

Abstract
The Polycomb group (PcG) proteins are essential for embryogenesis, and their expression is often found deregulated in human cancer. The PcGs form two major protein complexes, called polycomb repressive complexes 1 and 2 (PRC1 and PRC2) whose function is to maintain transcriptional repression. Here, we demonstrate that the chromodomain-containing protein, CBX8, which is part of one of the PRC1 complexes, regulates proliferation of diploid human and mouse fibroblasts through direct binding to the INK4A-ARF locus. Furthermore, we demonstrate that CBX8 is limiting for the regulation of INK4A-ARF, and that ectopic expression of CBX8 leads to repression of the Ink4a-Arf locus and bypass of senescence, leading to cellular immortalization. Gene expression and location analysis demonstrate that besides the INK4A-ARF locus, CBX8 also regulates a number of other genes important for cell growth and survival. On the basis of these results, we conclude that CBX8 is an essential component of one of the PRC1 complexes, which directly regulate the expression of numerous target genes, including the INK4A-ARF locus, involved in cell-fate decisions.
Keywords: ARF, CBX8, INK4A, Polycomb, senescence
Introduction
Cellular senescence is a fundamental program activated in normal cells in response to various types of stress such as telomere uncapping, oxidative stress, oncogene activity and DNA damage (Serrano and Blasco, 2001; Ben-Porath and Weinberg, 2005). Senescence can occur following a prolonged period of cellular proliferation (replicative senescence) or more instantly in response to acute stress. When cells have entered senescence, they cease to divide and undergo a series of morphologic and metabolic changes. Cellular senescence is thought to play an important role in tumor suppression and to contribute to aging of the organism (Campisi, 2000). Recent studies have provided important insights to how different types of stress and stimuli activate signaling pathways leading to senescence. It appears that these stress-signaling pathways are funneled through p53 and pRB (Narita et al, 2003), whose combined levels of activity determine whether cells enter senescence or remain in a state competent for proliferation (Dirac and Bernards, 2003).
BMI1, one of the Polycomb group (PcG) proteins, was originally identified as an oncogene collaborating with Myc in inducing B-cell lymphomas (Haupt et al, 1991; van Lohuizen et al, 1991). Several results have subsequently shown that the PcG proteins have a role in regulating normal proliferation and can contribute to the development of cancer (Bea et al, 2001; van Kemenade et al, 2001; Vonlanthen et al, 2001; Bracken et al, 2003; Kleer et al, 2003). BMI1 is overexpressed in different types of human cancer, and genetic studies have shown that it affects cell proliferation and senescence through repression of the Ink4a-Arf tumor suppressor locus (Jacobs et al, 1999).
The PcG proteins are part of two distinct protein complexes, named polycomb repressive complexes 1 and 2 (PRC1 and PCRC2). In humans, PRC2 contains the three PcG proteins (EZH2, EED and SUZ12), and RbAp48 (Cao et al, 2002; Czermin et al, 2002; Kuzmichev et al, 2002; Muller et al, 2002), whereas the PRC1 complex, which likely exists in many variant forms due to a large number of homologues in mammalian cells contains at least six different subunits: the polyhomeotic- (HPH1-3), the polycomb-/CBX (HPC1/CBX2, HPC2/CBX4, HPC3/CBX8, CBX6 and CBX7), the RING1- and 2-(RING1A/B), the posterior sex comb- (BMI1, MEL18, MBLR and NSPC1) and sex comb on midleg (SCML1–2) proteins (Levine et al, 2002).
EZH2 of the PRC2 complex possesses histone methyltransferase activity and di- and trimethylates lysine 27 on histone H3 (H3K27me2/3). Trimethylated H3K27 is believed to facilitate the recruitment of the PRC1 complex by serving as a docking site recognized by the chromodomains of a subset of the CBX proteins (Fischle et al, 2003; Bernstein et al, 2006). The recruitment of PRC1 is essential for the maintenance of repression of target genes during differentiation and development (Valk-Lingbeek et al, 2004).
Despite their potential important regulatory function, the five CBX proteins are poorly characterized. Interestingly, CBX7 was recently shown to extend the lifespan of human primary epithelial cells via repression of the INK4A-ARF locus by a BMI1-independent mechanism (Gil et al, 2004). In contrast to the growth promoting role of CBX7, CBX4 appears to work as a tumor suppressor through repression of the Myc promoter (Satijn et al, 1997).
Here we demonstrate that CBX8 is part of a BMI1-containing PRC1 complex required for growth of human and mouse diploid fibroblasts. Furthermore, we show that CBX8 directly binds to and represses the INK4A-ARF locus. Ectopic expression of CBX8 prevents oncogene- and stress-induced senescence, and we show that CBX8 regulates proliferation through p16Ink4a and p19Arf dependent and -independent mechanisms. Consistent with this, we have identified a number of known and putative tumor suppressor genes as being bound and regulated by CBX8 in human diploid fibroblasts. On the basis of these results, we conclude that CBX8 is a growth-promoting gene and an essential component of a PRC1-type complex.
Results
CBX8 is required for proliferation of human diploid fibroblasts
BMI1, a component of the PRC1 complex, and the three core members of the PRC2 complex, EZH2, EED and SUZ12, are all essential for cell proliferation in human diploid fibroblasts (Jacobs et al, 1999; Bracken et al, 2003; Pasini et al, 2004; Liu et al, 2006). To determine whether CBX8, previously shown to interact with Ring1 in vitro and containing gene repressor activity (Bardos et al, 2000), is required for cell proliferation, we transduced telomerase-immortalized TIG3 (TIG3-T) human diploid fibroblasts with a shRNA construct targeting CBX8. Interestingly, we found that inhibition of CBX8 expression dramatically reduced the S-phase fraction of cells (Figure 1A) and led to a complete growth arrest (Figure 1B). A similar effect was observed when the expression of SUZ12 and BMI1 was inhibited. Furthermore, inhibition of CBX8 led to lower levels of the two S-phase cyclins, cyclin A2 and cyclin E1, and disappearance of hyperphosphorylated pRB (Figure 1C), indicative of a G1 block (Figure 1A). Because BMI1 has previously been shown to affect cell proliferation through repression of the INK4A-ARF locus, we tested whether CBX8 and SUZ12 would affect the same locus. Indeed, cell cultures with reduced CBX8 or SUZ12 expression showed increased levels of p16INK4A protein, and in agreement with previous results we found that downregulation of BMI1 expression also led to increased levels of p16INK4A (Figure 1C) (Jacobs et al, 1999; Liu et al, 2006).
Figure 1.
