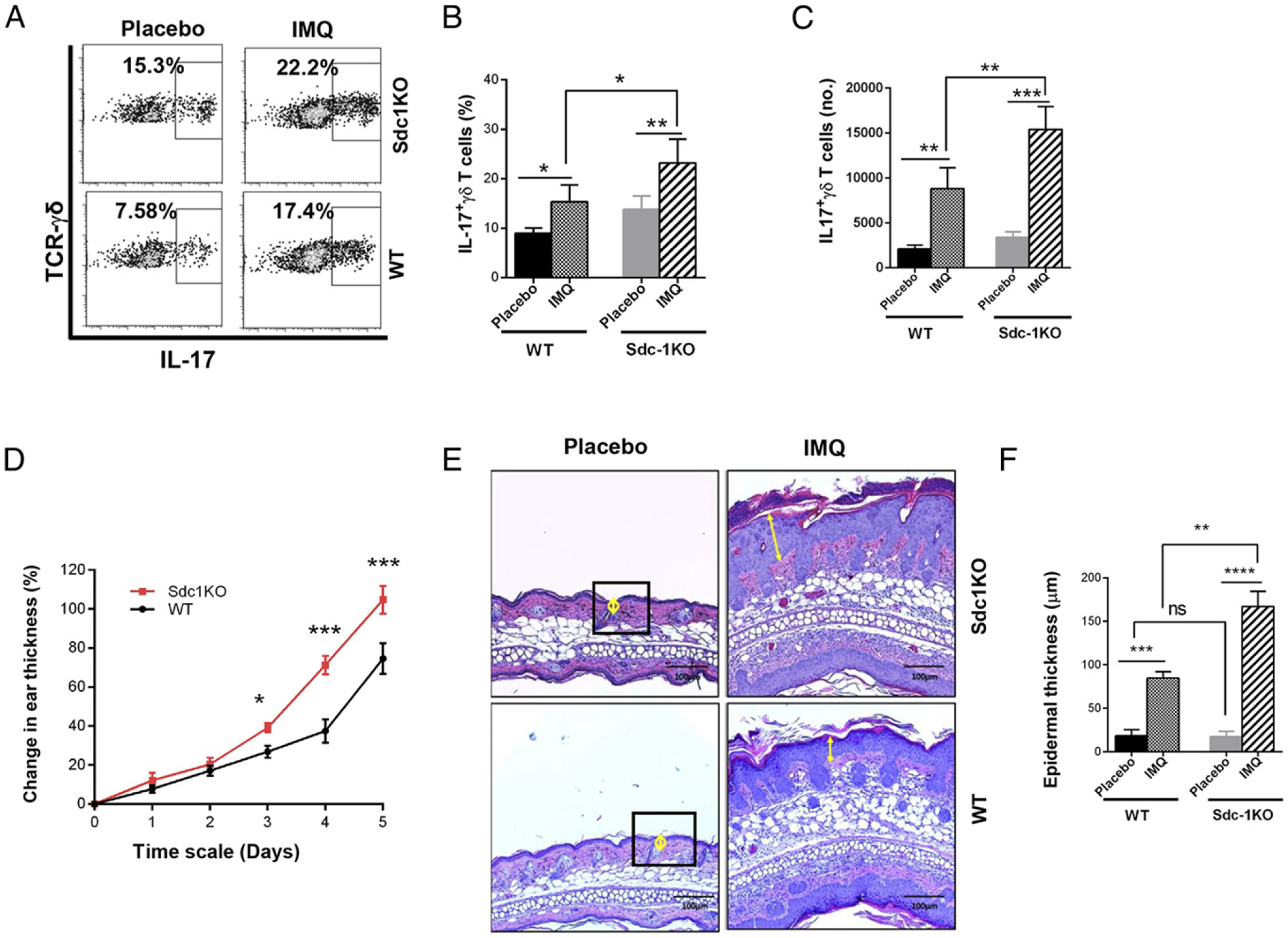
Deficiency of sdc1 promotes IMQ-induced psoriasiform dermatitis. sdc1KO or WT C57BL/6 mice received topical treatment with 5% IMQ cream on one ear and placebo on the other ear for five consecutive days as described in Materials and Methods. Skin thickness of each ear was measured daily. On day 6, γδ T cells in lymph nodes draining the IMQ- or placebo-treated ears were isolated and analyzed by FACS, and skin lesions were evaluated using H&E sections. (A) Representative dot plots show intracellular expression of IL-17 by γδ T cells in placebo and IMQ draining lymph nodes of WT and sdc1KO mice. Numbers indicate percentages positive for intracellular IL-17. (B and C) Graphs show cumulative data (mean ± SEM, n = 9 per group) pooled from three independent experiments. (D) Graph shows cumulative changes in thickness of placebo- or IMQ-treated ears of sdc1KO and WT mice. Results are expressed as mean ± SEM; n = 9 mice per group. (E) Representative images show histological skin lesions in IMQ-treated or placebo-treated ears of sdc1KO and WT mice. Yellow circles and arrows denote epidermal thickness. Scale bar, 100 μm. (F) Graph shows epidermal thickness (mean ± SEM, n = 9 per group) in IMQ- or placebo-treated ears measured and analyzed using ImageJ software. Data were compared using one-way ANOVA (Tukey multiple comparisons test). ***p < 0.001, **p < 0.01, *p < 0.05.
