

Abstract
Lysophosphatidylcholine (lysoPC) evokes diverse biological responses in vascular cells including Ca2+ mobilization, production of reactive oxygen species, and activation of the mitogen-activated protein kinases, but the mechanisms linking these events remain unclear. Here, we provide evidence that the response of mitochondria to the lysoPC-dependent increase in cytosolic Ca2+ leads to activation of the extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase through a redox signaling mechanism in human umbilical vein endothelial cells. ERK activation was attenuated by inhibitors of the electron transport chain proton pumps (rotenone and antimycin A) and an uncoupler (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), suggesting that mitochondrial inner membrane potential plays a key role in the signaling pathway. ERK activation was also selectively attenuated by chain-breaking antioxidants and by vitamin E targeted to mitochondria, suggesting that transduction of the mitochondrial hydrogen peroxide signal is mediated by a lipid peroxidation product. Inhibition of ERK activation with MEK inhibitors (PD98059 or U0126) diminished induction of the antioxidant enzyme heme oxygenase-1. Taken together, these data suggest a role for mitochondrially generated reactive oxygen species and Ca2+ in the redox cell signaling pathways, leading to ERK activation and adaptation of the pathological stress mediated by oxidized lipids such as lysoPC.
Lysophosphatidylcholine (lysoPC) is a naturally occurring bioactive lipid molecule thought to be important in the pathophysiology of atherosclerosis. It can be formed as a by-product of lecithin cholesterol acyltransferase-mediated cholesterol esterification with phosphatidylcholine in high-density lipoprotein or produced through phospholipase A2-mediated hydrolysis of phosphatidylcholine in platelet membranes during inflammation.1 LysoPC can also be generated from oxidation of low-density lipoprotein (LDL), accounting for nearly 50% of the phosphatidylcholine equivalents in oxidized LDL.2 This is particularly important because accumulation of oxidized LDL in the arterial intima has been suggested to play a role in the pathogenesis of atherosclerosis.3 In addition to the pro-atherogenic responses to lipid oxidation products, the endothelium adapts to this stress by induction of antioxidant protective pathways including glutathione-dependent pathways and enzymes such as heme oxygenase-1 (HO-1).4–6 HO-1 catalyzes the breakdown of pro-oxidant heme with the release of carbon monoxide and antioxidant products.7 The impact of lysoPC on these pathways or the mechanisms of signal transduction have not been examined in depth. Previously, lyso-lipids have been shown to causes cell lysis above their critical micelle concentration (40 to 50 μmol/L),8 but in blood, it is unlikely that these concentrations of the free lipid are achieved because of binding of albumin. In cell culture models, lysoPC exposure leads to increased expression of adhesion molecules, pro-inflammatory cytokines, and other potentially pro-atherogenic genes.9 Some biological actions of lysoPC have been ascribed to the activation of G protein-coupled receptors, namely, GPR 410,11 and G2A,12 as well as the platelet-activating factor receptor.13,14 However, recent reports have cast doubt on their role as lysoPC receptors.15–17 In addition, the ability of lysoPC to incorporate into cell membranes18 results in membrane perturbations that blunt a number of cellular responses to various vasoactive ligands.19–21 Previous studies have revealed that lysoPC can cause an increase in cytosolic Ca2+,21,22 induce reactive oxygen species (ROS),23 and activate a number of protein kinase pathways including the mitogen-activated protein kinase (MAPK) pathways,24 multiple isoforms of protein kinase C,25 and tyrosine kinases.26 Nevertheless, the mechanisms underlying those cellular signaling events and the mechanistic links between them remain unclear.
ROS can be generated in various cells in response to a variety of stimuli including growth factors, cytokines, and physicochemical stress. A growing body of evidence suggests that ROS can act as second messengers in a number of signal transduction pathways.27 The mechanisms are only now emerging and include modulation of the thiol proteome, thereby controlling the redox tone that regulates the sensitivity of redox signaling pathways to ROS-dependent activation, inactivation of phosphatases, and activation of tyrosine kinases.27,28 Several sources of vascular ROS production that could lead to cell signaling have been identified, including the NAD(P)H oxidases (NOXs), uncoupled endothelial nitric-oxide synthase, and most recently mitochondria.29 In this respect, the NOX enzymes are potential candidates because endothelial cells express subunits of the phagocyte NAD(P)H oxidase, including gp91 (NOX2) and its homologs, NOX1, NOX4, and NOX5.30 In contrast, ROS production by mitochondria, although long regarded as a pathological by-product of respiration, is increasingly being implicated in redox cell signaling pathways.31–38 Several mechanisms/molecules have been suggested as direct regulators of ROS production in mitochondria, including modulation of the membrane potential (ΔΨ),39 nitric oxide, and Ca2+.40,41 In isolated mitochondria, it has been shown that the mitochondrial complexes I, II, and III are potential sources of ROS formation. Precise molecular mechanisms remain unclear but could involve the posttranslational modification of mitochondrial proteins such as the recently described S-thiolation of complex I.42
In this study, we investigated the mechanism of ROS production by lysoPC, signaling pathway activation, and their pathophysiological significance in human umbilical vein endothelial cells (HUVECs). The data revealed that ROS production by lysoPC occurred predominantly in the mitochondrion and that this was associated with activation of extracellular signal-regulated kinase (ERK) but not c-Jun N-terminal kinase (JNK). The mechanisms of transduction of the ROS signal remain unclear but could involve the formation of a lipid peroxidation product within the organelle. Finally, we found that ERK activation by lysoPC leads to increased levels of the anti-atherogenic enzyme HO-1, consistent with a cytoprotective role of this mitochondrial ROS signaling pathway.
Materials and Methods
Cells and Reagents
HUVECs were purchased from Clonetics (San Diego, CA). Cells were maintained in endothelial basal medium 2 (Clonetics) supplemented with 2% fetal bovine serum, growth factors, and antibiotics, all of which were supplied by the manufacturer (Clonetics), under a humidified atmosphere containing 5% CO2 at 37°C. l-α-Palmytoyl-lysophosphatidylcholine (16:0) was purchased from Sigma (St Louis, MO). Unless otherwise noted, all other chemicals were used as obtained from the manufacturer.
1-[2-(5-Carboxyoxazol-2-yl)-6-aminobenzofuran-5-oxy]-2-(2′-amino-5′-methyl phenoxy)-ethane-N,N,N′N′-tetraacetic acid pentaacetoxymethyl ester (Fura-2-AM), Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP), and PD98059 were from Calbiochem (San Diego, CA). U0126 was from Promega (Madison, WI). MitoSOX and 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were from Invitrogen (Carlsbad, CA). Phospho-specific antibodies against ERK, JNK, and pan-ERK were obtained from BD Biosciences (San Jose, CA). Anti-HO-1 antibody and anti-CuZnSOD antibody were from StressGen (Victoria, BC, Canada) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. The monoclonal anti-phosphotyrosine antibody 4G10 was provided by Dr. Tanaka at the University of Tokyo. All other chemicals were of analytical grade.
