

Abstract
The hepatitis C virus (HCV) glycoproteins E1 and E2 should be anchored in the viral membrane by their C-terminal domains. During synthesis, they are translocated to the endoplasmic reticulum (ER) lumen where they remain. The 31 C-terminal residues of the E1 protein and the 29 C-terminal residues of the E2 protein are implicated in the ER retention. Moreover, the E1 and E2 C termini are implicated in E1-E2 heterodimerization. We studied the E1 and E2 C-terminal sequences of 25 HCV strains in silico using molecular modeling techniques. We conclude that both C-terminal domains should adopt a similar and peculiar configuration: one amphipathic α-helix followed by a pair of transmembrane β-strands. Several three-dimensional (3-D) models were generated. After energy minimization, their ability to interact with membranes was studied using the molecular hydrophobicity potentials calculation and the IMPALA procedure. The latter simulates interactions with a membrane by a Monte Carlo minimization of energy. These methods suggest that the β-hairpins could anchor the glycoproteins in the ER membrane at least transiently. Anchoring could be stabilized by the adsorption of the nearby amphipathic α-helices at the membrane surface. The 3-D models correlate with experimental results which indicate that the E1-E2 transmembrane domains are involved in the heterodimerization and have ER retention properties.
The hepatitis C virus (HCV) has been recognized as the major etiological agent of human non-A, non-B hepatitis since 1989 (2, 7, 29). HCV is a small enveloped virus of the Flaviviridae family which contains a positive-stranded RNA genome of approximately 9,500 nucleotides (7, 8). This genome codes for a single 3010- to 3033-amino-acid polyprotein which is processed by cell and viral proteases to produce the mature structural and nonstructural proteins C, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (38, 51). E1 and E2 are membrane-associated envelope glycoproteins. They are cleaved from the polyprotein by host signal peptidases (16, 23, 31, 48). E1 is the fragment between amino acids 192 and 383, and E2 is the fragment between amino acids 384 and 746. Native E1 and E2 interact to form heterodimers that are stabilized by noncovalent interactions (15, 17). These E1-E2 complexes should be subunits of the HCV envelope spikes.
The E1 and E2 proteins have hydrophobic carboxy-terminal (C-terminal) regions that presumably act as membrane anchors (51). Indeed, deletion of this region of E2 leads to its secretion (41, 53). The E2 membrane anchor should begin at amino acid 718 of the polyprotein (42). The situation is not as clear for E1, since a truncated form ending at amino acid 340 is secreted only if it contains an internal deletion between amino acids 262 and 290, suggesting the presence of a second membrane anchor (39). However, for Hussy et al. (26) and Michalak et al. (41), truncated forms ending at amino acid 311 and 334 with no additional deletion can be secreted. E1, like E2, is therefore probably anchored only by its C-terminal hydrophobic domain. The limits of this transmembrane domain have not yet been clearly determined.
Some data suggest that the E1 and E2 truncated proteins are secreted as complexes in the bathing medium of cell cultures (39), while others indicate that the C-terminal part of E2 is required to form the E1-E2 complexes (53). These contradictions were reanalyzed by Michalak et al. (41). Using immunoprecipitation with anti-E1 or anti-E2 monoclonal antibodies, they detected no E1-E2 dimer when deletions were introduced into the C terminus of E2. They therefore confirmed the observations by Selby et al. (53) and pointed out that the C-terminal domain of E2 should be important for heterodimerization (41). More recently, the crucial role of E1 and E2 C-terminal domains in heterodimerization was demonstrated through mutagenesis studies (11, 45).
Since no efficient cell culture replication system is available, the assembly and release of HCV particles have not been directly studied. Some observations such as the lack of modification of the glycans by the Golgi enzymes, the location of the HCV glycoproteins in the endoplasmic reticulum (ER), and their absence at the cell surface suggest that the mature E1-E2 heterodimer does not leave the ER and that E1 and/or E2 contains an ER retention signal (15, 16, 48, 55). The ER retention is static rather than due to a recycling from the cis-Golgi region (19). In 1998, Cocquerel et al. (10) showed that the E2 glycoprotein is retained in the ER as the E1-E2 heterodimer because of a specific signal in the C-terminal 29 amino acids. Replacement of this domain with the anchor domain of a protein normally targeted to the plasma membrane (the CD4 protein) is sufficient to drive E2 at the cell surface. Moreover, when the ectodomain of CD4 is fused to the transmembrane domain of E2, the chimeric protein is stored in the ER (10). The same authors (9) looked for an ER retention signal in E1 by making chimeric proteins with the ectodomain of CD4 or CD8 and the C-terminal hydrophobic sequence of E1 (C-terminal 31 amino acids). They showed that the transmembrane domain of E1 retains the chimera in the ER. In addition, the glycans of the chimeric proteins were not processed by the Golgi enzymes. This indicates that the transmembrane domain of E1 is responsible for a true retention in the ER. Charged residues of the E1 and E2 C-terminal domains have been shown to be crucial for the ER retention (11).
We studied the E1 and E2 C-terminal anchor domains in silico using molecular modeling. Analysis of 25 HCV strains, as well as calculation and characterization of three-dimensional (3-D) models, suggests a peculiar conformation of the E1 and E2 anchor regions. Both C-terminal domains should be one amphipathic α-helix followed by a β-hairpin. The β-strands could insert in the membrane and stay there, at least transiently. The expected properties of the 3-D models correlate with the heterodimerization and ER retention properties of E1 and E2.
MATERIALS AND METHODS
Materials.
Calculations were performed on RAMSES (Rapid Analysis Master/Slaves Extensible System) clusters of 320 Pentium processor PCs in parallel connected with a 100Mbits/s Ethernet network. The calculation softwares were developed in our laboratory (5, 18). Molecular views and hydrophobicity potentials were drawn with WinMGM 1.0 software (47) from Ab Initio Technology (Obernai, France).
HCV sequences.
The E1 and E2 sequences of the following 25 HCV strains were extracted from PIR, SwissProt, EMBL, and GenBank sequence databases: HCV-BEINNX11 (GenBank accession no. A48711), HCV-BK (SwissProt accession no. P26663), HCV-J (SwissProt accession no. P26662), HCV-J33 (GenBank accession no. D14484), HCV-J4/83 (GenBank accession no. D13558), HCV-J4/91 (GenBank accession no. D10750), HCV-JK1 (PIR accession no. S18030), HCV-JT (GenBank accession no. D11168), HCV-JT" (GenBank accession no. D11355), HCV-L2 (GenBank accession no. U01214), HCV-N (GenBank accession no. S62220), HCV-T (SwissProt accession no. P29846), HCV-Unkcds (EMBL accession no. M96362), HCV-K1-R1 (GenBank accession no. D50480), HCV-K1-R2 (GenBank accession no. D50481), HCV-K1-R3 (GenBank accession no. D50482), HCV-K1-S1 (GenBank accession no. D50483), HCV-K1-S2 (GenBank accession no. D50485), HCV-K1-S3 (GenBank accession no. D50484), HCV-1 (SwissProt accession no. P26664), HCV-H (SwissProt accession no. P27958), HCV-J1 (EMBL accession no. D10749), HCV-G9 (GenBank accession no. D14853), HCV-J6 (SwissProt accession no. P26660), and HCV-J8 (SwissProt accession no. P26661). Note that amino acid numbers always refer to the mature HCV-BEINNX11 glycoproteins. E1 extends from amino acid 1 (= amino acid 192 in the polyprotein) to amino acid 192 (= amino acid 383 in the polyprotein), and E2 extends from amino acid 1 (= amino acid 384 in the polyprotein) to amino acid 363 (= amino acid 746 in the polyprotein).