To determine the importance of T cell sdc1 expression in mediating IMQ-induced psoriasiform dermatitis, we reconstituted sdc1-sufficient TCR–βδ KO recipients, which lacked both αβ and γδ T cells (30), with WT or sdc1KO T cells. We subjected mice in each group to IMQ-induced skin inflammation as illustrated (Fig. 9A) Effect of the treatment was assessed daily by measuring ear thickness. In addition, on day 6, frequency and absolute numbers of Tγδ17 cells in placebo and IMQ draining lymph nodes were determined. The percentages and absolute numbers of Tγδ17 cells were significantly higher in lymph nodes of mice reconstituted with sdc1KO T cells than those reconstituted with WT T cells (Fig. 9B, 9C). Furthermore, psoriasis-like skin inflammation was significantly more severe in recipients of sdc1KO T cells as indicated by increased ear thickness and epidermal thickness (Fig. 9D–F). There were no major differences in frequency of Th17 cells (which do not express sdc1) in the two groups (Fig. 9G, 9H), consistent with previous reports that mice γδ T cells play a more prominent role than Th17 cells in driving the psoriasiform dermatitis (22, 29, 31, 32). In addition, transferred T cells were able to reach the dermis as indicated by detection of CD3+ cells in skin sections of recipient mice (Fig. 9I). These results show that limiting sdc1 deficiency to T cells is sufficient to exacerbate IMQ-induced skin inflammation.
FIGURE 9.
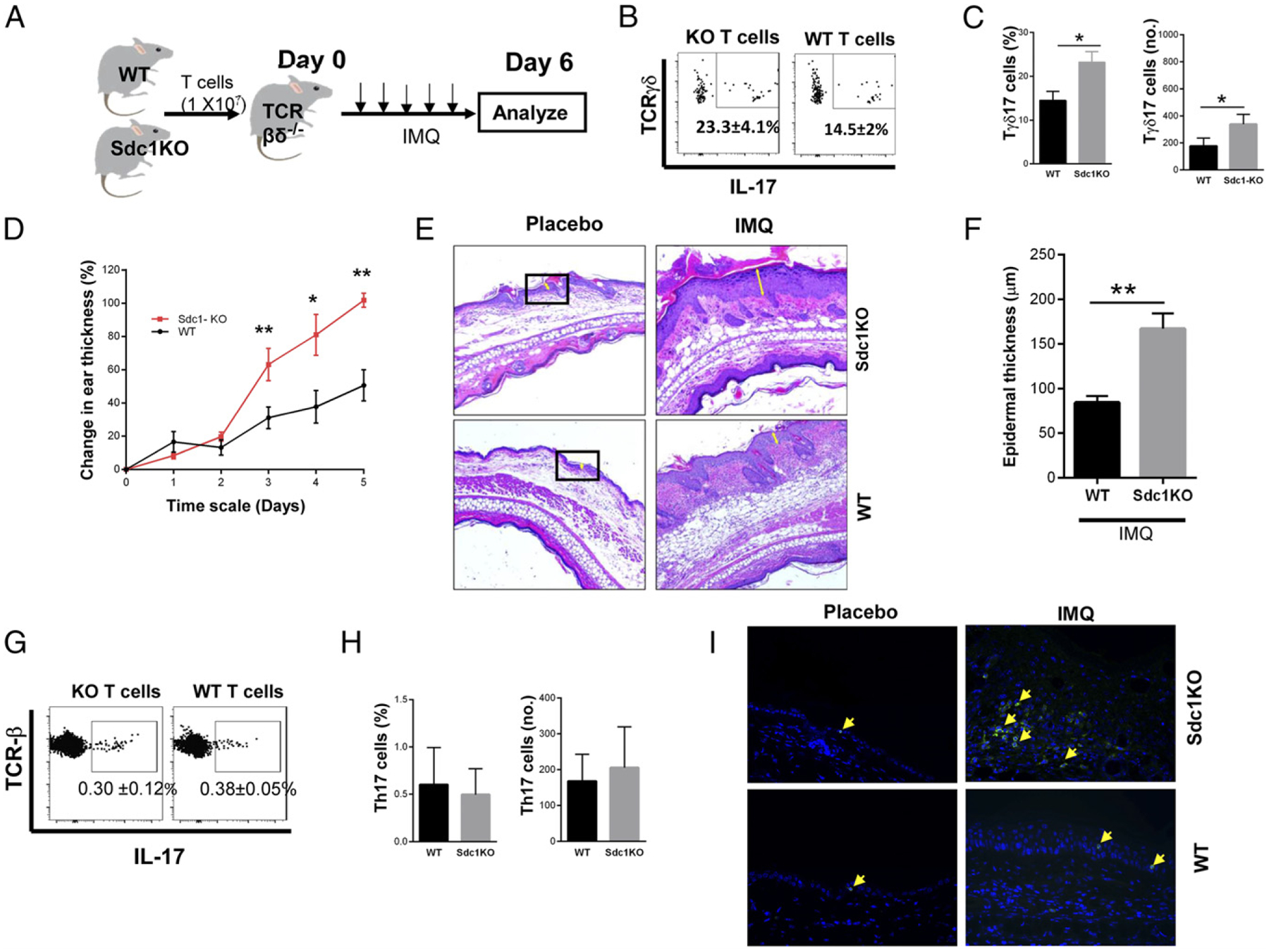