Western Blotting Analysis of the Activation State of MAPK and the Levels of HO-1
HUVECs were cultured in serum-containing medium until 50 to 70% confluency and then made quiescent by incubating with low-serum medium (0.5% fetal bovine serum in endothelial cell basal medium) for 14 to 16 hours. Next, the quiescent cells were incubated with lysoPC (0 to 40 μmol/L) for 0 to 90 minutes or 6 hours for the induction of HO-1. The inhibitors of mitochondrial respiratory chain, iron and calcium chelators, or lipophilic antioxidants, mito-E or diphenyleneiodonium (DPI), were applied for 15 to 60 minutes before exposure of the cells to lysoPC (unless otherwise indicated). Cells were then washed twice with phosphate-buffered saline and lysed (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 0.5% [v/v] Triton X-100, 1 mmol/L ethylenediamine tetraacetic acid, 1 mmol/L Na3VO4, 50 mmol/L NaF, 0.5 mmol/L phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, and 2 μg/ml aprotinin). The cell lysates were collected in centrifuge tubes, vigorously vortexed, and centrifuged at 10,000 × g for 15 minutes at 4°C. Protein concentration was determined by Bradford reagent (BioRad, Hercules, CA) with bovine serum albumin (BSA) as a standard. Samples were mixed with Laemmli sample buffer and boiled for 5 minutes. Equal amount of proteins were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Immobilon P; Millipore, Billerica, MA). The membranes were probed with respective anti-phospho-MAPK antibody, anti-phosphotyrosine antibody, or an anti-HO-1 antibody, followed by detection with horseradish peroxidase-conjugated anti-mouse or goat anti-rabbit IgG followed by visualization by enhanced chemiluminescence (ECL plus; Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Protein bands were quantified by densitometry, and each experiment was performed three or more times.
Measurement of Cellular ROS
Quiescent cells in four-well chambered coverslip (Nalge, Naperville, IL) were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (20 μmol/L) for 60 minutes followed by treatment with various concentrations of lysoPC for 30 minutes at 37°C. Cells were also incubated with MitoTracker Deep Red 633 (0.5 μmol/L) for 15 minutes. To examine the effects of iron or calcium chelators or mitochondrial complex inhibitors, cells were pre-incubated with N,N′-bis-(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid (HBED) for 60 minutes or 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), ruthenium red (RuRed) or rotenone, thenoyltrifluoroacetone (TTFA), or DPI for 30 minutes before applying lysoPC. Subsequently, cells were washed twice with culture medium, and images were acquired from three or more randomly chosen fields using an inverted epifluorescence microscope (IX70; Olympus, Tokyo, Japan). The levels of 2′,7′-dichlorofluorescein (DCF) fluorescence were quantitated using SIMPLEPCI software (Compix, Cranberry Township, PA). The subcellular source of ROS generation was assessed by single bidirectional scans of live cells using a Leica DMIRBE inverted epifluorescence/Nomarski microscope with Leica TCS NT Laser Confocal optics. The levels of mitochondrial superoxide in HUVECs induced by lysoPC were also measured using the mitoSOX probe. Briefly, cells were grown in a 4-well chambered coverslip until 70 to 80% confluent. Cells were then loaded with the mitoSOX probe (5 μmol/L) for 15 minutes followed by treatment with or without lysoPC (5 μmol/L) or rotenone (5 μmol/L) for 60 minutes at 37°C. Images were merged and processed using IPLab Spectrum and Adobe Photoshop (Adobe System).
Ca2+ Assay
Subconfluent HUVECs were loaded with 5 μmol/L Fura-2-AM for 30 minutes at 37°C in Ringer buffer (150 mmol/L NaCl, 4 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 5.6 mmol/L glucose, and 5 mmol/L HEPES-NaOH, pH 7.4) containing 0.1% BSA, and cells were harvested by trypsinization. The cell density of the suspension was adjusted to 1 × 106/ml in Ringer buffer without BSA. Changes in the fluorescence emission were monitored on a spectrophotometer (CAF-100; Jasco, Tokyo, Japan) at 37°C with excitation wavelengths at 340 and 380 nm and an emission wavelength at 500 nm. Intracellular Ca2+ concentration was calculated according to the following equation
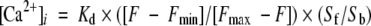 |
where Kd is 224 nmol/L, F is the ratio of the fluorescence emission at 340 nm/fluorescence emission at 380 nm, and Fmax and Fmin are obtained in the presence of 0.5% Triton X-100 and subsequently 37.5 mmol/L EGTA, respectively, at the end of each assay. Sb and Sf are emission values at 380 nm corresponding to Fmax and Fmin, respectively. In assays with EGTA, coefficients obtained without EGTA were used for calculation.
Small Interfering RNA (siRNA) Transfection
The siRNA double-strand targeting p22phox DNA sequence 5′-AAGTACATGACCGCCGTGGTG-3′ (211–231, GenBank accession number BT006861; sense, 5′-GUACAUGACCGCCGUGGUGdTdT-3′; antisense, 5′-CACCA-CGGCGGUCAUGUACdTdT-3′) and a control nonsilencing double-strand targeting DNA sequence 5′-AATTCTCCGAACGTGTCACGT-3′ were purchased from QIAGEN (Dusseldorf, Germany) Cells grown in 60-mm plates around 50% confluency were transfected with either nonsilencing RNA or siRNA against p22phox using Lipofectamine 2000 (Invitrogen). Briefly, siRNA (240 pmol) and Lipofectamine 2000 (12 μl) were mixed in OPTI-MEM (750 μl) to allow complex formation for 20 minutes at room temperature. Next, the complex was added dropwise into a HUVEC culture (3 ml of antibiotic-free serum containing medium). The medium was changed 8 hours after transfection, and all experiments were performed between 48 and 72 hours after transfection. p22phox could be completely suppressed at the protein level in the siRNA-transfected cells during this treatment.
Results
Induction of Phosphorylation of ERK, JNK, and Tyrosine Residues in High Molecular Proteins
Previous studies have shown that MAPK pathways are activated by lysoPC in endothelial cells,24 and in the case of smooth muscle cells, it is dependent on ROS production.43 In the first series of experiments lysoPC-dependent phosphorylation of ERK and JNK in endothelial cells was confirmed. Cells were incubated for a period of 14 to 16 hours under conditions of 0.5% serum to achieve a quiescent state before the addition of lysoPC for the range of concentrations and times shown (Figure 1). A significant increase in pERK1/2 phosphorylation was evident at a concentration of lysoPC of 10 μmol/L and reached a maximum at 40 μmol/L lysoPC over a 15- to 30-minute time period (Figure 1A). It is evident from Figure 1B that this transient ERK1/2 and JNK phosphorylation occurs over a similar time course and concentration. In contrast to ERK and JNK phosphorylation, lysoPC caused robust and prolonged tyrosine phosphorylation of high-molecular weight proteins (Figure 1B).