Kyte and Doolittle's method.
The mean hydrophobicity of a stretch of sequence is calculated by moving a seven-residue window along the sequence (30). The value is attributed to the central amino acid of the window, and the hydrophobicity of each amino acid is defined in the original work (30).
HCA.
The hydrophobic cluster analysis (HCA) plot (21) relies upon the 2-D helical representation of the sequence: the sequence is written on a classical α-helix (3.6 residues per turn) smoothed on a cylinder. To make the view easier to handle, the cylinder is then cut parallel to its axis and unrolled. As some adjacent amino acids are separated by the flattening of the cylinder, the plot is duplicated to restore all the connections of residues. The hydrophobic residues (F, V, L, I, M, Y, and W) are circled and hatched; proline is represented by a star, glycine is represented by a diamond, serine is represented by a square with a dot in it, and threonine is represented by an empty square. This method allows β-strands to be distinguished from α-helices, as the shape and morphology of the hydrophobic clusters are related to these secondary structures: long horizontal hydrophobic clusters denote hydrophobic or amphipathic α-helices, while short vertical or “mosaic” clusters are indicative of β-strands. This representation is also useful in predicting membrane or transmembrane domains.
Peptide structure and conformational search.
Helical peptides were constructed with standard values of φ and ψ angles (−60° and −40°) and subjected to a Simplex minimization (44). For the study of the putative β-pins, conformational searches of the turn were performed using the stereoalphabet procedure (14, 36). The analysis was carried out on 8 residues and in two steps. During the first step, the (φ, ψ) couples of the stereoalphabet were systematically attributed to the first five amino acids of the putative turn while the other residues were fixed as the β-strand. The couples of φ and ψ were as follows: −60°, −40° (right α-helix); −160°, 160° (β-sheet); −140°, 80° (intermediate structure between β-sheet and 310 helix); −80°, 160° (helix of the polyproline); −80°, 80° (310 helix); and 60°, 60° (left α-helix). From the 7,776 conformations tested, the 20 lowest-energy structures were subjected to a second analysis: the φ and ψ angles of the last five amino acids of the putative turn were tested using all couple-of-angle values of the stereoalphabet (the conformation of the fourth and the fifth amino acids were thus studied twice). For each of the 20 first-step structures, the 10 lowest-energy structures were conserved in the second step (200 structures out of a total of 155,520 calculated conformations). The energy of these 200 structures was minimized using the Simplex method (44). The structure with the lowest energy was considered to be the most-probable structure. The energy of all structures was calculated as the sum of the torsion potential and the Van der Waals, electrostatic, and hydrophobic interactions (35). All calculations take into account the presence of a hydrophobic/hydrophilic interface with the concomitant variation of the dielectric constant (5).
MHP.
The molecular hydrophobicity potentials (MHP) were used to describe the hydrophobic or hydrophilic environment of peptides as previously described (6). For the MHP calculation, the assumption is made that the hydrophobic potential decreases exponentially with the distance as follows:
 |
where N is the number of atoms; Etri and ri are the transfer energy and the Van der Waals radius of the i atom, respectively; and di is the distance between the i atom and the point where the potential is calculated.
Orientation towards a hydrophobic-hydrophilic interface.
A hydrophilic center is calculated using the following equation (5):
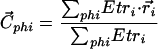 |
where r⃗i and Etri are the coordinate and the transfer energy of atom i when only the hydrophilic atoms of the molecule are taken into consideration (phi). A hydrophobic center (C⃗pho) is calculated using a similar equation taking into account only the hydrophobic atoms. The Cdelta⃗ = Cpho⃗ − Cphi⃗ vector is stated as perpendicular to the interface. The interface position (I⃗) is given by the equation:
 |
IMPALA.
In the IMPALA procedure (18), an empirical function, C, is used to describe the water-lipid interface. Considering that the properties of membranes are constant in the plane of the bilayer, C varies only along the z axis, perpendicular to that plane. The z axis has its origin at the center of the membrane, and the empirical function C is described by the following equation:
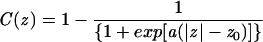 |
where α and z0 are defined so that C ≈ 1 for z < −18.5 and z > 18.5 and C ≈ 0 for −13.5 < z < 13.5. The interactions between the peptide and the bilayer are described by the sum of two restraint functions as follows:
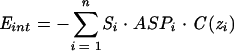 |
Eint simulates the hydrophobic effect by taking the accessible surface (Si), the free energy of transfer per accessible surface unit (ASPi), and the position (zi) of each atom i. The general evolution of this equation is an increase when accessible hydrophilic atoms (i.e., Etr > 0) penetrate the membrane and a decrease when accessible hydrophobic atoms do so. The more accessible the atoms are, the greater the effect is. The second function mimics the solvophobic effect in lipids:
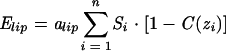 |
where alip is an empirical constant equal to 0.018. Elip tends to minimize the interactions between the peptide and the bilayer. Assuming a constant structure for all peptides, the position and orientation in the membrane were tested by running 10 Monte Carlo procedures of 105 steps at 310 K each. Maximal rotations of 5° and translations of 2 Å per step were allowed. The algorithm used to orient the structures with respect to a hydrophobic-hydrophilic interface (see above) was used to determine the starting position of the peptides. The position of minimal energy was retained.
RESULTS
Sequence analysis.
The hydropathy of E1 and E2 sequences was analyzed to determine which fragments of their C-terminal domains were likely to interact with membranes. From the HCA and the Kyte and Doolittle plots of the E1 protein (Fig. 1), we pointed out that a C-terminal hydrophobic domain extends from proline 137 to glycine 192 (position in the mature protein). For E2, a hydrophobic domain was detected from proline 281 to alanine 363 (position in the mature protein) (Fig. 2).
FIG. 1.

(A) HCA plot of the HCV-BEINNX11 E1 glycoprotein. (B) Minimal (dashed line), mean (solid line), and maximal (dotted line) values of the Kyte and Doolittle hydrophobicity along the E1 sequences of 25 HCV strains obtained by using a seven-residue window. Amino acid numbering refers to the position of the amino acids in the mature proteins.
