

Abstract
The leukocyte NADPH oxidase catalyzes the reduction of oxygen to O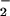 at the expense of NADPH. Extensive phosphorylation of the oxidase subunit p47PHOX occurs during the activation of the enzyme in intact cells. p47PHOX carrying certain serine-to-alanine mutations fails to support NADPH oxidase activity in intact cells, suggesting that the phosphorylation of specific serines on p47PHOX is required for the activation of the oxidase. Earlier studies with both intact cells and a kinase-dependent, cell-free system have suggested that protein kinase C can phosphorylate those serines of p47PHOX whose phosphorylation is necessary for its activity. Work with inhibitors suggested that a phosphatidylinositol 3-kinase-dependent pathway also can activate the oxidase. Phosphorylation of p47PHOX by Akt (protein kinase B), whose activation depends on phosphatidylinositol 3-kinase, could be the final step in such a pathway. We now find that Akt activates the oxidase in vitro by phosphorylating serines S304 and S328 of p47PHOX. These results suggest that Akt could participate in the activation of the leukocyte NADPH oxidase.
at the expense of NADPH. Extensive phosphorylation of the oxidase subunit p47PHOX occurs during the activation of the enzyme in intact cells. p47PHOX carrying certain serine-to-alanine mutations fails to support NADPH oxidase activity in intact cells, suggesting that the phosphorylation of specific serines on p47PHOX is required for the activation of the oxidase. Earlier studies with both intact cells and a kinase-dependent, cell-free system have suggested that protein kinase C can phosphorylate those serines of p47PHOX whose phosphorylation is necessary for its activity. Work with inhibitors suggested that a phosphatidylinositol 3-kinase-dependent pathway also can activate the oxidase. Phosphorylation of p47PHOX by Akt (protein kinase B), whose activation depends on phosphatidylinositol 3-kinase, could be the final step in such a pathway. We now find that Akt activates the oxidase in vitro by phosphorylating serines S304 and S328 of p47PHOX. These results suggest that Akt could participate in the activation of the leukocyte NADPH oxidase.
The antimicrobial activity of phagocytes depends in part on the cells' ability to reduce oxygen to reactive microbicidal oxidants by means of an NADPH oxidase. In resting phagocytes, the NADPH oxidase is in a dormant state, but exposure of the cell to any of a variety of stimuli can activate the enzyme, causing it to release large amounts of O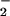 by reducing oxygen at the expense of NADPH. The oxidase is activated by the phosphorylation of one of its cytosolic subunits, p47PHOX, on particular serines (1). In whole cells, the stimulation of an appropriate receptor activates PKC and phosphatidylinositol 3-kinase (PI3-kinase) (2–4). Although NADPH oxidase activation by PKC has been well characterized (5, 6), the association between PI3-kinase and NADPH oxidase activation remains to be established. A possible connection is through the activation of Akt (7), whose role in oxidase activation is suggested by the finding that Akt is activated rapidly when neutrophils are treated with oxidase-activating agents (8) and by the finding that oxidase activation is inhibited by wortmannin (9). The experiments described here show that Akt is able to activate the oxidase by phosphorylating p47PHOX on serines S304 and S328.
by reducing oxygen at the expense of NADPH. The oxidase is activated by the phosphorylation of one of its cytosolic subunits, p47PHOX, on particular serines (1). In whole cells, the stimulation of an appropriate receptor activates PKC and phosphatidylinositol 3-kinase (PI3-kinase) (2–4). Although NADPH oxidase activation by PKC has been well characterized (5, 6), the association between PI3-kinase and NADPH oxidase activation remains to be established. A possible connection is through the activation of Akt (7), whose role in oxidase activation is suggested by the finding that Akt is activated rapidly when neutrophils are treated with oxidase-activating agents (8) and by the finding that oxidase activation is inhibited by wortmannin (9). The experiments described here show that Akt is able to activate the oxidase by phosphorylating p47PHOX on serines S304 and S328.
Experimental Procedures
Materials.