Figure 1.

LysoPC increases phosphorylation of ERK1/2, JNK, and tyrosine residues in high-molecular weight proteins. Representative immunoblots of total ERK, phospho-ERK1/2, phospho-JNK, and tyrosine-phosphorylated proteins in HUVECs induced by increasing concentrations of lysoPC (0 to 40 μmol/L) for 15 minutes (A) or the kinetics of their appearance (0 to 90 minutes) measured at 30 μmol/L (B).
LysoPC Induces Ca2+ Influx
Possible mechanisms for lysoPC-induced ERK and JNK activation may involve the reported elevation of intracellular calcium that occurs on exposure of cells to this lipid.21,22 Cells were loaded with Fura 2-AM (5 μmol/L) and harvested by trypsinization, and the suspensions were exposed to lysoPC. Exposure of HUVECs to lysoPC (20 to 30 μmol/L) provoked robust elevations of intracellular Ca2+, which persisted for at least 5 minutes (Figure 2). To examine whether the lysoPC-dependent increase in Ca2+ was derived from intracellular stores or an extracellular pool, cells were stimulated by lysoPC in the presence of EGTA. Under these conditions, the lysoPC-induced increase in intracellular Ca2+ level was significantly decreased, suggesting that the Ca2+ is derived from an extracellular source (Figure 2).
Figure 2.

Ca2+ mobilization by lysoPC in HUVECs. Representative trace of cytosolic levels of Ca2+ evoked by lysoPC treatment. HUVECs were loaded with Fura-2 (5 μmol/L) for 30 minutes, harvested by trypsinization, and then stimulated with indicated concentrations of lysoPC (10 or 30 μmol/L) at 37°C. A significant decrease in the level of lysoPC-induced Ca2+ mobilization was evident in the presence of EGTA (2.5 mmol/L; added 1 minute before treatment with 20 μmol/L lysoPC). Similar results were obtained from three independent experiments.
Ca2+ Influx by LysoPC Plays a Key Role in the Activation of the ERK1/2 Pathway
To determine whether lysoPC-induced ERK1/2 activation was associated with Ca2+ mobilization, the effects on ERK1/2 phosphorylation of the Ca2+ chelators and the inhibitor of the mitochondrial Ca2+ uniporter (RuRed) were assessed (Figure 3). The cytosolic Ca2+ chelator 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM; 20 μmol/L), extracellular Ca2+ chelator EGTA (2 mmol/L), and RuRed (10–50 μmol/L) significantly inhibited ERK1/2 phosphorylation (60 to 70%) (Figure 3, A–D). Simultaneous addition of equimolar Ca2+ abolished the inhibitory effect of EGTA (Figure 3C). Notably, these treatments had little effect on lysoPC-dependent JNK phosphorylation, suggesting that the activation of this signaling cascade proceeds through a different mechanism. Taken together, these results suggest that extracellular Ca2+ influx induced by lysoPC exposure leads to ERK1/2, but not JNK, activation in endothelial cells through a mechanism involving mitochondrial Ca2+ uptake.
Figure 3.

Effects of Ca2+ chelators on lysoPC-induced phosphorylation of ERK and JNK. HUVECs were pre-incubated with RuRed (50 μmol/L), BAPTA-AM (BAPTA, 20 μmol/L) for 30 minutes (A) or with EGTA (2 mmol/L) in the presence or absence of CaCl2 (2 mmol/L) for 15 minutes (C). Next, cells were stimulated with lysoPC (30 μmol/L) for 15 minutes. Supplementation of medium with CaCl2 reversed the inhibitory effect of EGTA (C). The histograms in B and D show the intensity of each band for p-ERK in A and C, respectively, obtained by densitometric analysis wherein lysoPC-dependent increase of p-ERK in the control group was set at 100%. *P < 0.05 compared with lysoPC treatment alone or lysoPC plus dimethylsulfoxide (DMSO); #P < 0.05 compared with EGTA alone (mean ± SEM, n ≥ 3).
Ca2+ Mobilization Regulates Mitochondrial ROS Generation
Previously, induction of DCF fluorescence in cells exposed to lysoPC has been reported,44 although the mechanism remains unclear. We hypothesized that mitochondrial Ca2+ uptake could lead to an increase in ROS formation in the organelle. To test this hypothesis, we used DCFH-DA (20 μmol/L) to measure ROS formation in the cell. This compound is converted to DCFH inside the cell, and on exposure to either hydrogen peroxide or peroxynitrite, it is oxidized to the fluorescent product DCF through mechanisms involving transition metal ions or cellular peroxidases as catalysts.45 In the first series of experiments, cells were exposed to lysoPC (20 μmol/L) for 30 minutes, then the medium was changed, and images were acquired. In agreement with previous studies, lysoPC treatment resulted in increased DCF fluorescence, and this was significantly (∼40%) inhibited by RuRed (50 μmol/L) (Figure 4). To further examine the relationship between Ca2+ influx and ROS/reactive nitrogen species generation by lysoPC, the effects of the Ca2+ chelators BAPTA-AM (20 μmol/L) and EGTA (2 mmol/L) on DCF fluorescence were measured. Treatment of cells with either of these compounds resulted in nearly a 40 to 60% decrease in the lysoPC-dependent DCF fluorescence (Figure 4). These data suggest that the mitochondrial lysoPC-dependent ROS generation is associated with an increase in mitochondrial Ca2+ uptake.
Figure 4.

Effects of Ca2+ chelators on lysoPC-induced ROS generation in HUVECs. A: HUVECs were loaded with DCFH-DA (20 μmol/L) in the presence or absence of RuRed (50 μmol/L), EGTA (2 mmol/L), or BAPTA-AM (20 μmol/L) for 30 minutes. Then, cells were further incubated with or without lysoPC (20 μmol/L) for 30 minutes. Cells were washed with culture medium, and live cells were imaged on an inverted fluorescence microscope. C: HUVECs were loaded with mitoSOX (5 μmol/L) followed by the treatment with lysoPC (5 μmol/L) or rotenone (5 μmol/L) for 60 minutes, and images of live cells were acquired. B and D: Average of DCF or mitoSOX fluorescence of cells treated as indicated in A or C was obtained from three randomly chosen fields and is expressed as pixel intensity/cell (mean ± SEM, n = 3; *P < 0.01 compared with lysoPC treatment alone.
As an alternative approach to measuring ROS generation induced by lysoPC, the mitoSOX Red probe was used. This compound is a mitochondrial targeted form of dihydroethidium. Exposure of cells to lysoPC or rotenone increased mitoSOX fluorescence six- and fivefold, respectively (Figure 4, C and D). These data support the findings with DCF that lysoPC can initially induce mitochondrial superoxide generation.