sdc1 expressed on nonhematopoietic cells does not mediate IMQ-induced psoriasiform dermatitis. T cells were purified from lymph nodes of WT or sdc1KO mice, and 1 × 107 cells were injected i.v. into syngeneic TCR–βδ KO B6 mice. Twenty-four hours later, mice in each group were subjected to IMQ treatment for five consecutive days, with skin thickness measured each day. On day 6, T cells in draining lymph nodes were analyzed by FACS, and skin lesions were evaluated using H&E sections. (A) Scheme of experimental strategy. (B) Representative dot plots show intracellular expression of IL-17 by gated γδ T cells of WT or sdc1KO origin. (C) Graphs show cumulative frequencies and absolute numbers of recovered WT or sdc1KO Tγδ17 cells. (D) Graph shows percentage changes in ear thickness (mean ± SEM) of mice that received WT or sdc1KO T cells. (E) Representative images show histological skin lesions in ears of mice that received sdc1KO or WT T cells. Yellow arrows denote epidermal thickness. (F) Graph shows cumulative epidermal thickness (mean ± SEM) measured and analyzed using ImageJ software. (G) Representative dot plots show expression of IL-17 by gated αβ T cells in recipients of WT or sdc1KO T cells. (H) Graphs show cumulative data of frequencies and absolute numbers of Th17 cells in both groups. Data (mean ± SEM) are from four individually analyzed mice per group. (I) Deparaffinized histologic sections of ear (sdc1ko and WT) for placebo control and IMQ were stained with CD3 Abs (see Materials and Methods for details), and nuclei were stained with DAPI. In all panels, arrow indicates positive signal for CD3 (green). Original magnification ×400. Unpaired t test (two-tailed). **p < 0.01, *p < 0.05.