Active Akt was obtained from Upstate Biotechnology (Lake Placid, NY) or from J.-H.L., whose recombinant material was 95% pure by SDS/PAGE. [γ-32P]ATP was purchased from New England Nuclear. Phosphorylated peptides were obtained from Sigma.
Isolation and Fractionation of Neutrophils.
Neutrophils were obtained from normal subjects by dextran sedimentation and Ficoll/Hypaque fractionation of freshly drawn citrate-anticoagulated blood (10). After treatment on ice for 10–20 min with 5 μl of 0.54 M diisopropyl fluorophosphate, the neutrophils, suspended at 108 cells per ml in a modified relaxation buffer (0.1 M KCl/3 mM NaCl/3.5 mM MgCl2/10 mM Pipes buffer, pH 7.3), were subjected to nitrogen cavitation. Membranes and cytosol were separated by centrifugation through a Percoll gradient. Aliquots of membrane and cytosol were stored at −70°C until use.
Recombinant Oxidase Components.
Recombinant fusion proteins composed of an upstream GST linked to a downstream p47PHOX, p67PHOX, or Rac2 were isolated from Escherichia coli transformed with pGEX-6P3 plasmids containing cDNA inserts encoding the downstream proteins. The fusion proteins were isolated by the addition of a prewashed 50% slurry of glutathione-Sepharose 4B (Amersham Biosciences) in PBS (133 mM NaCl/16 mM Na2HPO4/2.7 mM KCl/1.5 mM KH2PO4, pH 7.3). The tube was rotated end-over-end for 1 h at 4°C and then microcentrifuged for 3 min at 200 × g to sediment the glutathione-Sepharose beads. The beads were washed four times with 10 bead volumes of PBS, and the bound fusion proteins were eluted by incubating for 30 min at 4°C with three 1-ml washes of 50 mM Tris⋅HCl, pH 8.0/20 mM glutathione/0.2 M NaCl. Excess glutathione was removed from the purified recombinant protein by dialysis against relaxation buffer and concentrated with 10K Biomax Centrifugal Filter devices (Millipore). The concentration of protein was determined with the Bio-Rad Assay Kit, by using BSA as the standard.
P47phox S(304)A, S(328)A, and S(304,328)A Mutants.
pGEX-6P3 p47PHOX WT vector was prepared by using the GeneEditor kit (Promega). The mutant constructs S304A, S328A, and S304A/S328A were generated following the manufacturer's instructions and using the following oligonucleotides: S304A, GGCACCGCCCCGCAGGTCGGCCATCCG; and S328A, CCTATCGCCGGAATGCCGTCCG. The sequence of all mutant constructs was confirmed by dideoxynucleotide-based sequencing.
In Vitro Phosphorylation of GST-p47PHOX.
Labeling with Akt was performed by incubating a reaction mixture of 50 μg of GST-p47PHOX, 1 mM ATP, 10 mM magnesium, and 0.1 μg of His-c-Akt (11) or 0.1 unit of Akt in relaxation buffer (pH 7.3) for 30 min at 30°C. The reaction was terminated by the addition of a prewashed 50% slurry of glutathione-Sepharose and isolated as described above. The eluted fusion protein was dialyzed against relaxation buffer before use. To determine the locations of the phosphates in p47PHOX phosphorylated by Akt, the p47PHOX was incubated as described above with the addition of 100 μCi (1 Ci = 37 GBq) of [γ-32P]ATP.
Cell-Free Activation of the NADPH Oxidase.
NADPH oxidase activity was measured directly by the one-electron reduction of cytochrome c. Assay mixtures containing 106 cell equivalents of membrane, 1 mM ATP, 50 μM guanosine 5′-[γ-thio]triphosphate, and 250 nM calyculin A were incubated at 30°C for 10 min.‖ They then were combined with 0.1 mM cytochrome c, 5.3 μg of Rac2-guanosine 5′-[γ-thio]triphosphate, 9.9 μg of p67PHOX, and 7.1 μg of p47PHOX (unphosphorylated or phosphorylated with Akt) in a volume of 250 μl. Reactions were started with 0.16 mM NADPH. Cytochrome c reduction followed at 550 nm at room temperature in a Spectra-Max 190 plate reader (Molecular Devices), reading against a reference containing the same components plus 100 units of superoxide dismutase. The reference was subtracted automatically from the experimental values.