As a further test for the mitochondrial origin of lysoPC-dependent ROS formation, HUVECs were loaded with DCFH-DA (5 μmol/L) followed by treatment with lysoPC (5 μmol/L) for 30 to 60 minutes with the mitochondrial specific marker MitoTracker (500 nmol/L) applied for 15 minutes before imaging. After washing with serum-free medium, images were acquired with single bidirectional scanning. As shown in Figure 5, lysoPC-dependent DCF fluorescence colocalized with MitoTracker, consistent with a significant contribution of the organelle to lysoPC-dependent ROS formation. To gain some mechanistic insight into the site of mitochondrial ROS production by lysoPC, DCFH oxidation was also examined in the presence of inhibitors of the mitochondrial electron transport chain. In isolated mitochondria, inhibition of complex I by rotenone is known to increase ROS production at this site but would inhibit ROS formation arising from reversed electron transport through complex I.46 Rotenone treatment alone resulted in increased DCF fluorescence (Figure 6) but did not affect the ability of lysoPC to further induce DCF fluorescence. In addition to rotenone, we used the flavoprotein inhibitor DPI, which has been shown to inhibit mitochondrial complexes at low concentrations in endothelial cells.47 DPI substantially inhibited DCF fluorescence induced by lysoPC (Figure 6). In a recent study we have shown that the complex II inhibitor TTFA does not inhibit ROS formation by the extracellular source of hydrogen peroxide, glucose oxidase.48 In addition, the complex II inhibitor TTFA substantially inhibited lysoPC-induced ROS generation.
Figure 5.

LysoPC induces ROS generation in mitochondria. Cells were loaded with DCFH-DA (5 μmol/L) for 60 minutes followed by incubation with or without LysoPC (5 μmol/L) for 30 to 60 minutes. Cells were also incubated with MitoTracker (0.5 μmol/L) for 15 minutes before imaging. Images were acquired on a confocal scanning microscope (see Materials and Methods). The exposure of HUVECs to lysoPC increased the intensity of DCF fluorescence that colocalized with the mitochondrial marker.
Figure 6.

Effects of inhibitors of mitochondrial oxidative phosphorylation on lysoPC-induced ROS generation. Cells were pre-incubated with rotenone (10 μmol/L), TTFA (10 μmol/L), or DPI (20 μmol/L) for 30 minutes followed by treatment with lysoPC (20 μmol/L) for additional 30 minutes. The images were acquired (A), and DCF fluorescence was averaged (B) as described above (Figure 4). The rotenone treatment alone increased DCF fluorescence, which was additive to DCF fluorescence obtained after treatment with lysoPC. *P < 0.05 compared with lysoPC treatment alone; #P < 0.05 with rotenone compared with control cells (mean ± SEM, n ≥ 3).
Activation of the ERK1/2 Pathway by LysoPC Is Channeled through Mitochondria
To test whether NOXs participate in the regulation of the lysoPC-dependent ERK activation in HUVECs, an siRNA directed against p22phox was used. Previous studies have demonstrated that the abrogation of the function of NOX family of enzymes by transfection with a dominant-negative p47phox decreased lysoPC-induced ERK1/2 activation in vascular smooth muscle-like cells.43 The Western blotting analysis revealed essentially complete elimination of the level of p22phox subunit (22 kd) in HUVECs transiently transfected with p22phox siRNA (Figure 7A), whereas the nonsilencing siRNA had no effect. Next, the level of lysoPC-induced ERK1/2 phosphorylation was determined. As shown in Figure 7B, phosphorylation of ERK1/2 induced by lysoPC was not decreased in the p22phox-depleted cells. Thus, involvement of the NOX enzymes in lysoPC-induced ERK1/2 activation in HUVECs seems unlikely. In a further series of experiments, a role of endothelial nitric oxide synthase was examined by application of the inhibitor l-NAME (100 μmol/L) to the medium before activation by lysoPC. The NOS inhibitor showed no effect on either ROS formation or activation of the MAPK (data not shown). Moreover, we could observe no inhibitory effect on lysoPC-induced ERK activation in cells in which endothelial nitric-oxide synthase protein had been diminished by an siRNA directed against the enzyme (data not shown).
Figure 7.

Effect of siRNA-mediated knockdown of p22phox on the phosphorylation of ERK1/2 and JNK by lysoPC. HUVECs were transfected with either nonsilencing control siRNA (n.s.RNA) or p22phox siRNA (detailed in Materials and Methods). At 48 hours after transfection, cells were serum-starved for 16 hours and stimulated with lysoPC (30 μmol/L) for 15 minutes. A: Western blot analysis shows almost complete disappearance of p22phox in cells treated with p22phox siRNA compared with control or n.s.RNA-treated cells. The cell lysates of naïve and retinoic acid-treated HL60 cells were used as an indicator of the p22phox relative electrophoretic mobility. B: Effects of silencing of p22phox on lysoPC-induced phosphorylation of ERK1/2 or JNK. The Western blots shown are representative of the three independent experiments.
To investigate whether lysoPC-induced ERK1/2 activation depends on mitochondrial function, the inhibitors of mitochondrial oxidative phosphorylation were used. Inhibition of complex I or complex III by rotenone (2 μmol/L) or antimycin A (10 μmol/L), respectively, or dissipation of the proton gradient by the uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP; 10 μmol/L) significantly attenuated ERK1/2 phosphorylation induced by lysoPC (30 μmol/L) (Figure 8). In contrast, the complex II inhibitor TTFA (50 μmol/L) or the complex V (ATP synthase) inhibitor oligomycin (10 μg/ml) has no significant effect on ERK1/2 phosphorylation. Western blot analysis also demonstrated that none of the inhibitors significantly attenuated the lysoPC-dependent increase in JNK phosphorylation. These data suggest that the activity of complex I and complex III in the electron transport chain and the mitochondrial inner membrane potential (ΔΨ) contribute to lysoPC-dependent ERK1/2 phosphorylation.
Figure 8.

Effects of inhibitors of oxidative phosphorylation on lysoPC-induced MAPK activation. Representative immunoblots of phospho-ERK1/2, phospho-JNK and total ERK (A) and quantitative data obtained by densitometric analysis (B). A: Cells were pre-incubated with DMSO (0.1%), rotenone (2 μmol/L), TTFA (50 μmol/L), antimycin A (10 μmol/L), FCCP (10 μmol/L), or oligomycin (10 ng/ml) for 30 minutes and then exposed to lysoPC (30 μmol/L) for 15 minutes. *P < 0.05 compared with lysoPC treatment alone (mean ± SEM, n ≥ 3).
It is possible that mitochondrial inhibitors prevent ERK activation by an indirect mechanism, for example by ATP depletion.49 To test for this, the effects of the FCCP (10 μmol/L), RuRed (50 μmol/L), or rotenone on phorbol 12-myristate 13-acetate (PMA)-induced ERK activation were determined. No inhibitory effect could be observed with FCCP or RuRed in PMA-induced phosphorylation of ERK1/2 (Figure 9). The treatment with rotenone slightly attenuated ERK1/2 phosphorylation by PMA, but the extent was far less than that observed under lysoPC stimulation. Thus, it is unlikely that changes in the levels of ATP induced by the electron transport inhibitors or uncoupler were sufficient to suppress the activation of ERK induced by lysoPC.