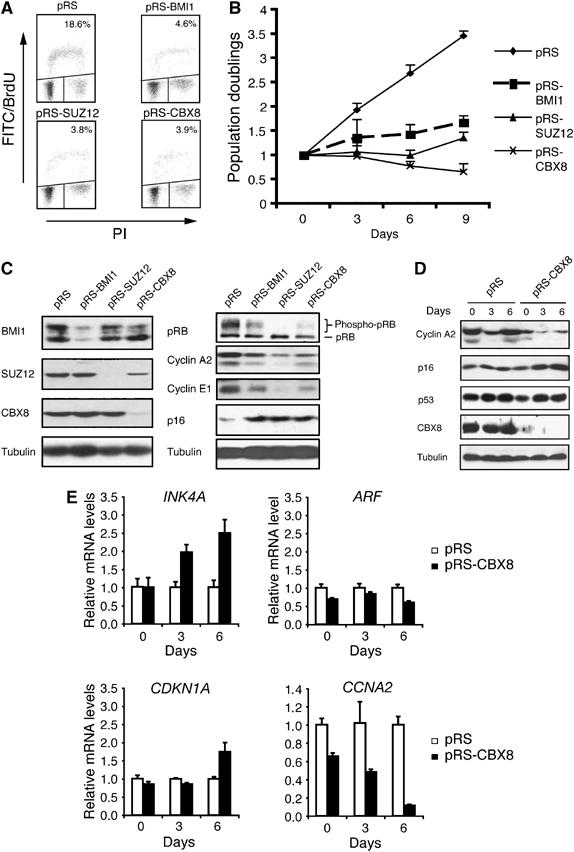
CBX8 is required for cell proliferation in human diploid fibroblasts. (A) TIG3-T cells were infected with shRNA constructs (pRS) targeting CBX8, SUZ12 and BMI1 as indicated and pulsed with BrdU at day 1, see panel B. The S-phase fraction of cells was evaluated by FACS analysis. (B) Growth curve of TIG3-T cells that were infected with shRNA constructs (pRS) targeting CBX8, SUZ12 and BMI1 are indicated. Cells were infected over 2 days, selected for 3 days (2 μg/ml puromycin) and plated for 3T3 assay (day 0 corresponds to 3 days after start of selection; the day of plating for the 3T3 assay). (C) Protein extracts were prepared at day 6 (depicted in (B)). Samples were processed for Western blot analysis using antibodies as indicated. (D, E) Relative protein (D) and mRNA (E) levels in TIG3-T cells treated with control (pRS) and shRNA targeting CBX8 (pRS-CBX8). Total protein and RNA was harvested at different time points after infection corresponding to days 0, 3 and 6 in (B). The relative expression levels of the indicated genes were determined by Western blot analysis (D) and RT-QPCR (E).
To reveal the dynamics of events after downregulation of CBX8, we determined the relative expression levels of cyclin A2, p16INK4A, p14ARF, p53 and p21CIP1 at different times after inhibiting CBX8 expression. Whereas inhibition of CBX8 expression led to a rapid decrease of cyclin A2 mRNA and protein levels (already observed at day 0), p16INK4A levels were increasing only after 3 and 6 days (Figure 1D and E). This indicated that the primary growth arrest induced by inhibition of CBX8 expression was not mediated by p16INK4A. In diploid fibroblasts, ARF is expressed at very low levels and we were therefore unable to detect the protein by Western blotting (Supplementary Figure 2A). However, the quantification of p14ARF mRNA levels showed that it was slightly decreased as a result of CBX8 downregulation (Figure 1E and Supplementary Figures 1 and 2). Moreover, we did not find any changes in p21CIP1 mRNA levels at day 0 and 3 (Figure 1E), suggesting that the p14ARF/MDM2/p53 pathway is not involved in the early growth arrest. By Western blotting we observed a small increase in p53 levels between days 0 and day 3, although this was identical between pRS-CBX8 and pRS control-treated cells (Figure 1D). Taken together, these results demonstrate that the inhibition of CBX8 expression in human fibroblasts does not lead to detectable activation of the p14ARF/MDM2/p53 pathway.
CBX8 and BMI1 directly target the INK4A-ARF locus
Knowing that loss of CBX8 leads to increased expression of p16INK4A in human TIG3-T cells, we next asked whether CBX8 was directly binding to the locus. The INK4A-ARF locus encodes two gene products, each having a unique first exon but share the second and third exons (Figure 2A). The locus covers more than 25 kb and to determine if and where CBX8 binds, we scanned the whole locus by chromatin immunoprecipitation (ChIP) experiments and location analysis. This showed that CBX8 binds to several regions along the locus with a peak after the first exon of INK4A (Bracken et al, submitted). Using this knowledge, we performed a direct ChIP amplifying the precipitated DNA with primers downstream of the first exon of the INK4A gene (Figure 2A). Interestingly, we found that both CBX8 and BMI1 bound this region of the INK4A gene in human and mouse fibroblasts (Figure 2B). The specificity of the BMI1 and CBX8 antibodies in the ChIP assay was confirmed by inhibiting the expression of the two proteins (Figure 2C). Remarkably, downregulation of CBX8 led to an almost complete loss of BMI1 on the INK4A locus and, furthermore, downregulation of BMI1 led to about 50% reduction of the CBX8 binding. Because IP experiments showed that CBX8 and BMI1 are part of a common complex (Figure 3B and Supplementary Figure 3A and B), these results suggest that both proteins are needed in the complex to achieve binding and repression of the INK4A gene. Affinity purification of Flag-Myc epitope-tagged CBX8 expressed in 293T cells, furthermore, led to the identification of a number of previously characterized PRC1 components such as Ring1A/B, Polyhomeotic-like 1-3, and reconfirmed the interaction with BMI1 (Supplementary Figure 4A and Supplementary Tables IV and V). Together with the fact that the endogenous CBX8 protein elutes in high-molecular-weight fractions of approximately 2 MDa by size-exclusion chromatography (Supplementary Figure 4B), as has been reported for other PRC1 complexes, these data strongly support that CBX8 is part of a PRC1-like repressor complex. Interestingly, two other members of CBX family, CBX4 and CBX7, were also found to bind the INK4A gene locus (Figure 2D), suggesting that several CBX family members contribute to the regulation of the locus.
Figure 2.