For cell-free assays requiring the removal of ATP, the membranes, cytosol, and prephosphorylated p47PHOX were processed as described below and run in the assay as described previously, with the exception that ATP was not added. To remove the ATP from the membranes, 100 μl of membranes was layered over 2.8 ml of 20% sucrose, which, in turn, was layered over 0.2 ml of 50% sucrose in a 3.5-ml ultracentrifuge tube and spun at 100,000 × g for 30 min at 4°C. The sucrose solution above the floating membrane band was discarded and the floating band was saved. The cytosol and prephosphorylated p47PHOX were desalted by using a 3000 MWCO Microcon Centrifugal Filter Device (Millipore), and the protein concentration was recalculated by using the Bio-Rad Protein Assay.
Cytosol-containing, cell-free assays free of ATP contained 5 × 106 cell equivalents of membrane that had been incubated for 30 min at 30° with 50 μM guanosine 5′-[γ-thio]triphosphate, 1 μg BSA, and 250 nM calyculin, but no ATP. The incubated membranes then were combined with 107 cell equivalents of cytosol, 0.1 mM cytochrome c, and 3.0 μg of p47PHOX, unphosphorylated or phosphorylated with Akt. For sets of cell-free assays requiring the elimination of ATP and the substitution of cytosol for the three cytosolic components, 3 μg of p47PHOX (prephosphorylated or unphosphorylated), 6 μg of p67PHOX, and 2 μg of Rac2 were incubated with ATP-free membranes, 50 mM guanosine 5′-[γ-thio]triphosphate, and 250 nM calyculin for 10 min at 30°C and then added to relaxation buffer. Reactions were started with 0.16 mM NADPH and assayed as described above.
Tryptic Phosphopeptide Analysis.
Labeled GST-p47PHOX was detected by autoradiography. The band corresponding to GST-p47PHOX was cleaved chemically with 50 mg/ml CNBr in 70% formic acid for 90 min at room temperature or digested three times with 10 μg of trypsin in 50 mM ammonium bicarbonate (pH 8) for 2 h at 37°C each. The resulting peptides were separated by SDS/PAGE or HPLC. For HPLC, the digest was loaded onto a 25-cm analytical C-18 reverse-phase column equilibrated with 0.6% trifluoroacetic acid in water. The column was eluted at 1.5 ml/min with a linear water–acetonitrile gradient, beginning with 5% acetonitrile/aqueous trifluoroacetic acid (0.06%) and ending 60 min later with 95% acetonitrile/aqueous trifluoroacetic acid (0.054%). Fractions of 0.375 ml were collected, and 32P-labeled phosphopeptides were detected by Čerenkov counting. Where indicated, phosphopeptides were separated by high-voltage electrophoresis and chromatography on a cellulose thin-layer plate as described by van der Geer and colleagues (12) and were detected by exposing the thin-layer plates to film at −70°C with an intensifying screen. Phosphopeptides were identified by coelution on reverse-phase liquid chromatography or by colocalization on phosphopeptide maps, using phosphorylated peptide standards purchased from Sigma.
Modification of the Phosphorylation Sites on p47PHOX During the Cell-Free Assay.
For these experiments, modified cell-free assays using 32P-labeled GST-p47PHOX were performed, and aliquots taken from the assays were run on a 10% Bis-Tris gel (NOVEX, San Diego) and exposed to film overnight. In this experiment, the cell-free assay was scaled down to 1/5 of the total volume while preserving the original concentrations, with the exception of the addition of 5 μg of p47PHOX and the addition of 1 μCi of [γ-32P]ATP to the assay before activation with NADPH as noted in Fig. 4. The assays were run for 5 min, followed by the addition of 5× sample buffer to a 20-μl (1 μg of p47PHOX) sample taken from the assay, and heated immediately at 70°C for 10 min before loading onto the gel.