FIG. 2.

(A) HCA plot of the HCV-BEINNX11 E2 glycoprotein. (B) Minimal (dashed line), mean (solid line), and maximal (dotted line) values of the Kyte and Doolittle hydrophobicity along the E2 sequences of 25 HCV strains obtained by using a seven-residue window. Amino acid numbering refers to the position of the amino acids in the mature proteins.
Alignment of the E1 and E2 C-terminal sequences from 25 HCV strains was performed in order to identify potent transmembrane stretches and/or other structural entities able to interact with membrane. In these alignments, conservation of residues, as well as conservation of their hydrophobicity, charges, etc., was studied.
In the E1 C-terminal domain, several stretches with conserved properties were identified from the C- to the N-terminal extremities (Fig. 3A) as follows. (i) The Val180-Ala188 stretch contains only hydrophobic residues and ends with an aromatic residue (Phe187). In addition, hydrophilic, charged, and coil-favorable residues are located on both sides (Gly175, Asn176, Lys179, Gly189, Asp191, and Gly192). (ii) The Trp162-Tyr171 region has no or few charged and polar residues (a lysine is present in one sequence). It is edged by aromatic residues on both sides (Trp162, Tyr170, and Tyr171) and preceded by a hydrophilic stretch containing a coil-favorable residue (Gly159). (iii) The Ala140-Ala158 region is rather amphiphilic. It contains hydrophobic and charged or polar residues and is preceded by a stretch containing polar residues that are often present in coil or turn structures (Pro137-Thr139).
FIG.3.

Sequence alignment of E1 (A) and E2 (B) C-terminal domains from 25 HCV strains. A dash indicates an amino acid identical to the amino acid of HCV-BEINNX11 strain in the alignment. Amino acids are indicated according to the following characteristics (13, 60). Hydrophobic amino acids (Val, Leu, Ile, Met, Phe, Tyr, and Trp) which are preferentially implicated in transmembrane stretches are hatched. Among these, the aromatic residues (Phe, Tyr, and Trp) prefer the water-membrane interface. Charged amino acids (Asp, Glu, Arg, and Lys) which are avoided in transmembrane stretches are highlighted on a black background. His, Asn, and Gln are polar amino acids and are generally located in the water phase or at the hydrophobic-hydrophilic interface. Pro, the well-known helix breaker, and Gly, Ser, and Thr are frequent in coil and turn structures. They are indicated in italic type. Ala can be both in the membrane and the water phase. Domains 1, 2, and 3 for E1 and domains 1, 2, 3, and 4 for E2 are discussed in the text. Positions of alanine inserted by Op De Beeck et al. (45) are indicated by arrows on the top of each alignment. Stars denote the positions of alanine which have a significant effect on heterodimerization.
From the C- to the N-terminal extremities, the following properties were found in the C-terminal domain of E2 (Fig. 3B). (i) The Leu352-Ile358 region contains only hydrophobic residues and an aromatic residue is present at the N-extremity (Trp353). This stretch is surrounded by polar and charged residues (Ala344-Cys351 and Ala359-Ala363). (ii) The Tyr335-Leu343 region contains only hydrophobic residues and starts with an aromatic residue (Tyr335). It is preceded by a charged stretch (Lys332-Glu334). (iii) The Ile313-Ile331 stretch is rather amphiphilic, since it contains hydrophobic and charged or polar residues. (iv) The N-terminal stretch (Pro281-Asn312) is also amphiphilic and is moreover rich in coil- or turn-favorable amino acids (Pro293 and -300; Ser285, -295, and -303; Thr286, -287, -297, -298, and -304; Gly305).
Based on these analyses, we suggest that the C-terminal domains of the E1 and E2 glycoproteins share a similar pattern (Fig. 3 and 4): a long amphiphilic region (18 or 19 residues) followed by two short hydrophobic stretches (7 to 10 residues) surrounded by hydrophilic residues.
FIG. 4.

Comparison of the HCA plots of the C-terminal domains of HCV-BEINNX11 E1 glycoprotein (A) and HCV-BEINNX11 E2 glycoprotein (B). (C) Schematic representation of the topological model. Putative transmembrane β-strands (1 and 2) and a putative amphipathic α-helix (3) are indicated.
An even number of transmembrane stretches is expected to anchor E1 and E2 in the membrane, because the N- and C-terminal extremities are in the lumen of the ER. The two hydrophobic stretches are the best candidates for these transmembrane stretches. It is usually thought that 18 to 20 hydrophobic residues are required to constitute transmembrane α-helices. The hydrophobic stretches of E1 and E2 are 7 to 10 residues long. This is inappropriate for classical transmembrane α-helices. In other words, since the mean distance between two consecutive residues of an α-helix is equal to 1.5 Å in the direction of the helix axis (13), these stretches should be 10.5 to 15 Å as α-helices. This is only half the width of a lipid bilayer. In a β-strand, the distance between two adjacent amino acids is 3.4 Å (13). Therefore, the E1 and E2 stretches would be 23.8 Å long for the shortest strand and 34 Å for the longest strand. This type of configuration seems, at least arithmetically, to be the most likely to cross the membrane (Fig. 4C).
In the amphiphilic region before the putative transmembrane β-strands, the HCA profile shows long horizontal hydrophobic clusters suggestive of amphipathic α-helices (21) (Fig. 4).
In order to test the hypotheses, several 3-D models were built for the HCV-BEINNX11 strain. These models were tested for membrane interaction.
3-D model of amphipathic α-helices.
Helices were built for every stretch of a minimal length of 13 amino acids between Ala140 and Ala158 for E1 and between Ile313 and Ile331 for E2. The energy of each α-helix was minimized using the Simplex method. Each structure was then oriented towards a hydrophobic-hydrophilic interface. Helices eliciting a tilt of less than 30° between their axis and this interface were considered to be amphipathic and were selected for a thorough study using the IMPALA procedure. The 10 IMPALA Monte Carlo simulations gave similar results for each peptide. The MHP were also calculated to check the hydropathy of each structure. The best candidate as amphipathic α-helix was selected as the stretch with a maximal length, an insertion angle close to 0°, and a maximal depth of penetration in the bilayer.
Table 1 summarizes the screening for putative amphipathic α-helices in E1. Most peptides behave as amphipathic α-helices, and Val142-Ala158 was selected as the best candidate (Fig. 5). This 17-residues stretch makes a 10° angle with respect to its hydrophobic-hydrophilic interface and with the IMPALA membrane surface (Fig. 5C). In this position, the mass center of the structure was located at 13.5 Å from the membrane center, indicating that the peptide is able to adsorb at the membrane surface by inserting between the phospholipid heads. The MHP confirm the amphipathy of the helix since the hydrophobic and hydrophilic isopotential envelopes are well segregated at the helix surface (Fig. 5B).
TABLE 1.