Figure 9.

Effects of selected inhibitors of oxidative phosphorylation on PMA-induced phosphorylation of ERK1/2. Representative immunoblots (A) and densitometric analysis (B) of phospho-ERK1/2 and total ERK. Cells were incubated with rotenone (2 μmol/L), RuRed (50 μmol/L), or FCCP (10 μmol/L) and then treated with PMA (1 μmol/L) in parallel with exposure to lysoPC (30 μmol/L) for 15 minutes. *P < 0.05 compared with lysoPC treatment alone (mean ± SEM, n ≥ 3).
Activation of the ERK Pathway by LysoPC Is ROS-Dependent
Involvement of ROS in ERK activation was examined. The level of ERK1/2 phosphorylation was almost completely inhibited by DPI (20 μmol/L) and the membrane-permeable SOD mimetic MnTBAP (250 μmol/L) (Figure 10, A and B). However, the effects of those compounds on JNK activation and tyrosine phosphorylation of high-molecular weight proteins were not significant (Figure 10). Ebselen, a glutathione peroxidase mimetic, also preferentially attenuated the lysoPC-induced phosphorylation of ERK1/2. Furthermore, as shown in Figure 10C, the lipid peroxyl radical scavengers α-tocopherol, γ-tocotrienol, and a synthetic radical scavenging antioxidant BO65350 could also mitigate ERK1/2 without affecting JNK phosphorylation. Thus, the activation of the ERK pathway is potentially associated with ROS production. Taking into account the specificity of the lipid radical scavengers and their minimal ability to scavenge both superoxide and hydrogen peroxide, a role for the formation of lipid peroxides in ERK activation is suggested.
Figure 10.

Effects of antioxidant agents on lysoPC-induced ERK or JNK phosphorylation. In A, cells were pre-incubated with or without DMSO (0.1%), DPI (20 μmol/L), MnTBAP (250 μmol/L), or ebselen (20 μmol/L) for 30 minutes and then treated with lysoPC (30 μmol/L) for 15 minutes. B: Quantitative data were obtained from three or more of independent experiments. *P < 0.05 compared with lysoPC treatment alone. C: Cells were pre-incubated with the lipophilic antioxidants (each 50 μmol/L) for 16 hours followed by treatment with lysoPC (30 μmol/L) for 0 to 20 minutes.
Targeting Mitochondria with Vitamin E Alters the Level of LysoPC-Dependent ROS Formation and ERK1/2 Phosphorylation
The results thus far suggest that activation of the ERK pathway by lysoPC involves the induction of mitochondrial ROS production, leading to intraorganelle lipid peroxidation. To further evaluate this possibility, cells were pretreated with mitochondrially targeted tocopherol derivative 2-(3,4-dihydro-6-hydroxy-2,5,7,8-tetramethyl-2H-1-benzopyran-2-yl)ethyl]triphenylphosphonium bromide (MitoVitE). This compound can accumulate in mitochondria according to the mitochondrial inner membrane potential (ΔΨ).51 Figure 11A demonstrates that level of lysoPC-induced DCF fluorescence was significantly inhibited in cells pretreated with MitoVitE (500 nmol/L) for 60 minutes. Under the same conditions, MitoVitE also inhibited lysoPC-dependent ERK activation.
Figure 11.

Effects of MitoVitE or HBED on lysoPC-induced DCFH oxidation and phosphorylation of ERK1/2. The level of DCF fluorescence was demonstrated in cells pre-incubated with MitoVitE (500 nmol/L) and HBED (500 nmol/L) for 60 minutes followed by treatment with lysoPC (20 μmol/L) for an additional 30 minutes. The representative images were acquired with epifluorescence microscopy (A), and quantitated analysis of DCF fluorescence (B). C: In parallel experiments, the level in ERK1/2 phosphorylation was determined, and results of densitometric analysis are shown. *P < 0.05 compared with lysoPC treatment alone (mean ± SEM, n ≥ 3).
Recently, it has been shown that DCF fluorescence induced by exogenous hydrogen peroxide can be inhibited by addition of an iron chelator such as HBED.52 In this study, lysoPC-induced DCF fluorescence was partially (∼30%) decreased in cells pre-incubated with HBED at 100 μmol/L (Figure 11A), suggesting a role for iron in lysoPC-induced DCF fluorescence. Additionally, pretreatment with HBED also decreased the level of ERK1/2 phosphorylation (Figure 11C). Taken together, these data suggest that generation of lipid peroxides, which results from mitochondrial ROS production and participation of iron, could preferentially regulate ERK activation by lysoPC in HUVECs.
ERK Activation by LysoPC Is Coupled with Induction of Heme Oxygenase 1
The regulation of pro-atherogenic genes in response to lysoPC is well documented, but these proteins are not known to have mitochondrial component in their regulation.9,24 In contrast, the antioxidant enzyme HO-1 has recently been shown to be regulated, in part, by ROS, and in a separate series of studies, ERK has also been implicated.5,35,53 These data suggest that an adaptation to the stress of lysoPC exposure that leads to the activation of ERK could result in increased levels of HO-1. In support of this hypothesis, we found that lysoPC was a potent inducer of HO-1 protein expression in HUVECs, and this effect could be attenuated by the inhibitors of MEK (PD98059 or U0126) (Figure 12).
Figure 12.

Effects of the inhibitors of the ERK pathway on HO-1 induction by lysoPC. Serum-starved cells were pre-incubated with PD98059 (30 μmol/L) or U0126 (20 μmol/L) for 30 minutes followed by treatment with lysoPC (25 μmol/L) for additional 6 hours. A: A representative blot; B: quantitative data obtained from five independent experiments. *P < 0.05 compared with lysoPC treatment alone.
Discussion
Recent evidence suggests that mitochondrially produced ROS may contribute to the regulation of various physiological processes including metabolic rate,31,32 tumor necrosis factor-α,54 leptin,33 endothelin cell signaling,55 and mechanical stress.34,56 A number of exogenous stimuli important in the pathophysiology of cardiovascular disease, such as lipid peroxidation products, have been shown to induce ROS in cells and to induce cell signaling, but a role for the mitochondrion has not been investigated.57,58 For example, involvement of ROS in a range of lysoPC-induced cellular responses, from activation of the MAPKs and tyrosine kinases to the expression of several genes, has been suggested.43,44,59 In this study, we confirm that lysoPC induces the transient activation of MAPKs in endothelial cells under conditions that do not induce cytotoxicity (Figure 1). One source of the ROS formed in response to lysoPC in HUVECs and vascular smooth muscle cells is thought to be the NADPH oxidases, which are members of the NOX family of proteins.43,44 Using an siRNA approach, it was shown that the p22phox subunit of the NOX enzymes does not significantly contribute to ERK activation in HUVECs (Figure 7).