Figure 4.

O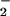 production by cell-free systems containing p47PHOX mutants phosphorylated with no kinase or with Akt, as indicated. “No Kinase” or “WT-AKT” refers to incubations when using WT p47PHOX. “304-AKT,” “328-AKT,” and “304/328-AKT” refer to incubations when using p47PHOX (S304A), p47PHOX (S328A), and p47PHOX (S304A, S328A), respectively. Incubations were carried out as described in the text. Results are the averages of four separate experiments, where values are expressed as mean ± SE.
production by cell-free systems containing p47PHOX mutants phosphorylated with no kinase or with Akt, as indicated. “No Kinase” or “WT-AKT” refers to incubations when using WT p47PHOX. “304-AKT,” “328-AKT,” and “304/328-AKT” refer to incubations when using p47PHOX (S304A), p47PHOX (S328A), and p47PHOX (S304A, S328A), respectively. Incubations were carried out as described in the text. Results are the averages of four separate experiments, where values are expressed as mean ± SE.
In a separate experiment, 5 μg of 32P-labeled GST-p47PHOX and 5 μg of unphosphorylated p47PHOX that were cut off the glutathione-Sepharose beads by using Prescission Protease (Amersham Biosciences) were added to a similar, scaled-down, cell-free assay and incubated for 5 min. Samples were run on a gel as described above. Both gels were stained, dried down, and exposed to autoradiography film at 70°C overnight (Fig. 6).
Figure 6.

Modification of the phosphorylation sites on p47PHOX is shown as both Coomassie stain (Left) and autoradiogram (Right).
Results
To determine whether Akt-directed phosphorylation of p47PHOX could be partly responsible for the observed increase in NADPH oxidase activity associated with PI3-kinase activation, we first examined whether Akt was able to phosphorylate p47PHOX at all. Previous studies using PKC showed that the phosphorylation of serines in the C-terminal quarter of p47PHOX was an important determinant of the activity of the NADPH oxidase (13–15). To ascertain whether a similar situation occurred with Akt, we phosphorylated p47PHOX with a constitutively active mutant of Akt and analyzed the CNBr cleavage products of the phosphorylated protein by SDS/PAGE. The results (Fig. 1) showed that, like PKC (lane 2), Akt (lane 1) phosphorylates hydroxyamino acids in the C-terminal quarter of p47PHOX. Two-dimensional phosphopeptide mapping showed that, unlike PKC, which targets multiple serines in the C-terminal portion of p47PHOX (17, 18), Akt-directed phosphorylation was limited chiefly to two tryptic peptides (Fig. 2). The specific serines phosphorylated by Akt initially were identified by a comparison with earlier peptide maps (5) and by the substrate consensus sequence BXXS/T, where B is a basic amino acid and X represents any amino acid. These considerations initially directed us to look at the two tryptic phosphopeptides containing S328 and S303/S304. For these experiments, p47PHOX first was labeled with [γ-32P]ATP by Akt. The tryptic peptides then were isolated by reverse-phase HPLC and mapped separately to identify the eluted peptides with the spots on the map shown in Fig. 3 (where peptide 1 corresponded to the lower spot and peptide 2 corresponded to the upper spot). Results obtained by using reverse-phase HPLC and tryptic phosphopeptide mapping with synthetic tryptic phosphopeptide standards for serine S328 (RNpSVR)** and serines S303/S304 (RpSpSIR, pSSIR, RSpSIR) showed that peptide 1 coeluted with the synthetic tryptic peptide containing phosphoserine S328 (Fig. 3A) and peptide 2 coeluted with the synthetic tryptic peptide containing phosphoserine S304 (Fig. 3B). In a second experiment, mixtures of radiolabeled peptide and the corresponding synthetic peptide were subjected to tryptic peptide mapping as described above. The ninhydrin-visualized spots corresponding to the peptides RSpSIR (containing serine S304) and RNpSVR (containing serine S328) exactly coincided with the autoradiographically visualized spots corresponding to the eluted radioactive peptides 1 and 2, respectively (data not shown). This confirmed S304 and S328 of p47PHOX as targets for phosphorylation by Akt. Further confirmation of the identities of the peptides was obtained by mapping the S304 and S328 mutants of phosphorylated p47PHOX. These results again established that peptide 1 was S328 and peptide 2 was S304 (data not shown).