Characterization of helical peptides found between Ala140 and Ala158a
| Start residue | End residue | Length (amino acids) | Orientation (°) | IMPALA angle (°) | IMPALA penetration (Å) |
|---|---|---|---|---|---|
| A140 | A158 | 19 | 35 | ||
| A140 | V157 | 18 | 25 | 10-15 | 14.5 |
| A140 | M156 | 17 | 35 | ||
| A140 | D155 | 16 | 40 | ||
| A140 | V154 | 15 | 20 | 10-15 | 14.5 |
| A140 | V153 | 14 | 30 | 10-15 | 14.5 |
| A140 | V152 | 13 | 40 | ||
| L141 | A158 | 18 | 35 | ||
| L141 | V157 | 17 | 25 | 10 | 14 |
| L141 | M156 | 16 | 35 | ||
| L141 | D155 | 15 | 40 | ||
| L141 | V154 | 14 | 25 | 5 | 16 |
| L141 | V153 | 13 | 35 | ||
| V142 | A158 | 17 | 15 | 10-15 | 13.5 |
| V142 | V157 | 16 | 5 | 15 | 13.5 |
| V142 | M156 | 15 | 20 | 15 | 13.5 |
| V142 | D155 | 14 | 25 | 5 | 15.5 |
| V142 | V154 | 13 | 10 | 5 | 15.5 |
| V143 | A158 | 16 | 0 | 10-15 | 14 |
| V143 | V157 | 15 | 0 | 15-20 | 14 |
| V143 | M156 | 14 | 0 | 15-20 | 14.5 |
| V143 | D155 | 13 | 20 | 15 | 15 |
| S144 | A158 | 15 | 10 | 10-15 | 14 |
| S144 | V157 | 14 | 15 | 15 | 14 |
| S144 | M156 | 13 | 10 | 15 | 14.5 |
| Q145 | A158 | 14 | 10 | 10-15 | 13.5 |
| Q145 | V157 | 13 | 0 | 15 | 14 |
| L146 | A158 | 13 | 35 |
Orientation towards a hydrophobic-hydrophilic interface and results of the IMPALA simulations are given. The data for the best putative amphipathic α-helix are shown in boldface type.
FIG. 5.

HCV-BEINNX11 E1 putative amphipathic α-helix (Val142 to Ala158). (A) Minimized 3-D structure. (B) MHP around the peptide (orange = hydrophobic; green = hydrophilic). (C) Best configuration found by the IMPALA procedure. The center of the phospholipid bilayer is indicated by an orange line, the limit between the phospholipid acyl chains and polar heads is indicated by a yellow line, and the limit between the water phase and the phospholipids is indicated by a green line.
Table 2 summarizes the screening for the E2 glycoprotein. As for E1, most α-helices are amphipathic. The 16-amino-acid Val316-Ile331 peptide was selected as the best candidate, as its angle with respect to its hydrophobic-hydrophilic interface or with the IMPALA modeled lipid bilayer surface is close to 0° (Fig. 6). The mass center of the helix in the membrane is at 12.5 Å from the membrane center. The MHP confirm the segregation of the hydrophobic and hydrophilic faces separately (Fig. 6B).
TABLE 2.
Characterization of helical peptides found between Ile313 and Ile331a
| Start residue | End residue | Length (amino acids) | Orientation (°) | IMPALA angle (°) | IMPALA penetration (Å) |
|---|---|---|---|---|---|
| I313 | 1331 | 19 | 35 | ||
| I313 | V330 | 18 | 45 | ||
| I313 | L329 | 17 | 65 | ||
| I313 | S328 | 16 | 70 | ||
| I313 | V327 | 15 | 65 | ||
| I313 | V326 | 14 | 75 | ||
| I313 | A325 | 13 | 80 | ||
| V314 | I331 | 18 | 25 | ||
| V314 | V330 | 17 | 5 | 5 | 12.5 |
| V314 | L329 | 16 | 30 | 5 | 12.5 |
| V314 | S328 | 15 | 50 | ||
| V314 | V327 | 14 | 30 | 20 | 14 |
| V314 | V326 | 13 | 45 | ||
| D315 | I331 | 17 | 40 | ||
| D315 | V330 | 16 | 15 | 30 | 17 |
| D315 | L329 | 15 | 5 | 30 | 17 |
| D315 | S328 | 14 | 35 | ||
| D315 | V327 | 13 | 10 | 0 | 18.5 |
| V316 | I331 | 16 | 5 | 0 | 12.5 |
| V316 | V330 | 15 | 20 | 0 | 12.5 |
| V316 | L329 | 14 | 35 | ||
| V316 | S328 | 13 | 50 | ||
| Q317 | I331 | 15 | 15 | ||
| Q317 | V330 | 14 | 10 | 20 | 15 |
| Q317 | L329 | 13 | 25 | 10 | 16 |
| Y318 | I331 | 14 | 25 | 20 | 14 |
| Y318 | V330 | 13 | 40 | ||
| L319 | I331 | 13 | 5 | 25 | 13.5 |
Orientation towards a hydrophobic-hydrophilic interface and results of the IMPALA simulations are given. The data for the best putative amphipathic α-helix is shown in boldface type.
FIG. 6.

HCV-BEINNX11 E2 putative amphipathic α-helix (Val316 to Ile331). (A) Minimized 3-D structure. (B) MHP around the peptide (orange = hydrophobic; green = hydrophilic). (C) Best configuration found by the IMPALA procedure. The center of the phospholipid bilayer is indicated by an orange line, the limit between the phospholipid acyl chains and polar heads is indicated by a yellow line, and the limit between the water phase and the phospholipids is indicated by a green line.
3-D model of transmembrane β-strands.
To test the hypothesis of a transmembrane β-pin, a β structure was imposed on the residues Trp162 to Tyr171 and Val180 to Ala188 of E1 and on Tyr335 to Leu343 and Leu352 to Ala359 of E2. The conformation of the eight residues located between the two β-strands was then calculated using the stereoalphabet method. For each protein, 155,520 conformations were tested.
The lowest energy structure obtained for E1 is shown in Fig. 7A. The analysis shows that the best conformation of the loop corresponds to an antiparallel pair of β-strands (Trp162 to Met173 and Trp177 to Ala188). Interstrand hydrogen bonds are implicated in the energy stabilization of the structure. MHP indicate that the final structure is highly hydrophobic except at the backbone contact (due to accessible main-chain C=O and N-H groups), in the turn, and at the peptide N- and C- terminal extremities (Fig. 7B). The hydrophobic part is longer than 35 Å, and this should be sufficient to cross a membrane. The results of the 10 IMPALA simulations confirmed this hypothesis. The most favorable position of this structure is transmembranous (Fig. 7C), with the soluble and adsorbed states being unlikely.
FIG. 7.