Although it is not clear at present whether lysoPC initially acts at the cell surface through binding to its proposed receptors,10–14 or other mechanisms such as membrane perturbation,60 lysoPC brings about a robust influx of extracellular Ca2+. In this study, a role for Ca2+ influx in lysoPC-dependent ERK activation is supported by the finding that EGTA, BAPTA, and RuRed all result in a substantial inhibition of ERK activation (Figure 3). This response is not due to a general dysfunction in cell signaling because JNK is not affected in a similar manner. Taken together, these data suggest that Ca2+ enters the cell via the plasma membrane and is taken up by the mitochondrion, which then leads to ERK activation. The fact that ERK can be activated by ROS and mitochondria are capable of generating ROS suggests that mitochondria may be a link between Ca2+ influx and ERK activation. When mitochondria accumulate cytosolic Ca2+, the Ca2+-dependent matrix dehydrogenases are activated, increasing NADH levels and the potential to form ROS.41,61,62 For the detection of intracellular ROS, we elected to use DCF-dependent fluorescence, which we have recently used to demonstrate that oxidized low-density lipoprotein is capable of eliciting mitochondrial ROS formation in endothelial cells.48 We next determined that the Ca2+ influx inhibitors that suppressed ERK activation also suppressed lysoPC-dependent DCF fluorescence (Figure 4). In addition we determined by confocal microscopy that this DCF formation was localized to the mitochondrion (Figure 5). Measuring ROS formation using DCF has a number of drawbacks that have been well recognized in the literature (reviewed in Ref. 45). Therefore, we confirmed mitochondrial ROS formation using MitoSOX, which detects superoxide through different mechanisms than DCF.
To test for the respiratory chain as a potential source of the mitochondrial ROS, inhibitors were used. First, we used the complex I inhibitor rotenone, which increased DCF fluorescence alone but did not prevent a further increase in response to lysoPC (Figure 6). In contrast, TTFA, which is a complex II inhibitor, decreased DCF fluorescence by approximately the same extent as the Ca2+ chelators, suggesting a role for complex II in lysoPC-dependent ROS formation. DPI, a flavoprotein inhibitor, also inhibited lysoPC-dependent ROS formation, but these results cannot be used to confirm the role of a specific protein because DPI acts via pleiotropic mechanisms.
ERK activation by lysoPC was blocked by rotenone, the complex III inhibitor antimycin A, and the protonophore FCCP. Unexpectedly, TTFA had no significant effect on ERK activation (Figure 8). Thus, lysoPC-dependent ERK activation requires a contributory activity from both complexes I and III and an inner-membrane impermeable to protons. To explain the apparently anomalous effect of TTFA, we considered the relationship between mitochondrial ROS formation under normal conditions and in the presence of respiratory chain inhibitors. In the normal functioning of the mitochondrion, the production of ROS increases as a function of ΔΨ,39 but this relationship is decoupled in the presence of inhibitors such as rotenone, TTFA, and antimycin A.63,64 For this reason, we cannot draw conclusions regarding the link between lysoPC-induced mitochondrial ROS formation and ERK activation on the basis of these data. However, it is possible to address the role of the mitochondrion in both ROS formation and ERK activation with the data using calcium influx inhibitors. Blocking Ca2+ uptake into the mitochondrion inhibits both lysoPC-dependent ERK activation and ROS formation, suggesting that these two events are mechanistically connected.
In the next series of experiments, we used selected antioxidants to determine the potential mechanism through which mitochondrial ROS formation contributes to ERK activation. Because it is difficult to rationalize how hydrogen peroxide, which is freely diffusible, can exert differential effects when formed at different sites in the respiratory chain, we propose the formation of a secondary mediator. A logical candidate would be lipid peroxidation, which can generate a family of molecules, such as 4-hydroxy-2-nonenal, that can initiate signal transduction events including MAPK activation.65
In support of this concept, the lipid peroxyl radical scavengers tocopherol, tocotrienol, and BO653 and the mitochondrially targeted α-tocopherol analog MitoVitE inhibited ERK activation to the same extent as the Ca2+ chelators. Because the iron chelator HBED inhibited ERK activation, then it is possible that the hydrogen peroxide and iron derived from the electron transport chain proteins initiate lipid peroxidation (Figure 11). Consistent with this interpretation, both ebselen and MnTBAP have also been shown to inhibit lipid peroxidation66,67 and lysoPC-dependent ERK activation (Figure 10). Overall, these data suggest that mitochondria transduce the hydrogen peroxide signal to a lipid-derived signaling molecule.
It is also now becoming clear that in addition to the cytotoxic pathways induced by lipid oxidation products, protective antioxidant pathways are also activated.4–6,53 These include the transcriptional regulation of antioxidant defenses such as glutathione and the antioxidant enzyme HO-1.4–6,53 This is important, because it has been demonstrated that HO-1 overexpression by an adenovirus-mediated gene transfer can attenuate atherosclerotic lesion formation in ApoE knockout mice,68 whereas genetic ablation of HO-1 exacerbates lesion formation in the same animal model.69 Interestingly, mitochondrial ROS has been shown to play a regulatory role in controlling levels of HO-1.35 In these experiments, mitochondrially generated hydrogen peroxide, but not ERK activation, was shown to be essential for HO-1 up-regulation. However, other studies have shown that ERK activation can play an important regulatory role in HO-1 expression in response to other oxidized lipids.5,53 These findings led us to test the potential of lysoPC to induce mitochondrial ROS formation in endothelial cells and examine the possible link to ERK activation and HO-1 expression. We found that lysoPC induced the expression of HO-1 in an ERK-dependent manner, suggesting that mitochondrial-dependent activation of ERK may be an im-portant cytoprotective pathway in vascular endothelial cells (Figure 12).
In conclusion, the present study revealed that the lysoPC-dependent activation of ERK has a significant contribution derived from Ca2+-dependent mitochondrial ROS formation, which can be readily detected on exposure of endothelial cells to the lipid (Figure 13). The mechanisms of transduction of the hydrogen peroxide signal appear to involve the formation of a lipid peroxidation product within the mitochondrion. The mitochondrial ROS-dependent activation of ERK in turn plays a key role in the induction of the anti-atherogenic enzyme HO-1.
Figure 13.

Potential signaling mechanism for ERK activation by lysoPC. Ca2+ influx from the extracellular stores induced by lysoPC exposure leads to uptake of the ion in mitochondria where it causes ROS production, possibly at multiple sites (respiratory chain complexes and TCA cycle dehydrogenases). Lipid peroxides generated as a result of membrane oxidation activate the ERK MAPK pathway. Impairment of electron transport with rotenone or antimycin A decreases mitochondrial Ca2+ uptake and thereby attenuates ERK activation.
Acknowledgments
We thank Dr. Kumiko Saeki of International Medical Center of Japan for providing HL60 cells and Dr. Keiko Horiuchi for technical supports for siRNA experiments.
