

Abstract
Quinolinic acid may be an important endogenous excitotoxin, but its concentrations in brain are low. We have therefore attempted to determine whether its neurotoxicity can be increased by the simultaneous presence of free radicals.
Quinolinic acid was injected into the hippocampus of anaesthetized rats at doses of 40 and 80 nmols which produced little neuronal loss, and 120 nmols which produced over 90% neuronal loss.
A mixture of xanthine and xanthine oxidase, a known source of free radical reactive oxygen species, also generated little damage alone, but killed over 80% of CA1 neurons when combined with 80 nmols of quinolinic acid. Similarly, the nitric oxide donor S-nitroso-N-acetylpenicillamine (SNAP) potentiated the damage produced by quinolinic acid.
The glutamate antagonist 5,7-dichlorokynurenic acid prevented the damage produced by 120 nmols of quinolinic acid, but not that produced by quinolinic acid plus xanthine/xanthine oxidase, indicating that damage was not simply the result of free radical enhancement of NMDA receptor activation.
Three chemically dissimilar antagonists at adenosine A2A receptors prevented the damage caused by quinolinic acid and xanthine/xanthine oxidase or by quinolinic acid plus SNAP.
It is concluded that reactive oxygen species can potentiate the neurotoxicity of quinolinic acid. The site of interaction is probably distal to the NMDA receptor. Blockade of adenosine A2A receptors can protect against this combined damage, suggesting potential value in the prevention of brain damage.
Keywords: Kynurenines, kynurenic acid, quinolinic acid, excitotoxicity, free radicals, reactive oxygen, hippocampus, neurodegeneration, neuroprotection
Introduction
The activation of several subtypes of glutamate receptor can produce damage to neurones in the central nervous system (CNS). As a result, it has been suggested that a rise in the level of an endogenous excitotoxin could contribute to the initiation or development of the neuronal damage which occurs in degenerative disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease or AIDS-dementia (Foster & Schwarcz, 1989; Kerr et al., 1998; Stone, 2001). One of the best studied compounds which could act as an endogenous excitotoxin at the N-methyl-D-aspartate (NMDA) receptors is quinolinic acid (Stone & Perkins, 1981; Schwarcz et al., 1983; 1984; Stone, 1993, 2001), a tryptophan metabolite produced by metabolism along the kynurenine pathway from tryptophan to nicotinic acid. Quinolinic acid has a potency comparable with that of NMDA in producing neurotoxicity but is relatively weak when producing neuronal excitation or displacement of glutamate receptor ligands in binding studies (Schwarcz et al., 1983; Perkins & Stone, 1983; Foster & Schwarcz, 1989; Stone, 1993). Furthermore, the concentrations of quinolinic acid in the CNS are normally substantially lower than those which can directly cause excitotoxicity (Moroni et al., 1984; Heyes et al., 1989). For these reasons, doubt has been expressed as to whether quinolinic acid could indeed contribute to any recognized CNS neuropathology.
There are, however, several reasons to believe that it can. Firstly, the concentration of quinolinic acid in the brain can increase several hundred-fold in response to pathological conditions and infection by agents such as HIV-1 (Heyes et al., 1989, 1991). Secondly, it has been demonstrated that even low micromolar concentrations of quinolinic acid can produce damage in cell cultures or in vivo, especially if those levels are maintained for several weeks (Khaspekov et al., 1989; Whetsell & Schwarcz, 1989; Galarraga et al., 1990; Kerr et al., 1998). Thirdly, neurones in some parts of the CNS are known to be exquisitely sensitive to injury by quinolinic acid, with damage produced at concentrations as low as 100 nM (Giulian et al., 1990, 1993).
In addition, it is possible that the excitotoxic damage produced by quinolinic acid, even at low concentrations, could be potentiated by other endogenous and injurious molecules. Quinolinic acid in the brain is produced mainly by the increased kynurenine pathway activity which is induced in microglia and invading macrophages by immune activation (Heyes et al., 1992a, 1996; Espey et al., 1997). These cells, when activated, also generate increased quantities of free radicals such as reactive oxygen species and we have therefore considered the possibility that free radicals generated by activated immune cells could interact with quinolinic acid to produce enhanced damage.
Methods
Intrahippocampal injections
Male Wistar rats weighing between 200 and 250 g were used. All animals were housed singly and provided with free access to food and water. Animals were anaesthetised with chloral hydrate (400 mg kg−1, intraperitoneally) and placed in a stereotaxic frame. The scalp was incised and a burr hole made through the skull to permit access of the injection needle into the hippocampus at the desired co-ordinates (anteroposterior: 3.0 mm behind the bregma suture, dorsoventral: 2.8 mm below the cortical surface and lateral: 3.0 mm from the midline suture; Paxinos & Watson, 1986). The needle was then lowered to these co-ordinates and left in place for 2 min before the injection of test agents. The compounds used in this study were all introduced through a 29-gauge needle, injections being made in a volume of between 1 and 2 μl at a constant rate of 0.3 ml min−1 using a Sage infusion pump (Jones et al., 1998a, 1998b). The injection needle was allowed to remain in place for 2 min after ending the injection so as to prevent leakage of drug along the needle track. The scalp was then sutured and the animals left to recover for 7 days.
Quinolinic acid was injected at doses of 40, 80 and 120 nmols. A mixture of xanthine (100 μM) and xanthine oxidase (0.1 U ml−1) was co-administered by mixing solutions of the individual agents with the stock quinolinic acid solution in the appropriate proportions. The dose of xanthine and xanthine oxidase was limited by the solubility of the compounds, so that higher dosage was not practicable. Quinolinic acid was dissolved in 1 N NaOH and then diluted with 165 mM NaCl solution. The pH of the solution was then adjusted using 1 N HCl to between 7 and 7.6 before making up to volume by the addition of further saline. S-nitroso-N-acetylpenicillamine (SNAP) was injected at doses of 100 and 500 nmols.