HCV-BEINNX11 E1 putative transmembrane β-strands (Trp162 to Ala188). (A) Best minimized 3-D structure obtained by the stereoalphabet procedure. (B) MHP around the peptide (orange = hydrophobic; green = hydrophilic). (C) Best configuration found by the IMPALA procedure. The center of the phospholipid bilayer is indicated by an orange line, the limit between the phospholipid acyl chains and polar heads is indicated by a yellow line, and the limit between the water phase and the phospholipids is indicated by a green line.
Figure 8Ashows the E2 transmembrane β-pins after the stereoalphabet procedure, and once again, the β-strands are antiparallel with interstrand hydrogen bonds (Tyr335 to Asp345 and Cys349 to Ile358). As for E1, the structure is highly hydrophobic, with hydrophilic patches in the turn, at the backbone contact, and at the N- and C-terminal ends, as shown with the MHP (Fig. 8B). The hydrophobic part of the structure is 35 Å long, and IMPALA simulations show that the position of minimal energy is transmembranous (Fig. 8C).
FIG. 8.

HCV-BEINNX11 E2 putative transmembrane β-strands (Tyr335 to Ala359). (A) Best minimized 3-D structure obtained by the stereoalphabet procedure. (B) MHP around the peptide (orange = hydrophobic; green = hydrophilic). (C) Best configuration found by the IMPALA procedure. The center of the phospholipid bilayer is indicated by an orange line, the limit between the phospholipid acyl chains and polar heads is indicated by a yellow line, and the limit between the water phase and the phospholipids is indicated by a green line.
DISCUSSION
The in silico study of protein transmembrane domains is limited by the number of experimental data already available. Indeed, prediction often works by analogy to templates, but there are very few elucidated membrane protein structures in the Protein Data Bank (fewer than 25). In most cases, the construction of topological models is thus based on sequence analysis to identify hydrophobic stretches with a minimum length of 18 to 20 residues, corresponding to transmembrane α-helices. Even if this method has already been tried and tested, it has limited utility for the study of nonhelical transmembrane domains or of membrane spans with unusual properties. This is the case for HCV glycoproteins which contain hydrophobic stretches shorter than 18 residues.
In the first part of our investigation, the analysis of E1 and E2 C-terminal sequences of 25 HCV strains showed that both envelope proteins share a similar pattern: two short hydrophobic stretches separated by charged or polar amino acids and, in the case of E1, by coil-favorable residues. These stretches follow a region likely to be an amphipathic helix. We postulate first that the E1 and E2 proteins could be anchored in the ER membrane by the two short hydrophobic C-terminal stretches. Stereoalphabet analysis, MHP calculation, and IMPALA simulations indicate that these stretches could form transmembrane β-pins. Second, these β-pins could interact more easily with the lipid bilayer because of the adsorption of the amphipathic α-helix. These two hypotheses are discussed below.
The E1 and E2 glycoproteins are cleaved from the HCV polyprotein by host signal peptidase (16, 23, 31, 48). Both extremities of the proteins are thus in the ER lumen, at least transiently, and this is the reason why an even number of transmembrane stretches is expected. Our first hypothesis is that the two required transmembrane spans are the short hydrophobic C-terminal segments of E1 and E2. Another configuration would present a significant number of hydrophobic residues in the water phase and charged and polar residues in the membrane. Moreover, the proposed configuration favorably places the aromatic residues at the water-phospholipid interface.
The E1 and E2 C-terminal domains signal for static retention in the ER (9, 10, 20). CD4 and CD8 are usually targeted to the plasma membrane. If the last 31 amino acids of E1 are coupled to the CD4 or CD8 ectodomain, the chimeric protein remains in the ER compartment (9). Cocquerel et al. (10) have also shown that E2 is retained in the ER by its transmembrane domain (the 29 C-terminal residues) and that this segment is able to confer the ER-retention property to the CD4 ectodomain. These “cut-and-paste” experiments prove the ER retention properties of the E1 31 C-terminal and the E2 29 C-terminal residues. Moreover, they demonstrate that these fragments are independent entities with native folding, which supports the idea that they are the E1 and E2 transmembrane segments, as proposed in our model.
It is generally admitted that the hydrocarbon region of a fluid bilayer is typically 30 Å thick (34, 58). Analyzing the cytochrome c oxidase from bovine mitochondria, Wallin et al. (57), however, showed that this bundle of 28 transmembrane α-helices extends about ±20 Å from the center of the membrane. They further emphasized that the hydrophobic residues are within ±10 Å from the bilayer center and that this slice is symmetrically flanked by regions rich in aromatic residues. In 1999, Monne et al. (43) determined that the minimum length of a poly-Leu helical hairpin is 31 residues. The shortest helical hairpin would thus be made by two α-helices of 14 amino acids separated by a three-residue turn. Since the standard translation per residue in an α-helix is 1.5 Å (13), the hydrophobic portion of these helices would be 21 Å. Taking these results into consideration, the hydrophobic core of a membrane would thus be at least ∼20 Å thick. As specified earlier, the E1 and E2 hydrophobic stretches are 7 to 10 residues long (aromatic residues included). An α-helix structure thus appears to be inappropriate to cross the membrane. In a β-strand conformation, the length of these stretches would be 23.8 or 34 Å. These lengths are adapted to a transmembrane topology and are consistent with the size of the strands of the porin β-barrels, which are 6 to 17 amino acids long. MHP confirm this hypothesis, since the transmembrane β-pins exhibit a hydrophobic span at least equal to 35 Å.
Extended structures are thought to be energetically unfavorable in a membrane environment because of the accessibility of main-chain polar groups (60). However, some peptides form β-sheet structures when associated to a lipid bilayer (1, 28, 32, 37). Hexapeptide made of tryptophan and leucine (acetyl-Trp-Leu5 peptides) has been shown to assemble into oligomers of 10 to 20 antiparallel β-strands in membranes (59). PATIR-FTIR measurements indicate that interstrand hydrogen bonds of this sheet are parallel to the membrane surface. The value of the dichroic ratio suggests that the β-sheet has a perpendicular orientation. When the length of the Ac-Trp-Leun peptides is increased to nine residues, β-sheets are still observed in POPC bilayers. The critical number of leucine residues for helix formation in a membrane should be between 10 and 14. These results reveal that the formation of β-sheet in a membrane is easier than expected and thus support our prediction.
The calculation of different stretches from the amphipathic region of E1 and E2 supports our second hypothesis. Indeed, most helices are amphipathic, with a clear segregation of hydrophobic and hydrophilic sides, and adsorb onto the IMPALA bilayer. The Val142-to-Ala158 fragment of E1, and the Val316-to-Ile331 fragment of E2 are the best candidates. Since these amphipathic α-helices are very close to the transmembrane domains, they must be close to the viral membrane as well and could thus probably adsorb to it. We suggest that the membrane adsorption of these amphipathic α-helices stabilizes the membrane insertion of the E1 and E2 β-pins. Characterization of truncated forms of E1 and E2 indicate that the truncation of the putative transmembrane domains is not sufficient to observe efficient secretion of products with native folding (26, 39, 41, 42, 53). This could be correlated to the presence of other membrane-interacting structures, such as adsorbed α-helices.