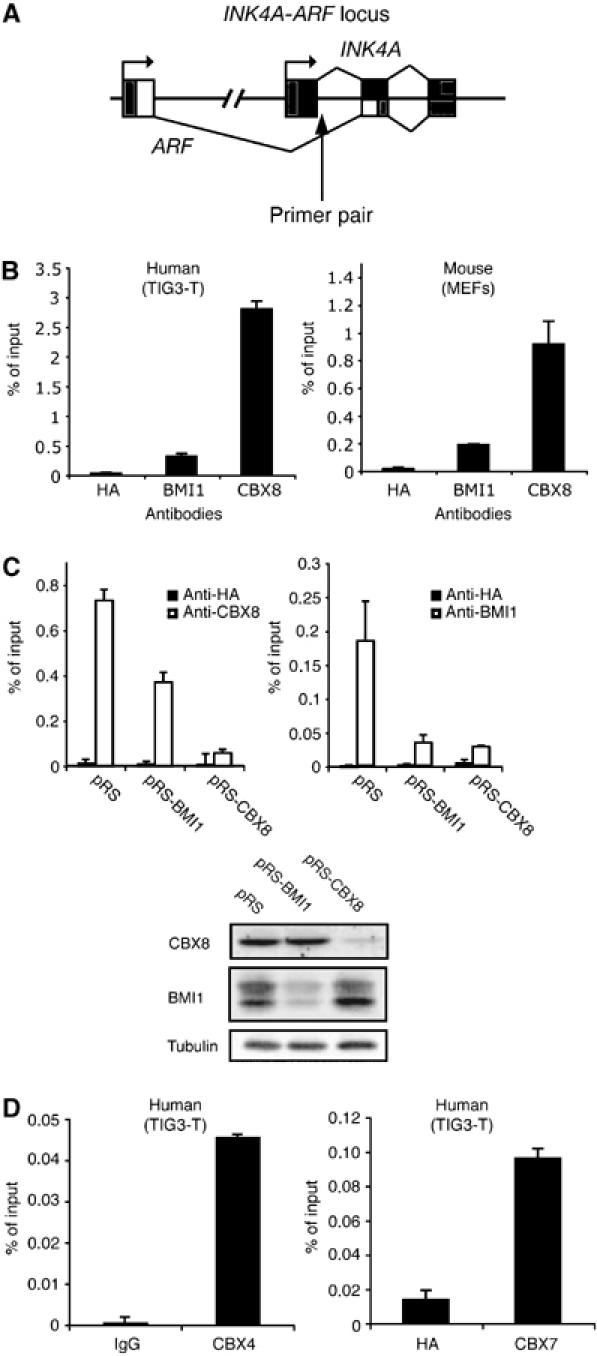
CBX8 and BMI1 directly bind the INK4A-ARF locus in human and mouse diploid fibroblasts. (A) Graphic illustration of the INK4A-ARF locus and the primer pair used for chromatin immunoprecipitation (ChIP) analysis in (B–D). (B) INK4A ChIP analysis in TIG3-T cells and MEFs using CBX8 and BMI1 antibodies. Anti-HA antibody was used as a negative control. (C) ChIP analysis of TIG3-T cells treated with pRS (control), pRS-CBX8 and pRS-BMI1. Left panel: anti-CBX8 and anti-HA. Right panel: anti-BMI1 and anti-HA. Lower panel: Western blot analysis of CBX8 and BMI1 in total protein extracts from the cells used for ChIP analysis in upper panels. (D) INK4A ChIP analysis in TIG3-T using either anti-CBX4 (left panel) or anti-CBX7 (right panel). ChIP data are presented as percentage of input DNA.
Figure 3.

Colocalization of CBX8 and BMI1 in Polycomb bodies. (A) TIG3-T cells were infected with control (pRS) or shRNA targeting CBX8 (pRS-CBX8). Puromycin-selected cells were fractionated into soluble and insoluble proteins. Input (total lysate), insoluble and soluble protein fractions were loaded as indicated, and the levels of CBX8, histone H3, GAPDH and BMI1 were revealed by Western blot analysis. (B) Immunodepletion of BMI1 and CBX8 from TIG3-T protein extracts. Three successive immunoprecipitations (IPs) were performed (IPs 1–3), followed by IP of CBX8 in the BMI1-depleted extract and of BMI1 in the CBX8-depleted extract. Extracts before depletion (BD) and after depletion (AD, after the third IP) were loaded next to each other to reveal differences in protein levels (*background, crosslinked IgG heavy and light chains, 75 kDa). (C) Colocalization of CBX8 with either BMI1, RING1B, HPH1 or HPH2 in pre-extracted U2OS cells. U2OS cells grown on coverslips were pre-extracted, fixed and incubated with rabbit anti-CBX8 in combination with either mouse anti-BMI1, anti-RING1B, anti-HPH1 or anti-HPH2. Anti-rabbit Alexa-594 and anti-mouse Alexa-488 were used for detection. Coverslips were stained with DAPI, mounted and analyzed by confocal microscopy. (D) U2OS cells grown on coverslips were cotransfected with H2B-GFP and the indicated shRNA constructs. Cells were pre-extracted and fixed as in (C). For detection of BMI1, RING1B or HPH1 and CBX8, anti-rabbit Alexa-650 and anti-mouse Alexa-568 were used as secondary antibodies. After staining, coverslips were mounted and analyzed by confocal microscopy.
BMI1 is partially dependent on CBX8 for chromatin association
As BMI1 displays a strong dependency on CBX8 for its association to a specific target gene (INK4A-ARF), we tested the impact of CBX8 downregulation on the amount of BMI1 associated with chromatin. We infected TIG3-T cells with a control (pRS) retrovirus or one expressing CBX8 shRNA and fractionated cells into soluble and insoluble protein fractions. The results showed that almost all CBX8 was present in the insoluble fraction (in the control), where histone H3 was also present (Figure 3A). As a control for the efficiency of extraction, we used GAPDH as a marker for soluble proteins. In TIG3-T cells, approximately one-third of the total BMI1 was in the soluble fraction, and this amount increased to about 50% in cells in which CBX8 expression was downregulated.
Because a substantial amount of BMI1 was still present in the insoluble fraction in the absence of CBX8, we reasoned that not all BMI1 was in a complex with CBX8 in the chromatin fraction. To analyze this in more detail, we immunodepleted either CBX8 or BMI1 from TIG3-T extracts followed by IP of BMI1 or CBX8, respectively. Western blot (WB) analysis of protein extracts before depletion (BD) and after depletion (AD) showed a reduction of about 60% of total CBX8 after BMI1 depletion and a reduction of about 20% of total BMI1 after CBX8 depletion. This confirmed our prediction that there was a considerable amount of BMI1, which was not in complex with CBX8 and a significant amount of CBX8 that does not appear to be associated with BMI1. We furthermore found that two other members of the CBX family, CBX4 and CBX7, co-immunoprecipitated with BMI1 in both non-depleted and CBX8-depleted extracts, indicating that BMI1 can exist in different subtypes of PRC1 complexes (Figure 3B).