Tissue fixing and slicing
Seven days after recovery from the intrahippocampal injections, rats were killed by an overdose of sodium pentobarbitone. The chest was opened to expose the heart and 10 ml of 0.9% sodium chloride solution was injected via a 26-gauge needle inserted into the left cardiac ventricle to wash blood from the cerebral vessels. This was immediately followed by 4% formaldehyde in phosphate buffered saline. The brain was then removed and stored in fixative for up to 1 week. A coronal slice of brain, approximately 3 mm thick, was prepared to include the location of the injection track, which was normally apparent from the residual dimpling of the cortical surface produced by the needle penetration. The block of brain was dehydrated and impregnated with paraffin wax throughout before embedding in wax. Sections were cut 6 μm thick, mounted on slides and stained with cresyl fast violet.
Sections were subsequently examined under a light microscope and areas CA1 and CA3 examined for damage. The damage was quantified in the CA1 region by selecting three sections approximately 2000 – 2500 μm from the site of the needle track and taking the average number of intact, surviving neurones at a magnification of 100×. A comparable count was made of neurons in the contralateral, unaffected side of the hippocampus, and the number of intact cells on the damaged side (a mean of the three sections counted) was then expressed as a percentage of the control side. As an indication of the number of cells per field counted for analysis, the number counted in a series of 10 control brains was 282×14. In all cases, the damaged and control sides were examined in the same coronal sections. Four animals were used for each data point, except for the preliminary data with quinolinic acid, where n=3. In a separate preliminary series of five brains, it was confirmed that there was no significant difference between the number of neurons in a CA1 field of view from an animal which had received no treatment or surgery, and a field from the contralateral side of a test animal.
Data analysis
The results are presented as mean±s.e.mean. Analysis of variance (ANOVA) followed by the Bonferroni post-test for multiple comparisons was used to determine any statistical significance. Significance refers to results where P<0.05 was obtained.
Drugs
All compounds were obtained from Sigma Chemicals, with the exception of 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3e]-1,2,4-triazolo[1,5c]pyrimidine (SCH58261) and 4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}(1,3,5}triazin-5-yl-amino]ethyl)phenol (ZM241385), which were partly obtained as gifts from Dr E. Ongini, Schering-Plough Research Institute, and Dr S. Poucher, Zeneca Pharmaceuticals respectively. The latter compound was also purchased from Tocris Chemicals. 8-(3-chlorostyryl)caffeine (CSC) was obtained from Research Biochemicals, Inc. All compounds were dissolved initially in dimethyl-sulphoxide and subsequently diluted to the required concentration for injection. The final concentration of dimethyl-sulphoxide was never more than 0.1%, which in earlier pilot experiments did not influence neuronal viability.
Results
Effects of quinolinic acid
Normal intact pyramidal neurones had a clearly rounded appearance with a clear nucleus and nucleolus (Figure 1). Quinolinic acid produced a dose-dependent effect in the CA1 and CA3 regions of the hippocampus with no obvious or significant damage being seen at the lower doses of 40 nmols (4.3% cell loss±2.4, n=3) and 80 nmols (14.1% loss±3.3, n=3) but a substantial and usually almost total destruction at the highest dose (91.1% loss±6.5, n=3). This relationship was consistent with our previous studies (Behan et al., 1999; Behan & Stone, 2000), in which a dose of 120 nmols was found to produce a substantial degree of damage against which neuroprotective agents could be assessed. The border between damaged areas of CA1 and adjacent undamaged areas was sharply defined, as noted by Foster & Schwarcz (1989). There was also an infiltration of the pyramidal cell layers and surrounding tissue by microglial cells after the higher dose. Of the damaged areas, the CA1 area was selected for the quantification of the damage and protection as described in Methods, and the intermediate 80 nmols dose of quinolinic acid was selected for some of the later combination studies with potentially neuroprotective agents.
Figure 1.

Photomicrographs of the CA1 area of the rat hippocampus. (A) Control section, showing the CA1 neurons as clear, rounded cells in a well-organized layer. (B) Following the administration of a combination of quinolinic acid 80 nmols with xanthine/xanthine oxidase (100 μM with 0.1 U ml−1), the CA1 layer is largely devoid of neurons, leaving a disorganized layer with a few neurons remaining intact (arrows). (C) The co-administration of ZM241385 together with quinolinic acid and the xanthine/xanthine oxidase mixture protects against the damage, leaving a largely intact cell layer in this example. Scale bar:-100 μM.
Effects of free radical generators
At the dose employed in this study, which was largely determined by the limited solubility of the compounds used, the mixture of xanthine and xanthine oxidase produced no obvious neuronal damage when injected alone (Figure 2). Similarly, the nitric oxide donor S-nitroso-N-acetylpenicillamine (SNAP) produced no significant neuronal loss when administered alone into the hippocampus at either of the doses tested (100 and 500 nmols). The higher dose was therefore selected for further experiments (Figure 3).
Figure 2.

Histogram illustrating the extent of neuronal damage induced by intrahippocampal injections of quinolinic acid at doses of 40 or 80 nmols (QN40, QN80) or a mixture of xanthine 100 μM with xanthine oxidase (0.1 U ml−1) (X/XO). When administered alone, neither injection induced a significant degree of cell loss, but when administered simultaneously, a highly significant loss of neurons was observed (QN+X/XO). The columns show the mean±s.e.mean (n=4) of the percentage of neurons surviving in the CA1 region of the counted sections. ***P<0.001.
Figure 3.