Footnotes
Address reprint requests to Noriko Noguchi, #34, Research Center for Advanced Science and Technology, University of Tokyo, 4-6-1 Komaba, Meguro, Tokyo 153-8904. E-mail: noguchi@lsbm.org.
Supported by “the special coordination funds for promoting science and technology” and the Academic Frontier Research Project on “New Frontier of Biomedical Engineering Research” from the Ministry of Education, Science, Culture, Sports, and Technology in Japan; the National Institutes of Health grants ES10167 and HL58031(to V.M.D.-U.); the Program of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation; and Focus 21 Project of New Energy and Industrial Technology Development Organization.
N.W. and J.W.Z. contributed equally to this work.
References
- Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- Steinbrecher UP, Zhang HF, Lougheed M. Role of oxidatively modified LDL in atherosclerosis. Free Radic Biol Med. 1990;9:155–168. doi: 10.1016/0891-5849(90)90119-4. [DOI] [PubMed] [Google Scholar]
- Noguchi N. Novel insights into the molecular mechanisms of the antiatherosclerotic properties of antioxidants: the alternatives to radical scavenging. Free Radic Biol Med. 2002;33:1480–1489. doi: 10.1016/s0891-5849(02)01114-0. [DOI] [PubMed] [Google Scholar]
- Moellering DR, Levonen AL, Go YM, Patel RP, Dickinson DA, Forman HJ, Darley-Usmar VM. Induction of glutathione synthesis by oxidized low-density lipoprotein and 1-palmitoyl-2-arachidonyl phosphatidylcholine: protection against quinone-mediated oxidative stress. Biochem J. 2002;362:51–59. doi: 10.1042/0264-6021:3620051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest. 1997;100:1209–1216. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Migita CT. Mechanism of heme degradation by heme oxygenase. J Inorg Biochem. 2000;82:33–41. doi: 10.1016/s0162-0134(00)00156-2. [DOI] [PubMed] [Google Scholar]
- Bergmann SR, Ferguson TB, Jr, Sobel BE. Effects of amphiphiles on erythrocytes, coronary arteries, and perfused hearts. Am J Physiol. 1981;240:H229–H237. doi: 10.1152/ajpheart.1981.240.2.H229. [DOI] [PubMed] [Google Scholar]
- Takabe W, Kanai Y, Chairoungdua A, Shibata N, Toi S, Kobayashi M, Kodama T, Noguchi N. Lysophosphatidylcholine enhances cytokine production of endothelial cells via induction of L-type amino acid transporter 1 and cell surface antigen 4F2. Arterioscler Thromb Vasc Biol. 2004;24:1640–1645. doi: 10.1161/01.ATV.0000134377.17680.26. [DOI] [PubMed] [Google Scholar]
- Zhu K, Baudhuin LM, Hong G, Williams FS, Cristina KL, Kabarowski JH, Witte ON, Xu Y. Sphingosylphosphorylcholine and lysophosphatidylcholine are ligands for the G protein-coupled receptor GPR4. J Biol Chem. 2001;276:41325–41335. doi: 10.1074/jbc.M008057200. [DOI] [PubMed] [Google Scholar]
- Lum H, Qiao J, Walter RJ, Huang F, Subbaiah PV, Kim KS, Holian O. Inflammatory stress increases receptor for lysophosphatidylcholine in human microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;285:H1786–H1789. doi: 10.1152/ajpheart.00359.2003. [DOI] [PubMed] [Google Scholar]
- Kabarowski JH, Zhu K, Le LQ, Witte ON, Xu Y. Lysophosphatidylcholine as a ligand for the immunoregulatory receptor G2A. Science. 2001;293:702–705. doi: 10.1126/science.1061781. [DOI] [PubMed] [Google Scholar]
- Ogita T, Tanaka Y, Nakaoka T, Matsuoka R, Kira Y, Nakamura M, Shimizu T, Fujita T. Lysophosphatidylcholine transduces Ca2+ signaling via the platelet-activating factor receptor in macrophages. Am J Physiol. 1997;272:H17–H24. doi: 10.1152/ajpheart.1997.272.1.H17. [DOI] [PubMed] [Google Scholar]
- Huang YH, Schafer-Elinder L, Wu R, Claesson HE, Frostegard J. Lysophosphatidylcholine (LPC) induces proinflammatory cytokines by a platelet-activating factor (PAF) receptor-dependent mechanism. Clin Exp Immunol. 1999;116:326–331. doi: 10.1046/j.1365-2249.1999.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Barak LS, Jolly PS, Liu H, Lynch KR, Lacana E, Suhr KB, Milstien S, Spiegel S. The G protein-coupled receptor GPR4 suppresses ERK activation in a ligand-independent manner. Biochemistry. 2003;42:12181–12191. doi: 10.1021/bi035051y. [DOI] [PubMed] [Google Scholar]
- Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–98. doi: 10.1038/nature01905. [DOI] [PubMed] [Google Scholar]
- Witte ON, Kabarowski JH, Xu Y, Le LQ, Zhu K. Retraction. Science. 2005;307:206. doi: 10.1126/science.307.5707.206b. [DOI] [PubMed] [Google Scholar]
- Stoll LL, Oskarsson HJ, Spector AA. Interaction of lysophosphatidylcholine with aortic endothelial cells. Am J Physiol. 1992;262:H1853–H1860. doi: 10.1152/ajpheart.1992.262.6.H1853. [DOI] [PubMed] [Google Scholar]
- Kugiyama K, Kerns SA, Morrisett JD, Roberts R, Henry PD. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature. 1990;344:160–162. doi: 10.1038/344160a0. [DOI] [PubMed] [Google Scholar]
- Flavahan NA. Lysophosphatidylcholine modifies G protein-dependent signaling in porcine endothelial cells. Am J Physiol. 1993;264:H722–H727. doi: 10.1152/ajpheart.1993.264.3.H722. [DOI] [PubMed] [Google Scholar]
- Inoue N, Hirata K, Yamada M, Hamamori Y, Matsuda Y, Akita H, Yokoyama M. Lysophosphatidylcholine inhibits bradykinin-induced phosphoinositide hydrolysis and calcium transients in cultured bovine aortic endothelial cells. Circ Res. 1992;71:1410–1421. doi: 10.1161/01.res.71.6.1410. [DOI] [PubMed] [Google Scholar]
- Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol. 2003;23:218–223. doi: 10.1161/01.atv.0000052673.77316.01. [DOI] [PubMed] [Google Scholar]
- Inoue N, Takeshita S, Gao D, Ishida T, Kawashima S, Akita H, Tawa R, Sakurai H, Yokoyama M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis. 2001;155:45–52. doi: 10.1016/s0021-9150(00)00530-x. [DOI] [PubMed] [Google Scholar]
- Murugesan G, Sandhya Rani MR, Gerber CE, Mukhopadhyay C, Ransohoff RM, Chisolm GM, Kottke-Marchant K. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J Mol Cell Cardiol. 2003;35:1375–1384. doi: 10.1016/j.yjmcc.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Bassa BV, Roh DD, Vaziri ND, Kirschenbaum MA, Kamanna VS. Lysophosphatidylcholine activates mesangial cell PKC and MAP kinase by PLCgamma-1 and tyrosine kinase-Ras pathways. Am J Physiol. 1999;277:F328–F337. doi: 10.1152/ajprenal.1999.277.3.F328. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Kawashima S, Takahashi T, Ueyama T, Ishido S, Inoue N, Hirata K, Yokoyama M. Regulation of tyrosine phosphorylation of PYK2 in vascular endothelial cells by lysophosphatidylcholine. Am J Physiol Heart Circ Physiol. 2001;281:H266–H274. doi: 10.1152/ajpheart.2001.281.1.H266. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M, Fukuto J. Redox signaling. Mol Cell Biochem. 2002;234–235:49–62. [PubMed] [Google Scholar]