The mammalian PRC1 complex (HPC2/CBX4, RING1, BMI1 and HPH1-2) has been shown to localize to heterochromatic areas in discrete foci, termed PcG bodies (Saurin et al, 1998; Isono et al, 2005). In U2OS cells pre-extracted to remove soluble nuclear proteins before fixation, CBX8 both localized in PcG bodies and, furthermore, displayed a more general distribution throughout the nucleus (Figure 3C). In PcG bodies, we found complete colocalization between CBX8 and several members of the PRC1 complex (BMI1, RING1B, HPH1 and HPH2). To test the effect of CBX8 downregulation on the formation of PcG bodies, we cotransfected U2OS cells with shRNA constructs targeting either BMI1 or CBX8 with a GFP-tagged version of histone H2B. We observed a drastic reduction of BMI1-positive PcG bodies in cells with downregulated CBX8 (Figure 3D). Downregulation of BMI1 expression also led to a reduction in CBX8-positive PcG bodies, suggesting that both BMI1 and CBX8 are required in a complex to form these structures. Consistent with CBX8 playing an important role in PcG bodies, we also observed that downregulation of CBX8 delocalized RING1B and HPH1 from the PcG body structures (Figure 3D).
Ectopic expression of CBX8 bypasses stress-induced senescence
The Ink4a-Arf locus is critical for stress-induced senescence in both human and mouse cells (Lundberg et al, 2000). To test whether ectopic expression of CBX8 leads to repression of the Ink4a-Arf locus and as a consequence immortalization of MEFs, we infected early-passage MEFs with human CBX8 and evaluated the growth potential of control and CBX8 overexpressing cultures. As shown in Figure 4A, overexpression of CBX8 bypassed senescence, and the growth rate of the CBX8 expressing cells did not change within the time frame of the experiment. Furthermore, ectopic expression of CBX8 still affected cell proliferation in late-passage MEFs, as demonstrated in Figure 4B, as pRS-CBX8 transduction of the FM-CBX8-expressing cells slowed down their growth.
Figure 4.
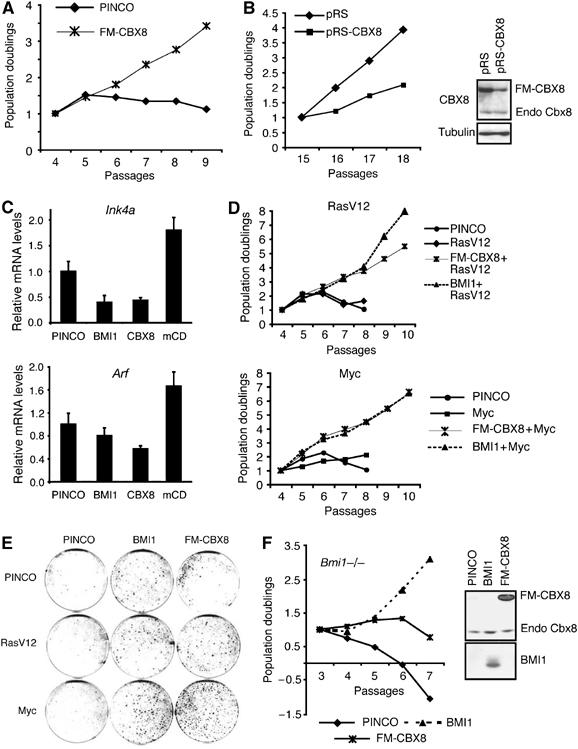
CBX8 overexpression bypasses stress-induced senescence and immortalize primary MEFs. (A) 3T3 assay of MEFs infected with control (PINCO) virus or FM-CBX8 expressing virus. Cell cultures were kept on a 3T3 protocol counting cells at each passage. (B) FM-CBX8 expressing cells kept in culture until passage 14 were infected with control or shRNA targeting the overexpressed human CBX8 mRNA and kept on a 3T3 protocol from passage 15 to 19. Left panel: 3T3 assay of pRS (control) and pRS-CBX8-infected cells. Right panel: Western blot analysis of CBX8 from pRS or pRS-CBX8 cultures at passage 18. (C) The relative mRNA levels of 16Ink4a and p19Arf were determined by quantitative real-time RT-QPCR in MEFs overexpressing BMI1, CBX8 or CBX8 mCD. (D) 3T3 assay of MEFs infected with CBX8 or BMI1 in combination with oncogenic Ras (RasV12) or Myc as indicated. (E) Colony assay of MEFs infected with CBX8 or BMI1 in combination with oncogenic Ras (RasV12) or Myc as indicated. (F) Left panel: 3T3 assay of Bmi1−/− MEFs infected with control (PINCO), BMI1 or FM-CBX8 virus. Right panel: Western blot analysis of the expression of BMI1 and CBX8.
Consistent with binding to the Ink4a-Arf locus, CBX8 expression led to a decrease in Ink4a and Arf mRNA levels (Figure 4C). We found that point mutations in the chromodomain of CBX8 (mCD) abrogated its repressive function (Figure 4C) and its ability to bypass stress-induced senescence (Supplementary Figure 3A). Interestingly, the CBX8 mCD mutant appeared to work as dominant negative, as it induced the expression of both Ink4a and Arf (Figure 4C). Taken together, these results demonstrate that the ability of Cbx8 to bypass senescence in MEFs correlates with the repression of the Ink4A-Arf locus.
Cooperation between CBX8 and the oncogenes c-Myc and RasV12
Inappropriate mitogenic stimulation owing to expression of oncogenes such as RasV12 and c-Myc leads to growth arrest and a senescence-like phenotype in MEFs, in the absence of other genetic lesions (Drayton et al, 2004). As this phenotype is dependent on p19Arf and p16Ink4a, we tested if CBX8 could immortalize MEF expressing RasV12 or c-Myc and thereby collaborate with these oncogenes. Indeed, as shown in Figure 4D and E, CBX8, as BMI1, immortalized MEFs expressing RasV12 or c-Myc and collaborated with these oncogenes, as seen by the increased growth rates of cultures expressing either RasV12 or c-Myc together with CBX8, compared with CBX8 alone.
Having established that CBX8 expression can bypass stress-induced senescence, and that CBX8 and BMI1 display mutual dependency for their binding to the INK4A gene, we next tested whether the growth promoting activity of CBX8 was dependent on BMI1. For this we expressed CBX8 or BMI1 in Bmi1−/− MEFs (Jacobs et al, 1999). As expected, BMI1 rescued the growth retardation of Bmi1−/− MEFs. In contrast, however, Bmi1−/− MEFs expressing CBX8 proliferated very slowly for two passages and then went into a similar crisis as observed in control cultures (Figure 4F). These results demonstrate that CBX8 cannot compensate for the lack of Bmi1; however, it contains some growth promoting or survival functions that are independent of BMI1.