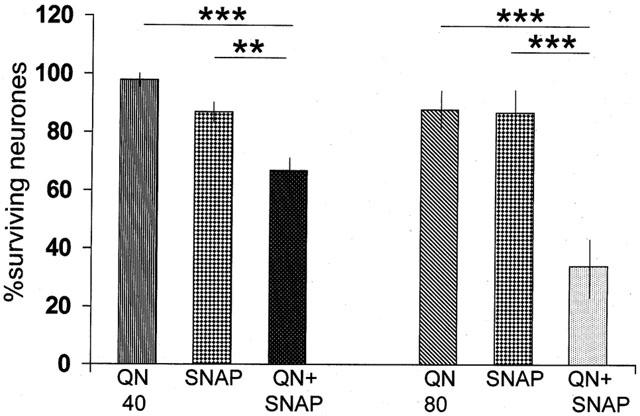
Histogram illustrating the extent of neuronal damage induced by intrahippocampal injections of quinolinic acid at doses of 40 or 80 nmols (QN40, QN80) or SNAP 500 nmols. When administered alone, neither injection induced a significant degree of cell loss, but when administered simultaneously, a highly significant loss of neurons was observed. The columns show the mean±s.e.mean (n=4) of the percentage of neurons surviving in the CA1 region of the counted sections. **P<0.01; ***P<0.001.
Combined administration
The two lower doses of quinolinate were tested in combination with the xanthine/xanthine oxidase mixture. With the lowest dose damage was apparent as a loss of healthy cells in the CA1 area when the two agents were administered simultaneously, with a proportion of cells showing general shrinkage, with enlarged and disrupted but darkly-staining nuclei or nuclear remnants (Figure 1). The damage induced by the combinations of xanthine/xanthine oxidase with either dose of quinolinic acid was very significantly greater than that produced by either agent alone (Figure 2), the combination based on quinolinic acid at 40 nmols producing a combined loss of 35% of neurons, and the combination involving 80 nmols of quinolinic acid yielding a loss of 81% of CA1 neurones.
The combined administration of quinolinate and SNAP (500 nmols) also produced significant damage with either the 40 or 80 nmol doses of the excitotoxin (Figure 3), the combination with the higher dose producing a loss of 66% of neurons in the CA1 region.
Effect of 5,7-dichlorokynurenic acid
The substantial degree of neuronal loss resulting from the administration of quinolinic acid at the high dose of 120 nmols is indicated in Figure 4. In this series of animals, only 3.5% of neurons remained intact after 7 days. 5,7-dichlorokynurenic acid was administered at a dose of 1 nmol, which had been found in preliminary experiments to block the damage produced by 120 nmols of quinolinic acid. This activity was confirmed in the present series, in which 5,7-dichlorokynurenate produced a highly significant protection when combined with 120 nmols of quinolinic acid compared with the administration of quinolinic acid alone (Figure 4).
Figure 4.
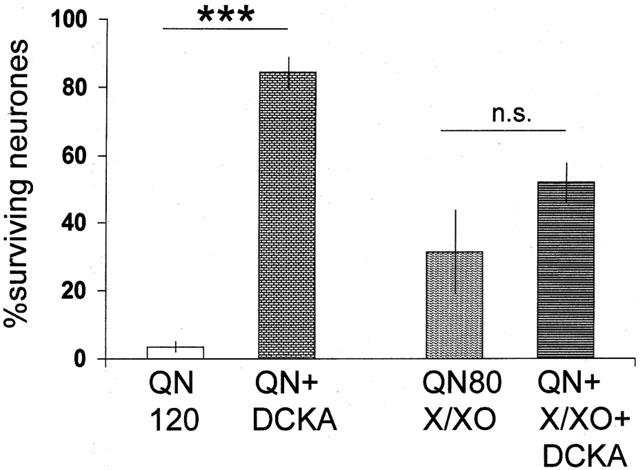
Histogram illustrating the extent of neuronal damage induced by intrahippocampal injections of quinolinic acid at a dose of 120 nmols (QN120) or a mixture of quinolinic acid 80 nmols with xanthine 100 μM and xanthine oxidase (0.1 U ml−1). Both these injections induced a highly significant degree of cell loss. When administered simultaneously, 5,7-dichlorokynurenic acid, 1 nmol (DCKA) was able to block completely the damage produced by quinolinic acid (QN+DCKA) but not that produced by the combination of quinolinic acid and the free radical-generating mixture (QN+X/XO+DCKA). The columns show the mean±s.e.mean (n=4) of the percentage of neurons surviving in the CA1 region of the counted sections. ***P<0.001.
A combination of 80 nmols of quinolinic acid with the xanthine/xanthine oxidase free-radical generating mixture generated a loss of 69% of the CA1 neurons, a value not significantly different from that seen in the experimental group illustrated in Figure 2. Against this combination, however, 5,7-dichlorokynurenate was not able to afford a significant degree of protection, yielding only a tendency towards protection and a neurone count of 52% of that in control sections (Figure 4).
Protection by purines
Using the combination of 80 nmols quinolinic acid with xanthine/xanthine oxidase, the potentially protective effects of antagonists at adenosine receptors were also examined. Administration of the A2A receptor antagonist ZM241385 at a dose of 2.5 pmols, which we have previously reported to protect against damage induced by kainic acid (Jones et al., 1998a, 1998b) produced a small change in the proportion of surviving neurons from 27%±5.4 to 40%±7.7 (n=3) which was not statistically significant. At a higher dose of 25 pmols, significant protection was seen compared with the control quinolinic acid and xanthine/xanthine oxidase mixture (Figure 5). Similarly, another non-xanthine compound SCH58261 had little effect at a dose of 5 pmols, increasing the proportion of surviving neurons from 22%±8.1 to 38%±6.4, whereas a higher dose of 50 pmols did produce a significant protection as illustrated in Figure 5. The xanthine-derived A2A receptor antagonist chlorostyrylcaffeine (CSC) also protected against the damage produced by the toxic combination, at a dose of 100 pmols (Figure 5).
Figure 5.

Histogram illustrating the extent of neuronal damage induced by intrahippocampal injections of quinolinic acid 80 nmols in combination with a mixture of xanthine 100 μM with xanthine oxidase (0.1 U ml−1). A highly significant degree of neuronal loss is produced by this combination (QN+X/XO), which is prevented by the simultaneous administration of chlorostyrylcaffeine, 100 pmols (CSC), ZM241385, 25 pmols (ZM) or SCH58261, 50 pmols (SCH). The columns show the mean±s.e.mean (n=4) of the percentage of neurons surviving in the CA1 region of the counted sections. *P<0.05; **P<0.01.