- Landar A, Darley-Usmar VM. Nitric oxide and cell signaling: modulation of redox tone and protein modification. Amino Acids. 2003;25:313–321. doi: 10.1007/s00726-003-0019-7. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol. 2000;20:7311–7318. doi: 10.1128/mcb.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- Chang SH, Garcia J, Melendez JA, Kilberg MS, Agarwal A. Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. Biochem J. 2003;371:877–885. doi: 10.1042/BJ20021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Thomas SR, Albano A, Murphy MP, Keaney JF., Jr Mitochondrial function is required for hydrogen peroxide-induced growth factor receptor transactivation and downstream signaling. J Biol Chem. 2004;279:35079–35086. doi: 10.1074/jbc.M404859200. [DOI] [PubMed] [Google Scholar]
- Fang Y, Han SI, Mitchell C, Gupta S, Studer E, Grant S, Hylemon PB, Dent P. Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology. 2004;40:961–971. doi: 10.1002/hep.20385. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Ng DC, Court NW, Draper KA, Dhillon A, Abas L. Intact mitochondrial electron transport function is essential for signalling by hydrogen peroxide in cardiac myocytes. J Mol Cell Cardiol. 2000;32:1469–1480. doi: 10.1006/jmcc.2000.1187. [DOI] [PubMed] [Google Scholar]
- Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- Yamakawa T, Tanaka S, Yamakawa Y, Kamei J, Numaguchi K, Motley ED, Inagami T, Eguchi S. Lysophosphatidylcholine activates extracellular signal-regulated kinases 1/2 through reactive oxygen species in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2002;22:752–758. doi: 10.1161/01.atv.0000015903.02749.71. [DOI] [PubMed] [Google Scholar]
- Wolfram Kuhlmann CR, Wiebke Ludders D, Schaefer CA, Kerstin Most A, Backenkohler U, Neumann T, Tillmanns H, Erdogan A. Lysophosphatidylcholine-induced modulation of Ca(2+)-activated K(+) channels contributes to ROS-dependent proliferation of cultured human endothelial cells. J Mol Cell Cardiol. 2004;36:675–682. doi: 10.1016/j.yjmcc.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Oxidized low density lipoprotein induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. Am J Physiol Heart Circ Physiol. 2005;289:H852–H861. doi: 10.1152/ajpheart.00015.2005. [DOI] [PubMed] [Google Scholar]
- Abas L, Bogoyevitch MA, Guppy M. Mitochondrial ATP production is necessary for activation of the extracellular-signal-regulated kinases during ischaemia/reperfusion in rat myocyte-derived H9c2 cells. Biochem J. 2000;349:119–126. doi: 10.1042/0264-6021:3490119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe W, Kodama T, Hamakubo T, Tanaka K, Suzuki T, Aburatani H, Matsukawa N, Noguchi N. Anti-atherogenic antioxidants regulate the expression and function of proteasome alpha-type subunits in human endothelial cells. J Biol Chem. 2001;276:40497–40501. doi: 10.1074/jbc.M104882200. [DOI] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- Tampo Y, Kotamraju S, Chitambar CR, Kalivendi SV, Keszler A, Joseph J, Kalyanaraman B. Oxidative stress-induced iron signaling is responsible for peroxide-dependent oxidation of dichlorodihydrofluorescein in endothelial cells: role of transferrin receptor-dependent iron uptake in apoptosis. Circ Res. 2003;92:56–63. doi: 10.1161/01.res.0000048195.15637.ac. [DOI] [PubMed] [Google Scholar]
- Iles KE, Dickinson DA, Wigley AF, Welty NE, Blank V, Forman HJ. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic Biol Med. 2005;39:355–364. doi: 10.1016/j.freeradbiomed.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RJ, Monnier JM, Nick HS. Tumor necrosis factor-alpha selectively induces MnSOD expression via mitochondria-to-nucleus signaling, whereas interleukin-1beta utilizes an alternative pathway. J Biol Chem. 2001;276:20419–20427. doi: 10.1074/jbc.M008915200. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–L496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- Cominacini L, Garbin U, Pasini AF, Davoli A, Campagnola M, Pastorino AM, Gaviraghi G, Lo Cascio V. Oxidized low-density lipoprotein increases the production of intracellular reactive oxygen species in endothelial cells: inhibitory effect of lacidipine. J Hypertens. 1998;16:1913–1919. doi: 10.1097/00004872-199816121-00010. [DOI] [PubMed] [Google Scholar]
- Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation: 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- Oka H, Kugiyama K, Doi H, Matsumura T, Shibata H, Miles LA, Sugiyama S, Yasue H. Lysophosphatidylcholine induces urokinase-type plasminogen activator and its receptor in human macrophages partly through redox-sensitive pathway. Arterioscler Thromb Vasc Biol. 2000;20:244–250. doi: 10.1161/01.atv.20.1.244. [DOI] [PubMed] [Google Scholar]
- Colles SM, Chisolm GM. Lysophosphatidylcholine-induced cellular injury in cultured fibroblasts involves oxidative events. J Lipid Res. 2000;41:1188–1198. [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- Usatyuk PV, Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J Biol Chem. 2004;279:11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- Noguchi N, Yoshida Y, Kaneda H, Yamamoto Y, Niki E. Action of ebselen as an antioxidant against lipid peroxidation. Biochem Pharmacol. 1992;44:39–44. doi: 10.1016/0006-2952(92)90035-h. [DOI] [PubMed] [Google Scholar]
- Day BJ, Batinic-Haberle I, Crapo JD. Metalloporphyrins are potent inhibitors of lipid peroxidation. Free Radic Biol Med. 1999;26:730–736. doi: 10.1016/s0891-5849(98)00261-5. [DOI] [PubMed] [Google Scholar]
- Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- Yet SF, Layne MD, Liu X, Chen YH, Ith B, Sibinga NE, Perrella MA. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17:1759–1761. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]