Ink4a-Arf-dependent and -independent functions of CBX8 in cell proliferation
To map genetically the critical targets for CBX8 in MEFs, we inhibited Cbx8 expression in wild type, Ink4a−/−, Arf−/−, (Ink4a−/−, Arf−/−) and p53−/− MEFs. As for human diploid fibroblasts, inhibition of Cbx8 expression in wild-type MEFs led to a proliferative arrest (Figure 5A–C), which was accompanied by reduced expression of cyclin A2 and a slight increase in p16Ink4a and p19Arf (Supplementary Figure 5A). In contrast, inhibition of Cbx8 expression in Arf−/−, (Ink4a−/−, Arf−/−) or p53−/−, but not in Ink4a−/−, MEFs resulted in minor changes in proliferation rates (Figure 5A–C). In conclusion, these results show that the loss of an intact Arf-p53 pathway allows proliferation of MEFs without Cbx8, and suggest that Arf is a critical target for Cbx8 in MEFs.
Figure 5.
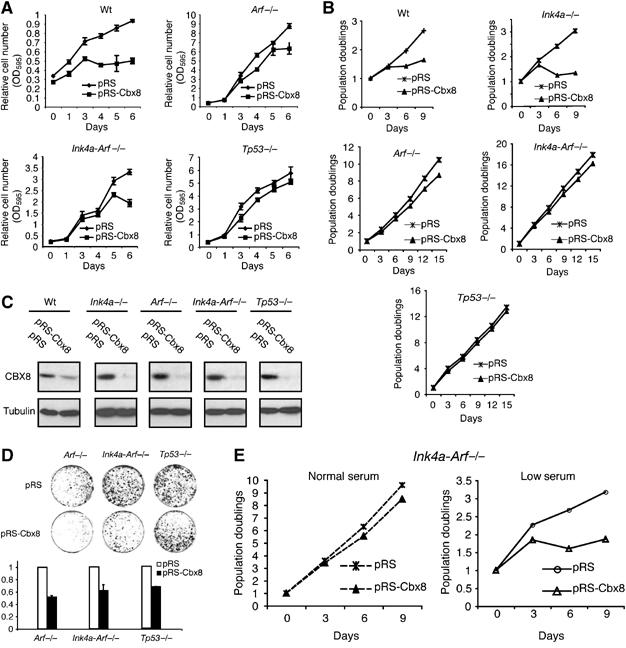
Ink4a-Arf-dependent and -independent functions of CBX8. (A) Growth curves of wild type (Wt), Arf−/− (Ink4, Arf−/−) and p53−/− MEFs infected with control (pRS) or pRS-Cbx8 shRNA constructs. Selected cultures were plated in triplicates (50 000 cells in each well of a six-well dish) and stained with crystal violet at the indicated time points after plating. Relative cell number is presented as OD595/ml. (B) 3T3 assays of wild type (Wt), Ink4a−/−, Arf−/−, (Ink4, Arf−/−) and p53−/− MEFs infected with control (pRS) or the pRS-Cbx8 shRNA construct. (C) Western blot showing the efficiency of the RNAi mediated Cbx8 downregulation in MEFs with the indicated genotypes. (D) Colony assay of Arf−/− (Ink4, Arf−/−) and p53−/− MEFs infected with control (pRS) or pRS-Cbx8 virus. Puromycin-selected cultures were plated in triplicates (10 000 cells/10-cm dish) and stained with crystal violet. Upper panel: representative plate image after 10 days in culture. Lower panel: color extracted from dishes shown above was measured by OD595. (E) 3T3 assay of (Ink4, Arf−/−) MEFs infected with control (pRS) or pRS-Cbx8 virus culture under normal (10% FBS) or low (2% FBS) serum conditions.
It is known that cells secrete factors that affect the proliferation of neighboring cells and that a hallmark of cellular immortalization is the ability to form colonies from single cells. When primary cells are plated sparsely they become stressed, stop proliferating and change morphology, displaying a senescence phenotype (Lundberg et al, 2000). Loss of a functional Arf-p53 pathway leads to cellular immortalization and higher cloning efficiency. To test if Cbx8 is required for the high- cloning efficiency of MEFs without a functional Arf-p53 pathway and therefore may have critical targets in addition to Arf, Cbx8 expression was inhibited in Arf−/−, (Ink4a−/−, Arf−/−) or p53−/− MEFs. Remarkably, we found that Cbx8 downregulation led to a significant reduction in the number of colonies in all the genetic backgrounds tested (Figure 5D). In support of this, we obtained similar results in U2OS cells (Supplementary Figure 6), in which the INK4A-ARF locus is silenced by DNA methylation (Park et al, 2002). Furthermore, (Ink4a−/−, Arf−/−) MEFs do not proliferate in low serum, when Cbx8 expression is inhibited (Figure 5E). Taken together, these results suggest that Cbx8 controls the expression of genes, in addition to Ink4a-Arf, which are involved in regulating the cellular response to stress.
Identification of target genes whose expression is regulated by CBX8
To identify genes whose expression changes upon ectopic expression of CBX8, microarray gene expression analysis was performed on MEFs overexpressing CBX8. As our experiments suggest that CBX8 to a large extent exerts its function by forming a complex with BMI1, we also included MEFs overexpressing BMI1. The results from the expression profiling showed CBX8 and BMI1 significantly repressed 748 and 337 genes, respectively. In agreement with CBX8 and BMI1 being part of a common PRC1 complex, about 50% of the genes repressed by BMI1 were also repressed by CBX8 and about 20% of the genes repressed by CBX8 were also repressed by BMI1 (Figure 6A). Recently, we published a genome-wide location analysis screen, which revealed 3000 potential PcG target genes in TIG3-T (Bracken et al, 2006). Combining the expression profile analysis with the location analysis, we found 93 genes, which are repressed by CBX8 or BMI1 and which were bound by CBX8 and/or possessing the H3K27me3 mark. Interestingly, nine (one of them being the INK4A-ARF locus) of the 93 genes are known as potential tumor suppressors, found to be deleted or silenced in cancer and shown to be associated with either decreased cell survival or proliferation (Table I).
Figure 6.

Identification of genes regulated by CBX8 and BMI1. (A) A Venn diagram showing the overlap between genes found to be repressed >1.2-fold by microarray expression profile analysis and 3366 genes identified by ChIP on a chip analysis (Bracken et al, 2006) to be positive for CBX8 binding or the H3K27me3 mark. (B) Direct ChIP in TIG3-T cells using BMI1, CBX8 and H3K27me3 antibodies for the genes listed in Table I. (C) ChIP using anti-CBX8 in TIG3-T treated with RNAi targeting BMI1 (pRS-BMI1) or CBX8 (pRS-CBX8). (D) RT–QPCR showing the relative mRNA levels of the selected genes from Table I in TIG3-T after RNAi-mediated downregulation of CBX8.
Table 1.