Discussion
This study shows that sdc1 plays a critical role in regulating homeostasis of and controlling pathogenesis of psoriasiform dermatitis in mice. Our results show that the majority of RORγt+ Tγδ17 cells in thymus and lymph nodes and ~30% of Tγδ17 cells in dermis expressed sdc1, whereas significantly less percentage of IL-17− cells expressed sdc1. Importantly, absence of sdc1 significantly increases overall frequencies and absolute numbers of Tγδ17 cells by increasing their proliferation and reducing apoptosis, with minimal effect on homeostasis of IL-17− γδ T cells. Furthermore, adoptive transfer experiments ruled out a major role for sdc1 expressed on nonhematopoietic cells in modulating responses or pathogenic roles of sdc1-deficient Tγδ17 cells. Absence of sdc1 also enhances proliferative responses of Tγδ17 cells to IMQ and exacerbates ensuing psoriasiform dermatitis. Furthermore, limiting sdc1 expression to nonhematopoietic cells did not alleviate the pathogenic role of sdc1-deficient Tγδ17 cells transferred into sdc1-sufficient TCR–βδ KO recipients. If future studies proved a similar regulatory role for sdc1 in humans, sdc1 targeting could then offer novel strategies to keep Tγδ17 cells in check and control their pathogenic roles in psoriasis and perhaps other autoimmune diseases, where innate-like T cells are major players.
The results revealed an intriguing triangular relationship between IL-17, sdc1, and γδ T cells. IL-17 is a potent cytokine that is required for host defense against bacterial and fungal infections (1, 33) but whose overproduction is implicated in causing autoimmune conditions at epithelial and mucosal barriers (3). sdc1 is a heparan sulfate proteoglycan that is predominantly expressed and involved in regulating homeostasis of epithelia and other nonadherent cells, including regulating their proliferation and apoptosis (8, 9). Tγδ17 cells are part of the regulatory network that protects dermis and other epithelial barriers against invading pathogens by producing IL-17. Our results show that Tγδ17 cells, similar to epithelia, use sdc1 to regulate their homeostasis and prevent exuberant IL-17 production in response to IMQ-induced skin inflammation.
Expression of sdc1 by Tγδ17 cells is not a part of peripheral tissue imprinting process. Instead, sdc1 is expressed by Tγδ17 cells while developing in the thymus. In fact, the percentage of Tγδ17 cells bearing sdc1 is the lowest in the dermis than in the thymus and lymph nodes. In contrast, sdc1 was minimally expressed on IL-17− γδ T cells, and sdc1 deficiency had no discernable effect on IL-17− γδ T cells whose proliferation and absolute numbers were comparable in sdc1KO and age-matched WT mice. Thus, the effect of sdc1 deficiency was clearly limited to Tγδ17 cells, including the dermis, which despite having the lowest sdc1 expression, appeared to be the most affected by sdc1 deficiency as reflected in expansion of dermal Tγδ17 cells in the steady state or in response to IMQ. Given that the dermis is an effector site with active exposure to environmental Ags, one interpretation for the low percentage of sdc1-expressing Tγδ17 cells in the dermis could be due to its shedding by stimuli in milieu (34). Thus, the ability of sdc1 to regulate γδ T cells could also be tissue-specific. For example, sdc1 absence significantly increased proliferation of Tγδ17 cells (RORγt+) in the lymph nodes and dermis but had no effect on proliferation of Tγδ17 cells in the thymus. Thus, the ability of sdc1 to regulate proliferation of Tγδ17 cells is limited to those in the periphery. In contrast, survival of thymic γδ T cells remained regulated by sdc1 as indicated by increased numbers and reduced apoptosis of thymic γδ T cells in sdc1KO mice. It is also noteworthy that sdc1 deficiency significantly reduced apoptosis of endogenous native Tγδ17 cells (Fig. 5) but had no significant effect on apoptosis of adoptively transferred γδ T cells (Fig. 6D). We speculate this could be related to the fact that, unlike endogenous γδ T cells, adoptively transferred cells showed high background apoptosis, which is expected given that their exposure to in vitro isolation procedure followed by injection into less than ideal microenvironment in the sublethally lymphopenic recipients.