Our model (Fig. 9) is compatible with available experimental data. Several features explain the retention of proteins in the ER (reviewed in reference 22). “KDEL,” di-lysine, or di-arginine patterns have been identified. Another signal for ER retention is the occurrence of one or more hydrophilic residues in the middle of a hydrophobic transmembrane domain (4, 33, 50). Recent studies suggest that more than one signal is required. The participation of both soluble and transmembrane domains has been illustrated with cytochrome P450 (56), cytochrome b5 (25), Sec12p (52), and the rubella virus E1 glycoprotein (24). Honsho et al. (25) showed that mutation of all or part of the transmembrane domain of cytochrome b5 with hydrophobic residues has almost no effect on the intracellular distribution. However, an increase in the length of the transmembrane domain over 22 amino acids suppresses the retention. Thus, the transmembrane domain length directly functions as a retention signal of cytochrome b5. These results are consistent with those of Pedrazzini et al. (46), who showed that the insertion of five amino acids into the transmembrane domain of cytochrome b5 resulted in cell surface localization. The length of the transmembrane domain is critical for retention of other proteins in the ER (49, 62) and in the Golgi apparatus (reviewed in references 12 and 22). According to these results, our model suggests that E1 and E2 anchor domains contain a bipartite signal responsible for ER retention. The length of E1 and E2 transmembrane stretches should be the first critical determinant for retention. Since there is a relation between the length and the structure of transmembrane spans (see above), we further propose that a β structure could be correlated to the ER retention properties of these anchor domains. This would also be consistent with Flint and McKeating's results (20), which suggest that the HCV E1 and E2 glycoproteins are located in the ER because of the structural properties of their transmembrane segments rather than of specific patterns of sequences. The other part of the retention signal should be in the turn between the two β-strands. Indeed, these stretches contain charged residues which are crucial for ER retention (11).
FIG. 9.

Topological models of the E1 and E2 C-terminal anchor domains.
The heterodimerization of E1 and E2 anchor domains which has now been demonstrated (11, 41, 45, 53) should be governed by several elements. First, E1 and E2 transmembrane β-pins should hide their accessible main-chain polar atoms from the membrane hydrophobicity through multimerization. In our model, these interactions are required to stabilize the transmembrane spans. They should mostly implicate the main chain NH and CO atoms, but the residue side chains could also be determinant, since pairing of interstrand residues has been shown to differ from random (61). Finally, the amino acids of the turn between the β-strands that should be close in the quaternary structure could also be crucial for the heterodimerization.
The final topological models of E1 and E2 anchor domains thus consist of an adsorbed α-helix followed by two interacting transmembrane β-strands (Fig. 9). This partly contradicts the results from Op De Beeck et al. (45), who used alanine insertion to demonstrate that the E1 and E2 C-terminal domains are implicated in E1-E2 dimerization. They further analyzed the 3-D structure of the half of the E1 putative transmembrane domain in membrane mimetic solvents by circular dichroism (CD) and nuclear magnetic resonance (NMR). CD spectra in 2,2,2-trifluoroethanol (TFE), sodium dodecyl sulfate, and lysophosphatidylcholine indicated an α-helical content equal to 48, 44, and 52%, respectively. In TFE, the 21-residue-long peptide (vol/vol) is more frequently helical as determined by NMR analysis.
If the alanine insertion (Fig. 3) clearly proves that the fragment is involved in heterodimerization, it does not prove which structure is required for that dimerization. Indeed, the insertion of one residue in the middle of a secondary-structure element would perturb the spatial distribution of the other residues in an α-helix as well as in a β-strand. In both cases, the perturbation could prevent the dimerization. According to our model, alanine insertion should prevent heterodimerization if the insertion modifies the structure of a β-strand or if it generates unfavorable cross-strand pairs of amino acids. Mutant proteins unable to form heterodimers could also occur if an alanine modifies the structure of the turn of one β-pin or the interactions between the turns. The addition of one alanine in a β-strand should not modify its secondary structure since the length of the transmembrane stretch should not be sufficiently increased to form an α-helix (see above). The insertion of alanine in a β-strand should thus only affect favorable or unfavorable cross-strand pairs of residues. Only three of the alanines inserted in the β-strands have an effect on heterodimerization which is reduced to less than 10% of the wild-type level (45). They are located in the first β-strand of E1 (Fig. 3). Alanines inserted in the other putative β-strands do not have such a drastic effect. Interestingly, the first β-strand of E1 has fewer hydrophobic residues than the others (Fig. 3). It contains two glycines, both on the same side of the strand. This peculiar composition could explain its sensitivity to alanine insertion. Indeed, glycine has particular requirements related to cross-strands pairs (40). On the other hand, the introduction of one alanine in the other strands should not drastically modify the interactions between strands since they contain exclusively hydrophobic amino acids (Fig. 3). In the case of alanine insertion close to the turn between E1 and E2 strands, three mutants are impaired in heterodimerization (Fig. 3). The effect is less dramatic for E2, since the heterodimerization is reduced to about 35% of the wild-type level. This could be due to a modification of the turn structure, less compatible with the interaction of the two β-pins.
CD and NMR analyses by Op de Beeck et al. tend to prove that the E1 transmembrane domain is one α-helix (45). These results must be considered with caution since they were not obtained with the complete E1 putative transmembrane domain. Furthermore, the use of membrane mimetic solvents could have induced an artifact, as TFE is a well-known helix-inducing solvent (3, 27, 37, 54). Finally, the model of Op De Beeck et al. does not take into account the requirement of crossing the membrane twice. They indeed suggest that the E1 and E2 transmembrane domains would first form hairpin structures and then reorganize into single transmembrane helices after the processing of E1 and E2 proteins. We support the argument that the E1 and E2 transmembrane domains adopt a β-hairpin configuration and that this configuration could be more stable than expected.
Conclusions.
In addition to their role in membrane association, the E1 and E2 transmembrane domains (the 31 and 29 last amino acids, respectively) are responsible for heterodimerization and retention of the two HCV envelope glycoproteins in the ER compartment. This is crucial for the HCV life cycle. We put forward the hypothesis that the E1 and E2 transmembrane domains adopt, at least transiently, a transmembrane β-pin conformation downstream of an amphipathic α-helix. The β-structure, the multimerization, and the ER retention properties of the transmembrane stretches would be correlated to their length. To our knowledge, this model is in agreement with results from the literature. Further experimental results will be needed to validate or invalidate our model.
Acknowledgments
The work of B.C. was partly supported by Innogenetics, Belgium.
We thank the National Funds for Scientific Research of Belgium (FNRS), where B.C. is research fellows, L.L. is research associate, and R.B. is research director.