Figure 1.

Phosphopeptides produced by CNBr cleavage of p47PHOX phosphorylated with Akt. SDS/PAGE of the labeled phosphopeptide obtained after CNBr cleavage of p47PHOX phosphorylated with Akt (lane 1) or PKC (lane 2, for comparison). The identical mobilities of the phosphopeptides indicate that the C-terminal region of the protein (residues 279–390) is phosphorylated (13, 14, 16).
Figure 2.

Detection of p47PHOX tryptic peptides phosphorylated by Akt. One-tenth of a unit of Akt was used to phosphorylate 100 μg of p47PHOX with 2 μCi [32P]ATP. The phosphorylated p47PHOX was digested with trypsin as described (5). In some experiments, the phosphopeptides initially were purified by reverse-phase HPLC and then comapped with authentic phosphopeptide standards. For purposes of identification, the lower spot is peptide 1 (per HPLC elution) and the upper spot is peptide 2.
Figure 3.

Identification of tryptic peptides phosphorylated by Akt. 32P-labeled p47PHOX tryptic phosphopeptides were separated by HPLC over a C-18 reverse-phase column and were detected by Čerenkov counting. The peptides were identified as peptide 1 and peptide 2 by two-dimensional phosphopeptide mapping. For identification purposes, each peptide was combined with 10 μg of the phosphorylated standard peptides for pS328, pS303, pS304, and pS303/pS304 (see text). The mobilities of the pS303, pS304, and pS303/pS304 peptides were all different. (A) Peptide 1 coeluted with the standard for pS328 (RNpSVR). (B) Peptide 2 coeluted with the standard for p304 (RSpSIR). For confirmation, each identified standard was mixed with its corresponding labeled peptide, spotted onto a cellulose plate, and subjected to two-dimensional peptide mapping. In both cases, the standard (ninhydrin staining) and the labeled (autoradiography) peptide were superimposable.
To investigate whether p47PHOX phosphorylated by Akt was capable of supporting NADPH oxidase activity by using a cell-free system, we examined O production by the cytochrome c assay, using recombinant p47PHOX phosphorylated by Akt. The results indicated that Akt-phosphorylated WT p47PHOX showed significant activity. On the other hand, the activity of Akt-phosphorylated p47PHOX (S304A) and p47PHOX (S328A) was negligible (Fig. 4). These findings confirm S304 and S328 as the functional targets of Akt in the phosphorylation of p47PHOX.
production by the cytochrome c assay, using recombinant p47PHOX phosphorylated by Akt. The results indicated that Akt-phosphorylated WT p47PHOX showed significant activity. On the other hand, the activity of Akt-phosphorylated p47PHOX (S304A) and p47PHOX (S328A) was negligible (Fig. 4). These findings confirm S304 and S328 as the functional targets of Akt in the phosphorylation of p47PHOX.