In contrast, our results ruled out a major role for sdc1 expressed on epithelia in influencing proliferation of Tγδ17 cells as demonstrated by the ability of adoptively transferred sdc1KO Tγδ17 cells to expand better than their WT counterparts in sdc1-sufficient recipients. However, because our preparations also contained αβ T cells, we cannot formally rule out cotransferred αβ T cells had some effects on the inflicted pathologic condition. This possibility, however, is unlikely because sdc1 is not expressed by αβ T cells, and its absence did not alter homeostasis IL-17–producing αβ Th cells. Future studies should examine mechanisms by which sdc1 regulates proliferation and survival of Tγδ17 cells. Identification of mechanisms by which sdc1 signals in T cells and its binding partners will be crucial in understanding its role in controlling homeostasis of Tγδ17 cells.
In summary, our results identify a novel role for sdc1 in regulating overall homeostasis of Tγδ17 cells and alleviating pathogenesis of IMQ-induced psoriasiform dermatitis in mice. The results extend our recent findings that sdc1 also marks NKT17 cells and that its deletion significantly increases their frequencies in sdc1KO mice (11). Thus, it appears that sdc1 regulates IL-17 production by both innate lymphoid cells, γδ T cells, and NKT cells, with important implications for both host defense and autoimmune diseases.
Supplementary Material
Acknowledgments
We thank The Johns Hopkins Medical Institutions Reference Histology Laboratory for preparing histology slides and performing H&E staining.
Abbreviations used in this article:
- IMQ
imiquimod
- NKT17
IL-17–producing subset of NKT cells
- PI
propidium iodide
- RORγt
retinoid orphan receptor γ t
- sdc1
syndecan-1
- sdc1KO
sdc1 knockout
- WT
wild type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest 120: 1762–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conti HR, Peterson AC, Brane L, Huppler AR, Hernández-Santos N, Whibley N, Garg AV, Simpson-Abelson MR, Gibson GA, Mamo AJ, et al. 2014. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J. Exp. Med 211: 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch EZ, Anderson KL, and Feldman SR. 2015. Interleukin 17 deficiency and implications in cutaneous and systemic diseases. J. Dermatol. Dermatol. Surg 19: 73–79. [Google Scholar]
- 4.Malik S, Want MY, and Awasthi A. 2016. The emerging roles of gammadelta T cells in tissue inflammation in experimental autoimmune encephalomyelitis. Front. Immunol 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin B, Hirota K, Cua DJ, Stockinger B, and Veldhoen M. 2009. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31: 321–330. [DOI] [PubMed] [Google Scholar]
- 6.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, et al. ; Secukinumab in Crohn’s Disease Study Group. 2012. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, and Lubberts E. 2009. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol 182: 5836–5845. [DOI] [PubMed] [Google Scholar]
- 8.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, and Zako M. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem 68: 729–777. [DOI] [PubMed] [Google Scholar]
- 9.Khotskaya YB, Dai Y, Ritchie JP, MacLeod V, Yang Y, Zinn K, and Sanderson RD. 2009. Syndecan-1 is required for robust growth, vascularization, and metastasis of myeloma tumors in vivo. J. Biol. Chem 284: 26085–26095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhodapkar MV, Abe E, Theus A, Lacy M, Langford JK, Barlogie B, and Sanderson RD. 1998. Syndecan-1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood 91: 2679–2688. [PubMed] [Google Scholar]
- 11.Dai H, Rahman A, Saxena A, Jaiswal AK, Mohamood A, Ramirez L, Noel S, Rabb H, Jie C, and Hamad AR. 2015. Syndecan-1 identifies and controls the frequency of IL-17-producing naïve natural killer T (NKT17) cells in mice. Eur. J. Immunol 45: 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Georgiev H, Ravens I, Benarafa C, Förster R, and Bernhardt G. 2016. Distinct gene expression patterns correlate with developmental and functional traits of iNKT subsets. Nat. Commun 7: 13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georgiev H, Ravens I, Shibuya A, Förster R, and Bernhardt G. 2016. CD155/CD226-interaction impacts on the generation of innate CD8(+) thymocytes by regulating iNKT-cell differentiation. Eur. J. Immunol 46: 993–1003. [DOI] [PubMed] [Google Scholar]