List of genes deleted or silenced in cancer and directly regulated by CBX8
| Gene name | CBX8 versus mock (fold change) | P-value | BMI1 versus mock (fold change) | P-value | Function | References |
|---|---|---|---|---|---|---|
| CDKN2A | −1.77 | 0.035 | −1.66 | 0.053 | Growth arrest | Lundberg et al (2000) |
| PERP | −2.25 | 0.001 | −1.56 | 0.005 | Apoptosis | Hildebrandt et al (2000) |
| GADD45G | −1.22 | 0.049 | −1.15 | 0.119 | Growth arrest | Ying et al (2005) |
| UNC-5B | −1.49 | 0 | 1.14 | 0.349 | Apoptosis | Thiebault et al (2003) |
| MTUS1 | −1.82 | 0.03 | −1.42 | 0.644 | Growth arrest | Seibold et al (2003) |
| GJB2 | −2.1 | 0.007 | −1.39 | 0.052 | Cell-to-cell signaling | Miyamoto et al (2005) |
| SORBS1 | −1.55 | 0.006 | −1.34 | 0.035 | Insulin signaling | Vanaja et al (2006) |
| ALDH1A2 | −1.64 | 0.011 | 1.29 | 0.081 | Involved in RA synthesis | Kim et al (2005) |
| PTGIS | −1.35 | 0.043 | 1.19 | 0.001 | Involved in prostacyclin synthesis | Frigola et al (2005) |
To confirm that CBX8 regulates the expression of the eight potential tumor suppressors, we performed direct ChIP experiments. These data showed that all eight genes are directly bound by CBX8 and contained the H3K27me3 repressive mark. The enrichment of BMI1 was less pronounced than CBX8 and only half of the genes displayed significant enrichment (Figure 6B). Nevertheless, on all of these targets, as for the INK4A, CBX8 binding was dependent on BMI1 (Figure 6C), showing that these genes are bona fide CBX8/BMI1 targets. By real-time quantitative PCR analysis, we found that CBX8 downregulation led to a significant increase in expression of four out of the eight genes tested (Figure 6D), whereas the others were either not detectable in TIG3-T cells or not significantly changed (data not shown).
Discussion
Here we show that the chromodomain containing polycomb protein CBX8 is a positive regulator of cell proliferation. Inhibition of CBX8 expression in mouse and human fibroblasts results in growth arrest, and ectopic expression of CBX8 bypasses stress-induced senescence in mice. The growth arrest can be separated into an early event that appears to be independent of p16INK4A and p14ARF and a later stable arrest that is dependent on the two tumor suppressor proteins. Consistent with this, we show that CBX8 in addition to binding to the INK4A-ARF locus, binds and represses the transcription of a number of other genes in diploid fibroblasts. We speculate that CBX8 in part regulates cell proliferation by binding to some of these additional genes.
CBX8 and BMI1 interact and bind the INK4A-ARF locus
Previous data have shown that inactivation of Bmi1 leads to increased levels of p16Ink4a/p19Arf expression resulting in premature senescence (Jacobs et al, 1999). In agreement with an earlier publication demonstrating that CBX8 colocalizes with BMI1 (Bardos et al, 2000), we show that CBX8 binds to BMI1 in human cells. Moreover, we demonstrate that both CBX8 and BMI1 associate with the INK4A-ARF locus in human and mouse cells, and that they are dependent on each other for binding. The finding that BMI1 is dependent on CBX8 supports the idea that the PRC1 complex is recruited to methylated lysines (H3K27me3) on the histone tail via its chromodomain-containing component(s). However, that CBX8 is dependent on BMI1 suggests that the chromodomain of CBX8 alone is not sufficient for its binding to the INK4A-ARF locus, and that a full complex is needed to achieve correct localization, conformation and/or stability of the complex. In addition, the incorporation of CBX8 in the PRC1 complex may lead to higher affinity of CBX8 for H3K27me3. Such a model may provide an explanation for the recent finding that the ‘isolated' chromodomain of CBX8 does not have any measurable affinity for the H3K27me3 mark (Bernstein et al, 2006), whereas CBX8 from nuclear extracts of HeLa cells is enriched on H3K27me3 peptides (KH Hansen, unpublished results).
Interestingly, the analysis of the CBX8 and BMI1 proteins showed that not all BMI1 is in complex with CBX8 and although CBX8 and BMI1 are mutually dependent on binding to the INK4A-ARF locus, they are only marginally dependent on each other for binding to chromatin. As both BMI1 and CBX8 have cellular homologues, a likely explanation is that they form part of other PRC1-like complexes. In agreement with this hypothesis, we have shown that both CBX4 and CBX7, independently of CBX8, associate with BMI1 in vivo, and likewise interact with the INK4A-ARF locus. The binding of CBX7 to BMI1 appears in contrast to a previous report (Gil et al, 2004), in which the authors found that CBX7 does not bind BMI1. The reason for this discrepancy is unknown, but may relate to the use of different antibodies and cell lines. Importantly, although BMI1 interacts with other CBX proteins than CBX8, we find that CBX8 is an essential repressor of the INK4A-ARF locus.
CBX8 overexpression bypasses cellular senescence
Senescence is considered to be a safeguard mechanism against cancer. The Ink4a-Arf tumor suppressor locus is central for the induction of senescence and both p16Ink4a and p19Arf levels are increased in senescent cells as compared with low-passage proliferating cells. BMI1 and CBX7 are both able to drive cells past replicative senescence through repression of the INK4A-ARF locus. Moreover, BMI1 can collaborate with oncogenic Ras and Myc in cell proliferation, and BMI1 is found amplified and overexpressed in cancer (Bea et al, 2001; Vonlanthen et al, 2001; Sanchez-Beato et al, 2006). Here, we have demonstrated that CBX8 is capable of bypassing oncogene- and stress-induced senescence in MEFs, through direct binding and repression of the Ink4a-Arf locus in a BMI1-dependent manner. Previously, it was shown that Cbx7 can mediate repression of the Ink4a-Arf locus and promote proliferation in the absence of Bmi1, indicating that CBX7 can be part of alternative complexes likely containing BMI1 homologues such as MEL18, MBLR or PCGF1. However, in contrast to CBX7 (Gil et al, 2004), we find that overexpression of CBX8 is not able to rescue the growth defects of Bmi1−/− MEFs, demonstrating differential functional features of the CBX proteins and the functional dependency of CBX8 on BMI1.