REFERENCES
- 1.Aggeli, A., N. Boden, Y. L. Cheng, J. B. Findlay, P. F. Knowles, P. Kovatchev, and P. J. Turnbull. 1996. Peptides modeled on the transmembrane region of the slow voltage-gated IsK potassium channel: structural characterization of peptide assemblies in the beta-strand conformation. Biochemistry 35:16213-16221. [DOI] [PubMed] [Google Scholar]
- 2.Alter, M. J., S. C. Hadler, F. N. Judson, A. Mares, W. J. Alexander, P. Y. Hu, J. K. Miller, L. A. Moyer, H. A. Fields, and D. W. Bradley. 1990. Risk factors for acute non-A, non-B hepatitis in the United States and association with hepatitis C virus infection. JAMA 264:2231-2235. [PubMed] [Google Scholar]
- 3.Arunkumar, A. I., T. K. Kumar, G. Jayaraman, D. Samuel, and C. Yu. 1996. Induction of helical conformation in all beta-sheet proteins by trifluoroethanol. J. Biomol. Struct. Dyn. 14:381-385. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino, J. S., P. Cosson, N. Shah, and R. D. Klausner. 1991. Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J. 10:2783-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brasseur, R. 1990. TAMMO: theoretical analysis of membrane molecular organization, p. 203-219. In R. Brasseur (ed.), Molecular description of biological membrane components by computer-aided conformational analysis. CRC Press, Boca Raton, Fla.
- 6.Brasseur, R. 1991. Differentiation of lipid-associating helices by use of three-dimensional molecular hydrophobicity potential calculations. J. Biol. Chem. 266:16120-16127. [PubMed] [Google Scholar]
- 7.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359-362. [DOI] [PubMed] [Google Scholar]
- 8.Choo, Q. L., K. H. Richman, J. H. Han, K. Berger, C. Lee, C. Dong, C. Gallegos, D. Coit, R. Medina-Selby, and P. J. Barr. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 88:2451-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cocquerel, L., S. Duvet, J. C. Meunier, A. Pillez, R. Cacan, C. Wychowski, and J. Dubuisson. 1999. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 73:2641-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cocquerel, L., J. C. Meunier, A. Pillez, C. Wychowski, and J. Dubuisson. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocquerel, L., C. Wychowski, F. Minner, F. Penin, and J. Dubuisson. 2000. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 74:3623-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colley, K. J. 1997. Golgi localization of glycosyltransferases: more questions than answers. Glycobiology 7:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Creighton, T. E. 1993. In Proteins: structures and molecular properties, 2nd ed. W. H. Freeman and Company, New York, N.Y.
- 14.Reference deleted.
- 15.Deleersnyder, V., A. Pillez, C. Wychowski, K. Blight, J. Xu, Y. S. Hahn, C. M. Rice, and J. Dubuisson. 1997. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 71:697-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubuisson, J., H. H. Hsu, R. C. Cheung, H. B. Greenberg, D. G. Russell, and C. M. Rice. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubuisson, J., and C. M. Rice. 1996. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70:778-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ducarme, P., M. Rahman, and R. Brasseur. 1998. IMPALA: a simple restraint field to simulate the biological membrane in molecular structure studies. Proteins 30:357-371. [PubMed] [Google Scholar]
- 19.Duvet, S., L. Cocquerel, A. Pillez, R. Cacan, A. Verbert, D. Moradpour, C. Wychowski, and J. Dubuisson. 1998. Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J. Biol. Chem. 273:32088-32095. [DOI] [PubMed] [Google Scholar]
- 20.Flint, M., and J. A. McKeating. 1999. The C-terminal region of the hepatitis C virus E1 glycoprotein confers localization within the endoplasmic reticulum. J. Gen. Virol. 80:1943-1947. [DOI] [PubMed]
- 21.Gaboriaud, C., V. Bissery, T. Benchetrit, and J. P. Mornon. 1987. Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett. 224:149-155. [DOI] [PubMed] [Google Scholar]
- 22.Gomord, V., E. Wee, and L. Faye. 1999. Protein retention and localization in the endoplasmic reticulum and the golgi apparatus. Biochimie 81:607-618. [DOI] [PubMed] [Google Scholar]
- 23.Grakoui, A., C. Wychowski, C. Lin, S. M. Feinstone, and C. M. Rice. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hobman, T. C., H. F. Lemon, and K. Jewell. 1997. Characterization of an endoplasmic reticulum retention signal in the rubella virus E1 glycoprotein. J. Virol. 71:7670-7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honsho, M., J. Y. Mitoma, and A. Ito. 1998. Retention of cytochrome b5 in the endoplasmic reticulum is transmembrane and luminal domain-dependent. J. Biol. Chem. 273:20860-20866. [DOI] [PubMed] [Google Scholar]
- 26.Hussy, P., G. Schmid, J. Mous, and H. Jacobsen. 1996. Purification and in vitro-phospholabeling of secretory envelope proteins E1 and E2 of hepatitis C virus expressed in insect cells. Virus Res. 45:45-57. [DOI] [PubMed] [Google Scholar]
- 27.Jayaraman, G., T. K. Kumar, A. I. Arunkumar, and C. Yu. 1996. 2,2,2-Trifluoroethanol induces helical conformation in an all beta-sheet protein. Biochem. Biophys. Res. Commun. 222:33-37. [DOI] [PubMed] [Google Scholar]
- 28.Krantz, D. D., R. Zidovetzki, B. L. Kagan, and S. L. Zipursky. 1991. Amphipathic beta structure of a leucine-rich repeat peptide. J. Biol. Chem. 266:16801-16807. [PubMed] [Google Scholar]
- 29.Kuo, G., Q. L. Choo, H. J. Alter, G. L. Gitnick, A. G. Redeker, R. H. Purcell, T. Miyamura, J. L. Dienstag, M. J. Alter, and C. E. Stevens. 1989. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244:362-364. [DOI] [PubMed] [Google Scholar]
- 30.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 31.Lanford, R. E., L. Notvall, D. Chavez, R. White, G. Frenzel, C. Simonsen, and J. Kim. 1993. Analysis of hepatitis C virus capsid, E1, and E2/NS1 proteins expressed in insect cells. Virology 197:225-235. [DOI] [PubMed] [Google Scholar]
- 32.Lee, S., H. Yoshitomi, M. Morikawa, S. Ando, H. Takiguchi, T. Inoue, and G. Sugihara. 1995. Homooligopeptides composed of hydrophobic amino acid residues interact in a specific manner by taking alpha-helix or beta-structure toward lipid bilayers. Biopolymers 36:391-398. [DOI] [PubMed] [Google Scholar]