Exactly comparable results were obtained when the adenosine receptor ligands were examined against the combination of quinolinic acid and SNAP (Figure 6). Chlorostyrylcaffeine and the higher doses of ZM241385 and SCH58261 were able to protect against cell loss in the CA1 region induced by the administration of the 500 nmols dose of SNAP (Figure 6).
Figure 6.

Histogram illustrating the extent of neuronal damage induced by intrahippocampal injections of quinolinic acid 80 nmols in combination with SNAP 500 nmols. A highly significant degree of neuronal loss is produced by this combination, which is prevented by the simultaneous administration of chlorostyrylcaffeine (CSC), ZM241385 (ZM) or SCH58261 (SCH). The columns show the mean±s.e.mean (n=4) of the percentage of neurons surviving in the CA1 region of the counted sections. ***P<0.001.
Discussion
Injury to the brain results in glial cell proliferation and infiltration both by activated glial cells, and by activated peripheral macrophages which can cross the blood – brain barrier. These activated cells exhibit a greatly enhanced secretion of quinolinic acid synthesized from tryptophan (Heyes et al., 1992a, 1996; Espey et al., 1997; Pemberton et al., 1997; see Stone, 1993, 2001). Since quinolinic acid is a selective agonist at NMDA-sensitive glutamate receptors (Stone & Perkins, 1981; Stone & Burton, 1988), one consequence of its activation of NMDA receptors is excitotoxicity (Schwarcz et al., 1983; 1984), and the massive elevation of its concentration within the brain in neuro-inflammatory states or in AIDS dementia (Heyes et al., 1989, 1991, 1992b), has raised the possibility that it may contribute to the brain damage and cognitive dysfunction that can occur in these and neurodegenerative conditions.
In general, however, the amount of quinolinate in brain is below the level which is sufficient to produce damage, and the question arises of whether quinolinate-induced damage may be potentiated by other compounds secreted during the neuro-inflammatory reaction. In this study, we have demonstrated that doses of quinolinate which alone produced no signs of overt cell damage, will destroy a large proportion of hippocampal neurones in the additional presence of xanthine and xanthine oxidase, a well-recognized generator of reactive oxygen species such as superoxide and hydroxyl radicals.
It has been suggested that the free radicals generated by the xanthine/xanthine oxidase combination can promote the release of excitatory amino acids in hippocampal slices (Pellegrini-Giampietro et al., 1988), so that the combination tested here could still be acting via glutamate receptors, partly by the direct action of quinolinic acid, and partly by the indirect action of free radicals releasing glutamate. However, the glutamate antagonist 5,7-dichlorokynurenate did not reduce significantly the mean level of neuronal damage, despite the fact that it could substantially reduce the damage produced by a higher dose of quinolinic acid alone. This suggests that the site of potentiation between quinolinic acid and free radical-induced damage is distal to activation of the NMDA receptor, and is not simply the result of free radical-enhanced glutamate release or a free radical-mediated enhancement of NMDA receptor toxicity. Alternatively, damage could result from a completely different mechanism of one or both of the agents. It is unlikely that non-NMDA receptors are involved, since there is no evidence for an action of quinolinic acid at such sites, and 5,7-dichlorokynurenic acid has high selectivity for the strychnine-resistant glycine site of the NMDA receptor (IC50 200 nM) compared with kainate (IC50>300 μM), quisqualate (IC50>30 μM) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (IC50 75 μM) (Leeson et al., 1991). Since the activation of NMDA receptors by NMDA itself or by quinolinic acid can increase the generation of free radicals (Hammer et al., 1993; Lafon-Cazal et al., 1993; Rios & Santamaria, 1991; Behan et al., 1999) the combination of agents used here may induce mutually potentiated form of oxidative stress.
The involvement of free radicals is supported by the combinations of nitric oxide and quinolinic acid, since nitric oxide and related species such as peroxynitrite are potent free radicals able to produce lipid peroxidation. Several studies have shown that nitric oxide donors can produce neuronal damage in vivo (Loiacono & Beart, 1992; Gross et al., 1994) and in vitro (Dawson et al., 1994b; Palluy & Rigaud, 1996). Similarly, despite some reports to the contrary, a majority of studies indicate that NOS inhibitors reduce ischaemic damage (Dawson et al., 1994a; Panahian et al., 1996; Margaill et al., 1997). These views are consistent with the present finding of increased damage in the presence of quinolinic acid and a nitric oxide donor.
The present data are also consistent with the report by Guidetti & Schwarcz (1999) showing potentiation of striatal damage by a combination of quinolinic acid and 3-hydroxykynurenine. The latter is another member of the kynurenine pathway which produces neuronal damage via the generation of free radicals (Eastman & Guilarte, 1990; Okuda et al., 1996). Taken together, all these data suggest that the neurotoxic properties of even low concentrations of quinolinic acid can be enhanced substantially by the simultaneous presence of reactive oxygen species. Since the production of both is raised in microglia and CNS macrophages following damage, inflammation or immune activation, this combination could account for a significant proportion of the delayed neurodegeneration which occurs after excitotoxic or ischaemic insults.
Neuroprotection by adenosine antagonists
Adenosine or its analogues can reduce excitotoxicity and ischaemic damage via A1 receptors (von lubitz et al., 1988; Macgregor & Stone, 1993; Heron et al., 1994) or A2A receptors (Sheardown & Knutsen, 1996; Jones et al., 1998a, 1998b). Evidence for neuroprotection by an A2A receptor antagonist was first suggested by Gao & Phillis (1994). This was later confirmed and extended to other ischaemic models and excitotoxic damage, using a range of A2A receptor antagonist ligands (Phillis, 1995; von lubitz et al., 1995; Ongini et al., 1997; Monopoli et al., 1998; Jones et al., 1998a, 1998b). The selective adenosine antagonist ZM241385, which is 80-fold selective for A2A versus A2B receptors, and 500 – 1000-fold selective for A2A versus A1 receptors (Palmer et al., 1995), was reported to protect the hippocampus against kainic acid to a similar degree as the agonist CGS21680 (Jones et al., 1998a, 1998b). In support, work using A2A receptor knockout mice has revealed that ischaemic brain injury is decreased compared with normal mice (Bona et al., 1997; Chen et al., 1999).