INK4A-ARF-dependent and -independent functions of CBX8 in mouse and human cells
The relative importance of p16INK4A-pRB and p14ARF-p53 tumor suppressor pathways seems to be dependent on cell type and species (Lundberg et al, 2000). The INK4A gene is more frequently mutated in human tumors than ARF, suggesting that p16INK4A is a more potent tumor suppressor in human cells than ARF (Gil and Peters, 2006). However, in mouse, inactivation of Arf alone leads to bypass of senescence, and Arf−/− MEFs are efficiently transformed by oncogenic Ras, suggesting that Arf is an essential tumor suppressor in mice. In vivo, genetic evidence in mice has shown that both Ink4a and Arf are critical targets of Bmi1 (Molofsky et al, 2003; Park et al, 2003; Bruggeman et al, 2005), whereas in vitro the growth defect observed in Bmi1-deficient MEFs was rescued by loss of Arf alone. In agreement with this, we have demonstrated that the proliferative arrest induced by Cbx8 knockdown in MEFs is largely dependent on the p19Arf-p53 pathway under normal growth conditions. However, when cells are plated at low density, the lack of Cbx8 affects the colony-forming ability of Arf−/−, (Ink4a−/−, Arf−/−) and p53−/− MEFs.
The question is: how Cbx8 can affect the proliferative potential under low-cell plating conditions and not under normal conditions? When cells are plated sparsely, paracrine signaling is inhibited by distance owing to low local concentration of secreted growth factors. It is therefore likely that under normal growth conditions, the effect of Cbx8 loss is rescued by paracrine growth stimulation. In support of this hypothesis, we have shown that (Ink4a−/−, Arf −/−) MEFs depend on the presence of Cbx8 when grown in low serum. Taken together, these results suggest that Cbx8 regulates genes in addition to the Ink4a-Arf locus and that these genes are involved in regulating cell proliferation. Further supporting this suggestion is our intriguing finding that downregulation of CBX8 leads to loss of proliferation and a decrease in cyclin A2 levels before a significant increase in p16INK4A levels was observed.
Gene expression profiling for identification of CBX8 target genes
To identify potential target genes of CBX8, which could be involved in regulating normal proliferation in addition to the INK4A-ARF locus, we have performed gene expression experiments and have compared these experiment with a previously performed CBX8/H3K27me3 location analysis. We identified 93 genes that were repressed by the ectopic expression of CBX8 or BMI1 and bound by CBX8 or enriched for H3K27me3. We analyzed in more detail nine putative tumor suppressors and showed that they are all bound by CBX8 and BMI1 and contain the H3K27me3 mark. Furthermore, expression of five of the nine genes (INK4A, MTUS1, PERP, GJB2/CX26 and SORBS1) was also increased by CBX8 downregulation, showing that they are physiological targets of CBX8. Interestingly, PERP is a p53 target involved in p53-induced apoptosis (Attardi et al, 2000), suggesting that Cbx8 in mouse cells, where Cbx8 represses Arf, works both up- and downstream of p53. Moreover, CBX8 does not repress ARF in human cells, but rather modulates p53 response by direct repression of some of its target genes. Thus CBX8, in addition to its function in proliferation control, may promote cell survival. The MTUS1 gene is a candidate tumor suppressor previously connected to cell proliferation, which could suggest that MTUS1 contributes to CBX8 proliferation control (Seibold et al, 2003). GJB2/CX26, a member of the connexin family and a mediator of gap junctional intercellular communication (GJIC), is found to be silenced by CpG methylation in breast cancer (Miyamoto et al, 2005) and loss of GJIC is associated with uncontrolled proliferation and cancer progression (Mesnil et al, 2005). Furthermore, SORBS1 is downregulated in prostate cancer and regulates insulin signaling (Vanaja et al, 2006). Although MTUS1, GJB2/CX26 and SORBS1 may contribute to the INK4A-ARF-independent functions of CBX8 in cell proliferation, we favor a model in which several CBX8 target genes contribute to the p16Ink4a/Arf-independent early arrest seen in response to CBX8 inhibition in TIG3-T cells (Figure 7).
Figure 7.

Proliferative control by CBX8. CBX8 regulates senescence and proliferation via INK4A-ARF-dependent and -independent pathways.
Regulation of CBX8 activity
Although it has been shown previously that BMI1 levels are reduced when cells approach senescence in human diploid fibroblasts (Itahana et al, 2003), we have not observed any such changes in Bmi1 or Cbx8 levels in senescent MEFs (data not shown). The question is then how the CBX8-containing PRC1 complex is displaced from the INK4A-ARF locus upon activation when cells become senescent. At least two mechanisms working either alone or in combination could be suggested: (1) post-translational modifications of one or more of the PRC1 proteins and (2) loss of the H3K27me3 mark. It has been shown previously that BMI1 is a phosphoprotein and that phosphorylation affects its ability to bind chromatin (Voncken et al, 1999). Whether other proteins in the complex such as CBX8 and Ring1 are also phosphorylated remains to be seen. The identification of casein kinase II (CKII) (Supplementary Figure 4A) in the CBX8-BMI1 PRC1-type complex suggests that CKII might play a function in a phosphorylation-dependent regulation of PRC1 function. Regarding a possible loss of the H3K27me3 mark, we envision that this might involve a H3K27-specific demethylase activity (reviewed in Klose et al, 2006) or a histone-exchange mechanism replacing the K27-methylated H3 with a K27-unmodified H3 molecule of the same or variant type (Jin et al, 2005; Hake and Allis, 2006). Future studies will be aimed at understanding how the CBX8-BMI1-PRC1 complex is regulated in response to replicative, oncogene- and stress-induced signals.
Materials and methods
Generation of antibodies
CBX7 ‘RELF' and CBX4 ‘PARN' antibodies were produced in rabbits using synthetic peptides RELF: (K)KFPPRGPNLESHSHRRELFLQEPP; PARN: (K)KPDLLAWDPARNTHPPSHHPH coupled to KLH. Antibodies were affinity-purified on the peptide antigens. Monoclonal antibodies for BMI1 were generated against full-length human BMI1 fused to the maltose-binding protein.
Generation of plasmids
Human CBX8 was PCR-amplified from a fetal brain cDNA library. A Flag-Myc-tagged version of CBX8 was cloned into the retroviral PINCO vector (Grignani et al, 1998). The mutant of CBX8, mCD (K31AW32A), was made using a QuickChange mutagenesis kit (Stratagene). For expression of BMI1, we used the LZRS-BMI1 (Jacobs et al, 1999). Vectors encoding shRNA for hBMI1 (pRS-BMI1), mCbx8 (pRS-Cbx8) and hCBX8 (pRS-CBX8) were constructed by oligo cloning into the retroviral pRetroSuper vector (Brummelkamp et al, 2002). Target sequences are provided in Supplementary data. pRS-SUZ12 has been described before (Pasini et al, 2004).