Two potential problems arise. The first is the possibility that the phosphorylation is due to kinases brought into the assay mixtures by the unfractionated cytosol and/or membranes used in these experiments. The second, less likely, is that sites of phosphorylation may change during the incubation. To rule out the former possibility, oxidase activity was measured after removal of ATP from the reagents. This was accomplished by using desalted cytosol, membranes, and phosphorylated p47PHOX. The results (Fig. 5A) indicate that phosphorylation is accomplished by Akt, not by kinases fortuitously included in the assay mixtures. In a set of similar assays, recombinant p47PHOX, p67PHOX, and Rac2 were used instead of cytosol, resulting in similar activity levels (Fig. 5B). Finally, 5 μg of 32P-labeled GST-p47PHOX and 5 μg of unphosphorylated Prescission Protease-cut p47PHOX were added to a  -scale cell-free assay and incubated for 5 min. A 20-μl aliquot then was taken, added to 5 μl of 5× sample buffer, and heated to 70°C for 10 min. The sample then was run on a 10% Bis-Tris gel, stained, dried, and exposed to film overnight. Samples of 1 μg of 32P-labeled GST-p47PHOX and 1 μg of Prescission Protease-cut p47PHOX were run as markers. No transfer of phosphates from the 32P-labeled GST-p47PHOX to the Prescission Protease-cut p47PHOX occurred (Fig. 6).
-scale cell-free assay and incubated for 5 min. A 20-μl aliquot then was taken, added to 5 μl of 5× sample buffer, and heated to 70°C for 10 min. The sample then was run on a 10% Bis-Tris gel, stained, dried, and exposed to film overnight. Samples of 1 μg of 32P-labeled GST-p47PHOX and 1 μg of Prescission Protease-cut p47PHOX were run as markers. No transfer of phosphates from the 32P-labeled GST-p47PHOX to the Prescission Protease-cut p47PHOX occurred (Fig. 6).
Figure 5.

(A) O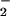 production by cytosol and membranes from which low-molecular-mass compounds (including ATP) have been removed, with phosphorylated p47PHOX from which excess ATP also has been removed. Values are expressed as mean ± 1 SE. (B) O
production by cytosol and membranes from which low-molecular-mass compounds (including ATP) have been removed, with phosphorylated p47PHOX from which excess ATP also has been removed. Values are expressed as mean ± 1 SE. (B) O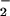 production by an assay mixture containing recombinant cytosolic components in an ATP-free, cell-free system. The assay was conducted as described in the text. Data are presented as mean ± 1 SE.
production by an assay mixture containing recombinant cytosolic components in an ATP-free, cell-free system. The assay was conducted as described in the text. Data are presented as mean ± 1 SE.
Discussion
We show here that Akt phosphorylates p47PHOX on S304 and S328 and that this phosphorylation supports NADPH oxidase activity. We previously have explored the manipulation of several serines identified as targets for phosphorylation during NADPH oxidase activation (19, 20), and, with the current findings, it will be important to explore the functional role of S304 and S328 further.
Another example of the importance of site-directed phosphorylation in the C terminus of p47PHOX is the recent observation characterizing the priming effect of granulocyte/macrophage colony-stimulating factor on whole neutrophils (21), where phosphorylation of a single tryptic peptide (not identified) does not act to support oxidase activation but greatly enhances the phosphorylation of p47PHOX and NADPH oxidase activity after stimulation with N-formyl-methionyl-leucyl-phenylalanine. Given that the activation of PI3-kinase results in Akt activation and NADPH oxidase activation, our demonstration here that NADPH oxidase is activated by the Akt-mediated phosphorylation of p47PHOX may explain these results.
In an earlier study in which phorbol myristate acetate, an activator of PKC, was used to stimulate the neutrophils, we found that phosphorylation of one of the pairs (S303, S304) together with another of the pairs (S359, S370) was necessary for oxidase activation (19, 20). PKC did not phosphorylate S328. In contrast, Akt phosphorylates S304 and S328. We believe that the phosphorylation of S328 by Akt but not by PKC explains why phosphorylation of S359 or S370 is not required for oxidase activation by Akt.
Acknowledgments
We thank Dr. U. Knaus for insightful discussions. C.R.H. is the recipient of a postdoctoral fellowship from the Arthritis Foundation. This work was supported in part by U.S. Public Health Service Grants AI-24227, AI-28479, and RR-00833.
Abbreviation
- PI3-kinase
phosphatidylinositol 3-kinase
Footnotes
We observed some differences among patients in the ability of their membrane fractions to sustain activity after incubation at 30°C. For an alternate activation method, see ref. 1.