In the present study three chemically distinct antagonists at A2A receptors, including the xanthine CSC and non-xanthines ZM241385 and SCH58261, protected against quinolinic acid. The doses used were chosen to target A2A receptors with some selectivity at the sections used for cell counting. The dilution factor of an agent injected in a volume of 1 μl but subsequently diluting into a sphere of radius 2500 μm (the distance selected for assessment, see Methods) is approximately 30. This would yield a concentration of around 4 μM for CSC, the Ki for which at A2A receptors is 54 nM, while the Ki at rat A1 receptors is 28 μM (Jarvis & Williams, 1989; Jacobson et al., 1993). Similarly, ZM241385 is likely to reach a concentration of about 1 μM, compared with its Ki at A2A receptors of approximately 1 nM, a Ki of 3 μM at A1 receptors and 100 μM at A3 receptors (Poucher et al., 1995). Both these compounds should, therefore, completely saturate A2A receptors, while leaving A1 receptors relatively unaffected. SCH58261 was used at a relatively high concentration which is expected to reach around 2 μM at the site or analysis, compared with a Ki of 1 nM at striatal A2A receptors and over 100 nM at A1 receptors (Cunha et al., 1996; Ongini et al., 1997). The high dose was chosen because of evidence that the hippocampal A2A receptor is less sensitive to SCH58261 than the striatal receptors (Johansson et al., 1993; Johansson & Fredholm, 1995; Lindstrom et al., 1996). In all cases, however, blockade of A1 receptors should increase cell damage, rather than protect, and would tend to oppose, rather than explain, the present data.
The most likely mechanism by which blockade of A2A receptors can produce protection is the suppression of glutamate release. Activation of A2A receptors has an excitatory action on neurones, partly via an increased release of glutamate (Simpson et al., 1992; Sebastião & Ribeiro, 1992; Popoli et al., 1995). The blockade of A2A receptors, therefore, may reduce the extracellular concentrations of glutamate below a threshold necessary for cell damage. In addition, since A2A receptors suppress responses mediated by A1 sites (Lopes et al., 1999; O'kane & Stone, 1998), A2A receptor antagonists will ‘unmask' the A1 receptors and thus permit a degree of protection by A1 receptors.
There may also be effects of A2A receptor antagonists on glial cell function. Activation of A2A receptors suppresses phagocytic activity (Zalavary et al., 1994); antagonists should therefore facilitate the removal of dying cells and toxic materials from a region of injury. In addition, although A2A receptors inhibit the production of several pro-inflammatory cytokines from cells (Dianzani et al., 1994), they can also potentiate the pro-inflammatory effects of those compounds (Scholz-Pedretti et al., 2001). The protective effect of A2A receptor antagonists may therefore reflect a net reduction of pro-inflammatory activity in the damaged region. Activation of A2A receptors can promote glial proliferation after brain injury (Rathbone et al., 1999; Hindley et al., 1994). Since these cells produce anti-inflammatory cytokines as well as pro-inflammatory compounds and kynurenines, the net balance of these may influence neuronal survival.
Whatever the mechanism, the ability of A2A receptor antagonists to protect not only aginst excitotoxins, but also against combined excitotoxic/free radical injury suggests that this class of compounds could be of value in preventing neuronal damage resulting from a variety of insults.
Acknowledgments
The authors are grateful to The Wellcome Trust for support (grant No 057604).
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CGS 15943
5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline
- CGS 21680
2-[4-(2-carboxyethyl)-phenylethylamino]-5′N-ethyl-carboxamido-adenosine
- CNS
central nervous system
- CSC
8-(3-chlorostyryl)caffeine
- DMPA
N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]-adenosine
- HIV
human immunodeficiency virus
- NMDA
N-methyl-D-aspartate
- NOS
nitric oxide synthase
- SCH58261
7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3e]-1,2,4-triazolo[1,5c]pyrimidine
- SNAP
S-nitroso-N-acetylpenicillamine
- ZM241385
4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}(1,3,5}triazin-5-yl-amino]ethyl)phenol
References
- BEHAN W.M.H., MCDONALD M., DARLINGTON L.D., STONE T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: protection by melatonin and deprenyl. Brit. J. Pharmacol. 1999;128:1754–1760. doi: 10.1038/sj.bjp.0702940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEHAN W.M.H., STONE T.W. The role of kynurenines in the neurotoxic action of kainic acid. Br. J. Pharmacol. 2000;129:1764–1770. doi: 10.1038/sj.bjp.0703250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONA E., ADEN U., GILLAND E., FREDHOLM B.B., HAGBERG H. Neonatal cerebral hypoxia-ischemia: the effect of adenosine receptor antagonists. Neuropharmacology. 1997;9:1327–1338. doi: 10.1016/s0028-3908(97)00139-1. [DOI] [PubMed] [Google Scholar]