- 33.Letourneur, F., and P. Cosson. 1998. Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J. Biol. Chem. 273:33273-33278. [DOI] [PubMed] [Google Scholar]
- 34.Levine, Y. K., and M. H. Wilkins. 1971. Structure of oriented lipid bilayers. Nat. New Biol. 230:69-72. [DOI] [PubMed] [Google Scholar]
- 35.Lins, L., and R. Brasseur. 1995. The hydrophobic effect in protein folding. FASEB J. 9:535-540. [DOI] [PubMed] [Google Scholar]
- 36.Lins, L., R. Brasseur, M. De Pauw, J. P. Van Biervliet, J. M. Ruysschaert, M. Rosseneu, and B. Vanloo. 1995. Helix-helix interactions in reconstituted high-density lipoproteins. Biochim. Biophys. Acta 1258:10-18. [DOI] [PubMed] [Google Scholar]
- 37.Mancheno, J. M., M. Gasset, J. P. Albar, J. Lacadena, d. P. Martinez, M. Onaderra, and J. G. Gavilanes. 1995. Membrane interaction of a beta-structure-forming synthetic peptide comprising the 116-139th sequence region of the cytotoxic protein alpha-sarcin. Biophys. J. 68:2387-2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuura, Y., and T. Miyamura. 1993. The molecular biology of hepatitis C virus. Semin. Virol. 4:297-304. [Google Scholar]
- 39.Matsuura, Y., T. Suzuki, R. Suzuki, M. Sato, H. Aizaki, I. Saito, and T. Miyamura. 1994. Processing of E1 and E2 glycoproteins of hepatitis C virus expressed in mammalian and insect cells. Virology 205:141-150. [DOI] [PubMed] [Google Scholar]
- 40.Merkel, J. S., and L. Regan. 1998. Aromatic rescue of glycine in beta sheets. Fold. Des. 3:449-455. [DOI] [PubMed] [Google Scholar]
- 41.Michalak, J. P., C. Wychowski, A. Choukhi, J. C. Meunier, S. Ung, C. M. Rice, and J. Dubuisson. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 78:2299-2306. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima, H., M. Hijikata, S. Asabe, M. Hirota, K. Kimura, and K. Shimotohno. 1994. Two hepatitis C virus glycoprotein E2 products with different C termini. J. Virol. 68:6215-6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monne, M., I. Nilsson, A. Elofsson, and G. von Heijne. 1999. Turns in transmembrane helices: determination of the minimal length of a “helical hairpin” and derivation of a fine-grained turn propensity scale. J. Mol. Biol. 293:807-814. [DOI] [PubMed] [Google Scholar]
- 44.Nelder, J. A., and R. A. Mead. 1965. A simplex method for functino minimization. Comput. J. 7:308-313. [Google Scholar]
- 45.Op De Beeck, A., R. Montserret, S. Duvet, L. Cocquerel, R. Cacan, B. Barberot, M. Le Maire, F. Penin, and J. Dubuisson. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 275:31428-31437. [DOI] [PubMed]
- 46.Pedrazzini, E., A. Villa, and N. Borgese. 1996. A mutant cytochrome b5. with a lengthened membrane anchor escapes from the endoplasmic reticulum and reaches the plasma membrane. Proc. Natl. Acad. Sci. USA 93:4207-4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rahman, M., and R. Brasseur. 1994. WinMGM: a fast CPK molecular graphics program for analyzing molecular structure. J. Mol. Graph. 12:212-218, 198. [DOI] [PubMed] [Google Scholar]
- 48.Ralston, R., K. Thudium, K. Berger, C. Kuo, B. Gervase, J. Hall, M. Selby, G. Kuo, M. Houghton, and Q. L. Choo. 1993. Characterization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia viruses. J. Virol. 67:6753-6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rayner, J. C., and H. R. Pelham. 1997. Transmembrane domain-dependent sorting of proteins to the ER and plasma membrane in yeast. EMBO J. 16:1832-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reth, M., J. Hombach, J. Wienands, K. S. Campbell, N. Chien, L. B. Justement, and J. C. Cambier. 1991. The B-cell antigen receptor complex. Immunol. Today 12:196-201. [DOI] [PubMed] [Google Scholar]
- 51.Rice, C. M. 1996. Flaviridae: the viruses and their replication, p. 931-959. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
- 52.Sato, M., K. Sato, and A. Nakano. 1996. Endoplasmic reticulum localization of Sec12p is achieved by two mechanisms: Rer1p-dependent retrieval that requires the transmembrane domain and Rer1p-independent retention that involves the cytoplasmic domain. J. Cell Biol. 134:279-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selby, M. J., E. Glazer, F. Masiarz, and M. Houghton. 1994. Complex processing and protein:protein interactions in the E2:NS2 region of HCV. Virology 204:114-122. [DOI] [PubMed] [Google Scholar]
- 54.Shiraki, K., K. Nishikawa, and Y. Goto. 1995. Trifluoroethanol-induced stabilization of the alpha-helical structure of beta-lactoglobulin: implication for non-hierarchical protein folding. J. Mol. Biol. 245:180-194. [DOI] [PubMed] [Google Scholar]
- 55.Spaete, R. R., D. Alexander, M. E. Rugroden, Q. L. Choo, K. Berger, K. Crawford, C. Kuo, S. Leng, C. Lee, and R. Ralston. 1992. Characterization of the hepatitis C virus E2/NS1 gene product expressed in mammalian cells. Virology 188:819-830. [DOI] [PubMed] [Google Scholar]
- 56.Szczesna-Skorupa, E., K. Ahn, C. D. Chen, B. Doray, and B. Kemper. 1995. The cytoplasmic and N-terminal transmembrane domains of cytochrome P450 contain independent signals for retention in the endoplasmic reticulum. J. Biol. Chem. 270:24327-24333. [DOI] [PubMed] [Google Scholar]
- 57.Wallin, E., T. Tsukihara, S. Yoshikawa, G. von Heijne, and A. Elofsson. 1997. Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci. 6:808-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of X-ray and neutron diffraction data. II. Distribution and packing of terminal methyl groups. Biophys. J. 61:428-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wimley, W. C., K. Hristova, A. S. Ladokhin, L. Silvestro, P. H. Axelsen, and S. H. White. 1998. Folding of beta-sheet membrane proteins: a hydrophobic hexapeptide model. J. Mol. Biol. 277:1091-1110. [DOI] [PubMed] [Google Scholar]
- 60.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842-848. [DOI] [PubMed] [Google Scholar]
- 61.Wouters, M. A., and P. M. Curmi. 1995. An analysis of side chain interactions and pair correlations within antiparallel beta-sheets: the differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins 22:119-131. [DOI] [PubMed] [Google Scholar]
- 62.Yang, M., J. Ellenberg, J. S. Bonifacino, and A. M. Weissman. 1997. The transmembrane domain of a carboxyl-terminal anchored protein determines localization to the endoplasmic reticulum. J. Biol. Chem. 272:1970-1975. [DOI] [PubMed] [Google Scholar]