In peptides with the sequence RXpS, the R–X bond is resistant to tryptic hydrolysis. The three phosphorylated peptides used to analyze serines S303/S304 migrated separately on HPLC.
References
- 1.Park J W, Hoyal C R, El Benna J, Babior B M. J Biol Chem. 1997;272:11035–11043. doi: 10.1074/jbc.272.17.11035. [DOI] [PubMed] [Google Scholar]
- 2.Ding J, Vlahous R, Liu R, Brown R F, Badwey J A. J Biol Chem. 1995;270:11684–11691. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- 3.Heyworth P G, Matter W F, Brown R F, Traynor-Kaplan A E, Prossnitz E R, Ye R D, Marder P, Schelm J A, Rothfuss K J, Serlin B S, et al. J Immunol. 1999;154:2413–2422. [PubMed] [Google Scholar]
- 4.Didichenko S A, Tilton B, Hemmings B A, Ballmer-Hofer K, Thelen M. Curr Biol. 1996;6:1271–1278. doi: 10.1016/s0960-9822(02)70713-6. [DOI] [PubMed] [Google Scholar]
- 5.El Benna J, Faust L P, Johnson J L, Babior B M. J Biol Chem. 1996;271:6374–6378. doi: 10.1074/jbc.271.11.6374. [DOI] [PubMed] [Google Scholar]
- 6.Nauseef W M, Volpp B D, McCormick F, Leidal S, Clark R A. J Biol Chem. 1991;266:5911–5917. [PubMed] [Google Scholar]
- 7.Coffer P J, Jin J, Woodgett J R. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilton B, Andjelkovic M, Didichenko S A, Hemmings B A, Thelen M. J Biol Chem. 1997;272:28096–28101. doi: 10.1074/jbc.272.44.28096. [DOI] [PubMed] [Google Scholar]
- 9.Dewald B, Thelen M, Baggiolini M. J Biol Chem. 1988;263:16179–16184. [PubMed] [Google Scholar]
- 10.Borregaard N, Heiple J M, Simmons E R, Clark R A. J Cell Biol. 1983;97:52–61. doi: 10.1083/jcb.97.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta K, Bellacosa A, Chan T O, Tsichlis P. J Biol Chem. 1999;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- 12.Boyle W J, van der Geer P, Hunter T. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 13.Dusi S, Bianca V D, Grzeskowiak M, Rossi F. Biochem J. 1993;290:173–178. doi: 10.1042/bj2900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy R, Dana R, Leto T L, Malech H L. Biochim Biophys Acta. 1994;1220:253–260. doi: 10.1016/0167-4889(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 15.Rotrosen D, Leto T L. J Biol Chem. 1990;265:19910–19915. [PubMed] [Google Scholar]
- 16.Laemmli U K. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.El Benna J, Faust L P, Babior B M. J Biol Chem. 1994;269:23431–23436. [PubMed] [Google Scholar]
- 18.Faust L P, El Benna J, Babior B M, Chanock S J. J Clin Invest. 1995;96:1499–1505. doi: 10.1172/JCI118187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson J L, Park J W, El Benna J, Faust L P, Inanami O, Babior B M. J Biol Chem. 1998;273:35147–35152. doi: 10.1074/jbc.273.52.35147. [DOI] [PubMed] [Google Scholar]
- 20.Inanami O, Johnson J L, McAdara J K, El Benna J, Faust L P, Newburger P E, Babior B M. J Biol Chem. 1998;273:9539–9543. doi: 10.1074/jbc.273.16.9539. [DOI] [PubMed] [Google Scholar]
- 21.Dang P M, Dewas C, Gaudry M, Fay M, Pedruzzi E, Gougerot-Pocidalo M-A, El Benna J. J Biol Chem. 1999;274:20704–20708. doi: 10.1074/jbc.274.29.20704. [DOI] [PubMed] [Google Scholar]