- CHEN J.F., HUANG Z., MA J., ZHU J., MOTATALLA R., STANDAERT D., MOSKOWITZ M.A., FINK J.S., SCHWARZSCHILD M.A. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA R.A., JOHANSSON B., CONSTANTINO MD., SEBASTIÃO A.M., FREDHOLM B.B. Evidence for high affinity binding sites for the adenosine A2A receptor agonist [3H] CGS21680 in the rat hippocampus and cerebral cortex that are different from striatal A2A receptors. Naunyn-Schmiedebergs Arch Pharmacol. 1996;353:261–271. doi: 10.1007/BF00168627. [DOI] [PubMed] [Google Scholar]
- DAWSON D.A., GRAHAM D.I., MCCULLOCH J., MACRAE I.M. Anti-ischemic efficacy of a nitric oxide synthase inhibitor and a NMDA receptor antagonist in models of transient and permanent focal cerebral ischaemia. Br. J. Pharmacol. 1994a;113:247–253. doi: 10.1111/j.1476-5381.1994.tb16201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWSON V.L., BRAHMBHATT H.P., MONG J.A., DAWSON T.M. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994b;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- DIANZANI C., BRUNELLESCHI S., VIANO I., FANTOZZI R. Adenosine modulation of primed human neutrophils. Eur. J. Pharmacol. 1994;263:223–226. doi: 10.1016/0014-2999(94)90547-9. [DOI] [PubMed] [Google Scholar]
- EASTMAN C.L., GUILARTE T.R. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynyrenine. Neurochem. Res. 1990;15:1101–1107. doi: 10.1007/BF01101711. [DOI] [PubMed] [Google Scholar]
- ESPEY M.G., CHERNYSHEV O.N., REINHARD J.F., JR, NAMBOODIRI M.A.A., COLTON C.A. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8:431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- FOSTER A.C., SCHWARCZ R.Neurotoxic effects of quinolinic acid in the mammalian CNS Quinolinic acid and the Kynurenines 1989Boca Raton, CRC Press; 73–192.ed.Stone, T.W. pp [Google Scholar]
- GALARRAGA E., SURMEIER D.J., KITAI S.T. Quinolinate and kainate neurotoxicity in neostriatal cultures is potentiated by co-culturing with neocortical neurons. Brain. Res. 1990;512:269–276. doi: 10.1016/0006-8993(90)90636-P. [DOI] [PubMed] [Google Scholar]
- GAO Y., PHILLIS J.W. CGS 15943, an adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the mongolian gerbil. Life Sci. 1994;55:PL61–PL65. doi: 10.1016/0024-3205(94)00889-2. [DOI] [PubMed] [Google Scholar]
- GIULIAN D., VACA K., NOONAN C.A. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- GIULIAN D., WENDT E., VACA K., NOONAN C.A. The envelope glycoprotein of human immunodeficiency virus type-1 stimulates release of neurotoxins from monocytes. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2796–2773. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSS P.M., WEAVER D.F., BOWERS R.J., NAG S., HO L.T., PANG J.J., ESPINOSA F.J. Neurotoxicity in conscious rats following intraventricular SNAP, a nitric oxide donor. Neuropharmacology. 1994;33:915–927. doi: 10.1016/0028-3908(94)90190-2. [DOI] [PubMed] [Google Scholar]
- GUIDETTI P., SCHWARCZ R. 3-hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999;11:3857–3863. doi: 10.1046/j.1460-9568.1999.00806.x. [DOI] [PubMed] [Google Scholar]
- HAMMER B., PARKER W.D., BENNETT J.P. NMDA receptors increase hydroxyl radicals in vivo by using nitric oxide synthase and protein kinase C. Neuroreport. 1993;5:72–74. doi: 10.1097/00001756-199310000-00018. [DOI] [PubMed] [Google Scholar]
- HERON A., LEKIEFFRE D., LE PEILLET E., LASBENNES F., SEYLEZ J., PLOTKINE M., BOULU R.G. Effects of an A1 adenosine receptor agonist on the neurochemical, behavioural and histological consequences of ischaemia. Brain Res. 1994;641:217–224. doi: 10.1016/0006-8993(94)90149-x. [DOI] [PubMed] [Google Scholar]
- HEYES M.P., ACHIM C.L., WILEY C.A., MAJOR E.O., SAITO K., MARKEY S.P. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEYES M.P., BREW B.J., MARTIN A., PRICE R.W., SALAZAR A.M., SIDTIS J.J., YERGEY J.A., MOURDIAN M.M., SADLER A.E., KEILP J., RUBINOW D., MARKEY S.P. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationhip to clinical and neurologic status. Ann. Neurol. 1991;29:202–209. doi: 10.1002/ana.410290215. [DOI] [PubMed] [Google Scholar]
- HEYES M.P., RUBINOW D., LANE C., MARKEY S.P. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann. Neurol. 1989;26:275–277. doi: 10.1002/ana.410260215. [DOI] [PubMed] [Google Scholar]
- HEYES M.P., SAITO K., CROWLEY J.S., DAVIS L.E., DEMITRACK M.A., DER M., DILLING L.A., ELIA J., KRUESI M.J.P., LACKNER A., LARSEN S.A., LEE K., LEONARD H.L., MARKEY S.P., MARTIN A., MILSTEIN S., MOURDIAN M.M., PRANTAZELLI M.R., QUEARRY B.J. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain. 1992b;115:1249–1273. doi: 10.1093/brain/115.5.1249. [DOI] [PubMed] [Google Scholar]
- HEYES M.P., SAITO K., MARKEY S.P. Human macrophages convert L-trptophan into the neurotoxin quinolinic acid. Biochem. J. 1992a;283:633–635. doi: 10.1042/bj2830633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HINDLEY S., HERMAN M.A.R., RATHBONE M.P. Stimulation of astrogliosis in vivo by extracellular ADP or an adenosine A2 receptor agonist. J. Neurosci. Res. 1994;38:399–406. doi: 10.1002/jnr.490380405. [DOI] [PubMed] [Google Scholar]