- 14.Lee YJ, Starrett GJ, Lee ST, Yang R, Henzler CM, Jameson SC, and Hogquist KA. 2016. Lineage-specific effector signatures of invariant NKT cells are shared amongst γδ T, innate lymphoid, and Th cells. J. Immunol 197: 1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drees C, Vahl JC, Bortoluzzi S, Heger KD, Fischer JC, Wunderlich FT, Peschel C, and Schmidt-Supprian M. 2017. Roquin paralogs differentially regulate functional NKT cell subsets. J. Immunol 198: 2747–2759. [DOI] [PubMed] [Google Scholar]
- 16.Jaiswal AK, Sadasivam M, and Hamad ARA. 2017. Syndecan-1-coating of interleukin-17-producing natural killer T cells provides a specific method for their visualization and analysis. World J. Diabetes 8: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stepp MA, Gibson HE, Gala PH, Iglesia DD, Pajoohesh-Ganji A, Pal-Ghosh S, Brown M, Aquino C, Schwartz AM, Goldberger O, et al. 2002. Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J. Cell Sci 115: 4517–4531. [DOI] [PubMed] [Google Scholar]
- 18.Szabo SK, Hammerberg C, Yoshida Y, Bata-Csorgo Z, and Cooper KD. 1998. Identification and quantitation of interferon-gamma producing T cells in psoriatic lesions: localization to both CD4+ and CD8+ subsets. J. Invest. Dermatol 111: 1072–1078. [DOI] [PubMed] [Google Scholar]
- 19.Mohamood AS, Bargatze D, Xiao Z, Jie C, Yagita H, Ruben D, Watson J, Chakravarti S, Schneck JP, and Hamad AR. 2008. Fas-mediated apoptosis regulates the composition of peripheral alphabeta T cell repertoire by constitutively purging out double negative T cells. PLoS One 3: e3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martina MN, Bandapalle S, Rabb H, and Hamad AR. 2014. Isolation of double negative αβ T cells from the kidney. J. Vis. Exp 87: e51192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strober W 2015. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol 111: A3.B1–A3.B.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, and Yan J. 2011. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 35: 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray EE, Ramírez-Valle F, Xu Y, Wu S, Wu Z, Karjalainen KE, and Cyster JG. 2013. Deficiency in IL-17-committed Vγ4(+) γδ T cells in a spontaneous Sox13-mutant CD45.1(+) congenic mouse substrain provides protection from dermatitis. Nat. Immunol 14: 584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas JD, González FH, Schmitz S, Chennupati V, Föhse L, Kremmer E, Förster R, and Prinz I. 2009. CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells. Eur. J. Immunol 39: 3488–3497. [DOI] [PubMed] [Google Scholar]
- 25.Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, and Silva-Santos B. 2009. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat. Immunol 10: 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramírez-Valle F, Gray EE, and Cyster JG. 2015. Inflammation induces dermal Vγ4+ γδT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc. Natl. Acad. Sci. USA 112: 8046–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang F, Meng G, and Strober W. 2008. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. [Published erratum appears in 2009 Nat. Immunol. 10: 223.] Nat. Immunol 9: 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shedlock DJ, Talbott KT, Morrow MP, Ferraro B, Hokey DA, Muthumani K, and Weiner DB. 2010. Ki-67 staining for determination of rhesus macaque T cell proliferative responses ex vivo. Cytometry A 77: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, and Becher B. 2012. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Invest 122: 2252–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts SJ, Smith AL, West AB, Wen L, Findly RC, Owen MJ, and Hayday AC. 1996. T-cell alpha beta + and gamma delta + deficient mice display abnormal but distinct phenotypes toward a natural, widespread infection of the intestinal epithelium. Proc. Natl. Acad. Sci. USA 93: 11774–11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilliet M, Conrad C, Geiges M, Cozzio A, Thürlimann W, Burg G, Nestle FO, and Dummer R. 2004. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol 140: 1490–1495. [DOI] [PubMed] [Google Scholar]
- 32.Becher B, and Pantelyushin S. 2012. Hiding under the skin: interleukin-17-producing γδ T cells go under the skin? Nat. Med 18: 1748–1750. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, and Perl A. 2014. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J. Immunol 192: 4134–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Götte M, Bernfield M, and Reizes O. 2005. Constitutive and accelerated shedding of murine syndecan-1 is mediated by cleavage of its core protein at a specific juxtamembrane site. Biochemistry 44: 12355–12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.