- JACOBSON K.A., NIKODIJEVIC O., PADGETT W.L., GALLO-RODRIGUEZ C., MAILLARD M., DALY J.W. 8-(3-chlorostyryl)caffeine (CSC) is a selective A2-adenosine antagonist in vitro and in vivo. FEBS Lett. 1993;323:141–144. doi: 10.1016/0014-5793(93)81466-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JARVIS M.F., WILLIAMS M. Direct autoradiographic localisation of adenosine A2 receptors in the brain using the A2-selective agonist, [3H] CGS21680. Eur. J. Pharmacol. 1989;168:243–246. doi: 10.1016/0014-2999(89)90571-2. [DOI] [PubMed] [Google Scholar]
- JOHANSSON B., FREDHOLM B.B. Further characterization of the binding of the adenosine receptor agonist [3H]CGS 21680 to rat brain using autoradiography. Neuropharmacology. 1995;34:393–403. doi: 10.1016/0028-3908(95)00009-u. [DOI] [PubMed] [Google Scholar]
- JOHANSSON B., GEORGIEV V., PARKINSON F.E., FREDHOLM B.B. The binding of the adenosine A2 receptor selective agonist [3H]CGS 21680 to rat cortex differs from its binding to rat striatum. Eur. J. Pharmacol. 1993;247:103–110. doi: 10.1016/0922-4106(93)90066-i. [DOI] [PubMed] [Google Scholar]
- JONES P.A., SMITH R.A., STONE T.W. Protection against intrahippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res. 1998a;800:328–335. doi: 10.1016/s0006-8993(98)00540-x. [DOI] [PubMed] [Google Scholar]
- JONES P.A., SMITH R.A., STONE T.W. Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience. 1998b;85:229–237. doi: 10.1016/s0306-4522(97)00613-1. [DOI] [PubMed] [Google Scholar]
- KERR S.J., ARMATI P.J., GUILLEMIN G.J., BREW B.J. Chronic exposure of human neurones to quinolinic acid results in neuronal changes consistent with AIDS dementia complex. AIDS. 1998;12:355–363. doi: 10.1097/00002030-199804000-00003. [DOI] [PubMed] [Google Scholar]
- KHASPEKOV L., KIDA E., VICTOROV I., MOSSAKOWSKI M.J. Neurotoxic effect induced by quinolinic acid in dissociated cell culture of mouse hippocampus. J. Neurosci. Res. 1989;22:150–157. doi: 10.1002/jnr.490220207. [DOI] [PubMed] [Google Scholar]
- LAFON-CAZAL M., CULCASI M., GAVEN F., PIETRI S., BOCKAERT J. Nitric oxide, superoxide and peroxynitrite–putative mediators of NMDA-induced cell death in cerebellar granule cells. Neuropharmacology. 1993;32:1259–1266. doi: 10.1016/0028-3908(93)90020-4. [DOI] [PubMed] [Google Scholar]
- LEESON P.D., BAKER R., CARLING R.W., CURTIS N.R., MOORE K.W., WILLIAMS B.J., FOSTER A.C., DONALD A.E., KEMP J.A., MARSHALL G.R. Kynurenic acid derivatices: structure-activity relationships for excitatory amino acid antagonism and identification of potent and selective antagonists at the glycine site on the NMDA receptor. J. Med. Chem. 1991;34:1243–1252. doi: 10.1021/jm00108a002. [DOI] [PubMed] [Google Scholar]
- LINDSTROM K., ONGINI E., FREDHOLM B.B. The selective adenosine A(2A) receptor antagonist SCH 58261 discriminates between two different binding sites for [3H]-CGS 21680 in the rat brain. Naunyn-Schmiedebergs Arch. Pharmacol. 1996;354:539–541. doi: 10.1007/BF00168448. [DOI] [PubMed] [Google Scholar]
- LOIACONO R.E., BEART P.M. Hippocampal lesions induced by microinjection of the nitric oxide donor nitroprusside. Eur. J. Pharmacol. 1992;216:331–333. doi: 10.1016/0014-2999(92)90381-d. [DOI] [PubMed] [Google Scholar]
- LOPES L.V., CUNHA R.A., RIBEIRO J.A. Cross-talk between A1 and A2A adenosine receptors in the hippocampus and cortex of young and adult old rats. J. Neurophysiol. 1999;82:3196–3203. doi: 10.1152/jn.1999.82.6.3196. [DOI] [PubMed] [Google Scholar]
- MACGREGOR D.G., STONE T.W. Inhibition by the adenosine analogue, (R-) -N6-phenylisopropyladenosine, of kainic acid neurotoxicity in rat hippocampus after systemic administration. Br. J. Pharmacol. 1993;109:316–321. doi: 10.1111/j.1476-5381.1993.tb13572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARGAILL I., ALLIX M., BOULU R.G., PLOTKINE M. Dose- and time-dependence of L-NAME neuroprotection in transient focal ischaemia in rats. Br. J. Pharmacol. 1997;120:160–163. doi: 10.1038/sj.bjp.0700889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONOPOLI A., LOZZA G., FORLANI A., MATTAVELLI A., ONGINI E. Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport. 1998;9:3955–3959. doi: 10.1097/00001756-199812010-00034. [DOI] [PubMed] [Google Scholar]
- MORONI F., LOMBARDI G., MONETI G., ALDINIO C. The excitotoxin quinolinic acid is present in the brain of several animal species and its cortical content increases during the ageing process. Neurosci. Lett. 1984;47:51–55. doi: 10.1016/0304-3940(84)90385-9. [DOI] [PubMed] [Google Scholar]
- O'KANE E.M., STONE T.W. Interactions between A1 and A2 adenosine receptor- mediated responses in the rat hippocampus in vitro. Eur. J. Pharmacol. 1998;362:17–25. doi: 10.1016/s0014-2999(98)00730-4. [DOI] [PubMed] [Google Scholar]
- OKUDA S., NISHIYAMA N., SAITO H., KATSUKI H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. U.S.A. 1996;93:12553–12558. doi: 10.1073/pnas.93.22.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ONGINI E., ADAMI M., FERRI C., BERTORELLI R. Adenosine A2A receptors and neuroprotection. Ann. N. Y. Acad. Sci. 1997;825:30–48. doi: 10.1111/j.1749-6632.1997.tb48412.x. [DOI] [PubMed] [Google Scholar]
- PALLUY O., RIGAUD M. Nitric oxide induces cultured cortical neuron apoptosis. Neurosci. Lett. 1996;208:1–4. doi: 10.1016/0304-3940(96)12532-5. [DOI] [PubMed] [Google Scholar]
- PALMER T.M., POUCHER S.M., JACOBSON K.A., STILES G.L. 125I-4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a} {1,3,5}triazin-5-yl-amino]ethyl)phenol, a high affinity antagonist radioligand selective for the A2A adenosine receptor. Mol. Pharmacol. 1995;48:970–974. [PMC free article] [PubMed] [Google Scholar]
- PANAHIAN N., YOSHIDA T., HUANG P.L., HEDLEY-WHITE E.T., DALKARA T., FISHMAN M.C., MOSKOWITZ M.A. Attenuated hippocampal damage after global cerebral ischaemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in Stereotaxic Co-ordinates. London, Academic Press; 1986. [Google Scholar]
- PELLEGRINI-GIAMPIETRO D.E., CHERICI G., ALESIANI M., CARLA V., MORONI F. Excitatory amino acid release from rat hippocampal slices as a consequence of free radical formation. J. Neurochem. 1988;51:1960–1963. doi: 10.1111/j.1471-4159.1988.tb01187.x. [DOI] [PubMed] [Google Scholar]
- PEMBERTON L.A., KERRS J., SMYTHE G., BREW B.J. Quinolinic acid production by macrophages stimulated with IFN-γ, TNF-α and IFN-α. J. Interferon. Cytokine Res. 1997;17:589–595. doi: 10.1089/jir.1997.17.589. [DOI] [PubMed] [Google Scholar]
- PERKINS M.N., STONE T.W. Pharmacology and regional variations of quinolinic acid–evoked excitations in the rat central nervous system. J. Pharmacol. Exp. Ther. 1983;226:551–557. [PubMed] [Google Scholar]
- PHILLIS J.W. The effects of selective A1 and A2A adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995;705:79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- POPOLI P., BETTO P., REGGIO R., RICCIARELLO G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur. J. Pharmacol. 1995;287:215–217. doi: 10.1016/0014-2999(95)00679-6. [DOI] [PubMed] [Google Scholar]
- POUCHER S.M., KEDDIE J.R., SINGH P., STOGGALL S.M., CAULKETT P.W.R., JONES G., COLLIS M.G. The in vitro pharmacology of ZM241385, a potent non-xanthine, A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RATHBONE M.P., MIDDLEMISS P.J., GYSBERS J.W, , ANDREWS C., HERMAN M.A.R., REED J.K., CICCARELLI R., DI IORIO P., CACIAGLI F. Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 1999;59:663–690. doi: 10.1016/s0301-0082(99)00017-9. [DOI] [PubMed] [Google Scholar]
- RIOS C., SANTAMARIA A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 1991;16:1139–1143. doi: 10.1007/BF00966592. [DOI] [PubMed] [Google Scholar]
- SCHOLZ-PEDRETTI K., PFEILSCHIFTER J., KASKIN M. Potentiation of cytokine induction of group IIA phospholipase A2 in rat mesangial cells by ATP and adenosine via the A2A adenosine receptor. Br. J. Pharmacol. 2001;132:37–46. doi: 10.1038/sj.bjp.0703774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWARCZ R., FOSTER A.C., FRENCH E.D., WHETSELL W.O., JR, KOHLER C. Excitotoxic models for neurodegenerative disorders. Life Sci. 1984;35:19–29. doi: 10.1016/0024-3205(84)90148-6. [DOI] [PubMed] [Google Scholar]
- SCHWARCZ R., WHETSELL W.O., JR, MANGANO R.M. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- SEBASTIÃO A.M., RIBEIRO J.A. Evidence for the presence of excitatory A2 adenosine receptors in the rat hippocampus. Neurosci. Lett. 1992;138:41–44. doi: 10.1016/0304-3940(92)90467-l. [DOI] [PubMed] [Google Scholar]
- SHEARDOWN M.J., KNUTSEN L.J.S. Unexpected neuroprotection observed with the adenosine A2A receptor agonist CGS21680. Drug Develop. Res. 1996;39:108–114. [Google Scholar]
- SIMPSON R.E., O'REGAN M.H., PERKINS L.M., PHILLIS J.W. Excitatory transmitter amino acid release from the ischaemic rat cerebral cortex: Effects of adenosine recptor agonists and antagonists. J. Neurochem. 1992;58:1683–1690. doi: 10.1111/j.1471-4159.1992.tb10041.x. [DOI] [PubMed] [Google Scholar]
- STONE T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- STONE T.W. Kynurenines in the CNS–from endogenous obscurity to therapeutic importance. Prog. Neurobiol. 2001;64:185–218. doi: 10.1016/s0301-0082(00)00032-0. [DOI] [PubMed] [Google Scholar]
- STONE T.W., BURTON N.R. NMDA receptors and their endogenous ligands in vertebrate CNS. Prog. Neurobiol. 1988;30:333–368. doi: 10.1016/0301-0082(88)90027-5. [DOI] [PubMed] [Google Scholar]
- STONE T.W., PERKINS M.N. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- VON LUBITZ D.K.J.E., DAMBROSIA J.M., KEMPSKI O., REDMOND D.J. Cyclohexyladenosine protects against neuronal death following ischaemia in the CA1 region of gerbil hippocampus. Stroke. 1988;19:1133–1139. doi: 10.1161/01.str.19.9.1133. [DOI] [PubMed] [Google Scholar]
- VON LUBITZ D.K.J.E., LIN R.C.-S., JACOBSON K.A. Cerebral ischaemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur. J. Pharmacol. 1995;287:295–302. doi: 10.1016/0014-2999(95)00498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHETSELL O., JR, SCHWARCZ R. Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci. Lett. 1989;97:271–275. doi: 10.1016/0304-3940(89)90609-5. [DOI] [PubMed] [Google Scholar]
- ZALAVARY S., STENDAHL O., BENGTSSON T. The role of cyclic AMP, calcium and filamentous actin in adenosine modulation of Fc-mediated phagocytosis in human neutrophils. Biochem. Biophys. Acta. 1994;1222:249–256. doi: 10.1016/0167-4889(94)90176-7. [DOI] [PubMed] [Google Scholar]