Cell culture
MEFs, telomerase-immortalized TIG3 cells expressing the Ecotropic receptor (TIG3-T) and U2OS cells were cultured in DMEM, 10% FBS, penicillin and streptomycin.
shRNA in TIG3-T and MEFs
TIG3-T or MEFs were repeatedly infected for 2 days with virus expressing shRNA constructs (pRetroSuper, puromycin). Transduced cells were selected with puromycin (2 μg/ml). The end of the third day of selection corresponds to day 0 in Figure 1.
3T3 and colony assays
MEFs were transduced with one of the retroviral vectors. 3T3 assays were performed as described previously by Todaro and Green (1963). For colony assays, 10 000 or 30 000 cells were plated on 90 or 150-mm plates, respectively. Cells were stained with crystal violet.
FACS analysis
TIG3-T were pulsed with BrdU (33 μM) for 20 min and recovered by trypsinisation. Cells were fixed and stained according to standard procedures and analyzed using a FACS machine.
Immunoflourescence
U2OS cells were plated on coverslips and cotransfected with shRNA constructs and pBos-H2B-GFP. Coverslips were pre-extracted (20 mM Hepes, pH 7.2, 0.5% IGEPAL-630, 50 mM NaCl, 3 mM MgCl2 and 300 mM sucrose) and fixed in 4% formaldehyde in PBS. Coverslips were incubated with rabbit anti-CBX8 and mouse anti-BMI1 (F6, Upstate, 05-637) in DMEM/10% FBS, incubated with biotin-conjugated anti-mouse IgG (Amersham), Alexa-647 anti-rabbit (Molecular Probes) and streptavidin-conjugated Alexa-568 (Molecular Probes), stained with DAPI and mounted. Between each incubation step, coverslips were washed with PBS.
WB analysis and IP
Protein extracts for WB analysis and IPs were performed using a high-salt lysis buffer (50 mM Tris, pH 7.2, 300 mM NaCl, 0.5% IGEPAL-630, 1 mM DTT, Leupeptin, Aprotinin and 1 mM PMSF). WBs and IPs were performed according to standard protocols and the antibodies used were anti-cyclin A2 (Santa Cruz, sc-751), anti-cyclin E1 (Santa Cruz, HE12), anti-pRB (Pharmingen, 245), anti-human p16 (DCS-50), anti-human p53 (DO-1), anti-Histone H3 (Abcam, ab1791), anti-GAPDH (Stressgene, A00084), anti-Tubulin (Sigma, T6074), anti-CBX8 (LAST and GALD; Bracken et al, 2006), anti-BMI1 (DC-9 for WB and AF27 for IPs) and anti-SUZ12 (Upstate, 07-379).
ChIPs
ChIPs were performed as previously described (Bracken et al, 2003). The antibodies used were a mixture of two rabbit polyclonal anti-CBX8 antibodies (Bracken et al, 2006), anti-BMI1 (AF27), anti-H3K27me3 (Upstate, 07-449), anti-CBX7 (RELF), anti-CBX4 (PARN) and anti-HA (Santa Cruz). The immunoprecipitated DNA was quantified by real-time PCR. ChIP primers for target genes were designed in the promoter regions and all primer pairs only amplify one amplicon. Sequences, melting temperature and reaction conditions are shown in Supplementary Table III.
Extractions
Cells were trypsinized, spun down at 1200 g in DMEM/10% FBS, washed in PBS and resuspended in pre-extraction buffer. After 30 min on ice, samples were spun at 1300 g for 2 min. Supernatants were saved and pellets were washed once in 1 ml cold pre-extraction buffer. Protein concentration was measured in the supernatants.
Real-time quantitative RT–PCR
RNA was purified using the RNeasy mini kit (Qiagen). cDNA synthesis was performed using the PE Applied Biosystems Taqman Reverse Transcription reagents. Quantification of mRNA levels was carried out using the SYBR Green I detection system (Applied Biosystems), on a ABI Prism 7300. All primer pairs used only amplify one amplicon. Sequences, melting temperature and reaction conditions are shown in Supplementary Table III.
Affymetrix expression array
RNA was purified from control (PINCO), BMI1 and FM-CBX8-infected MEFs. About 2 μg of purified RNA isolated from three independent experiments was processed for microarray expression analysis (for further technical information, see Supplementary data).
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank the members of the Helin laboratory for fruitful discussions and Rie Christensen for technical assistance. We also thank Rehannah Borup and the Microarray Center of Copenhagen University Hospital for technical support. Furthermore, we thank Dr Salek for assisting with mass spectrometric analysis and Dr Marten van Lohuizen for kindly providing the Bmi1−/− MEFs. This work was supported in part by grants from the Danish Natural Science Research Council, the Danish Medical Research Council, the Danish Cancer Society, the Novo Nordisk Foundation and the Danish National Research Foundation.
References
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev 14: 704–718 [PMC free article] [PubMed] [Google Scholar]
- Bardos JI, Saurin AJ, Tissot C, Duprez E, Freemont PS (2000) HPC3 is a new human polycomb orthologue that interacts and associates with RING1 and Bmi1 and has transcriptional repression properties. J Biol Chem 275: 28785–28792 [DOI] [PubMed] [Google Scholar]
- Bea S, Tort F, Pinyol M, Puig X, Hernandez L, Hernandez S, Fernandez PL, van Lohuizen M, Colomer D, Campo E (2001) BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res 61: 2409–2412 [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37: 961–976 [DOI] [PubMed] [Google Scholar]
- Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD (2006) Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol 26: 2560–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K (2006) Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20: 1123–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, Hulsman D, Leung C, Arsenijevic Y, Marino S, van Lohuizen M (2005) Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev 19: 1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Campisi J (2000) Cancer, aging and cellular senescence. In Vivo 14: 183–188 [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043 [DOI] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196 [DOI] [PubMed] [Google Scholar]
- Dirac AM, Bernards R (2003) Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem 278: 11731–11734 [DOI] [PubMed] [Google Scholar]
- Drayton S, Brookes S, Rowe J, Peters G (2004) The significance of p16INK4a in cell defenses against transformation. Cell Cycle 3: 611–615 [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J, Munoz M, Clark SJ, Moreno V, Capella G, Peinado MA (2005) Hypermethylation of the prostacyclin synthase (PTGIS) promoter is a frequent event in colorectal cancer and associated with aneuploidy. Oncogene 24: 7320–7326 [DOI] [PubMed] [Google Scholar]
- Gil J, Peters G (2006) Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 7: 667–677 [DOI] [PubMed] [Google Scholar]
- Gil J, Bernard D, Martinez D, Beach D (2004) Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol 6: 67–72 [DOI] [PubMed] [Google Scholar]
- Grignani F, Kinsella T, Mencarelli A, Valtieri M, Riganelli D, Grignani F, Lanfrancone L, Peschle C, Nolan GP, Pelicci PG (1998) High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res 58: 14–19 [PubMed] [Google Scholar]
- Hake SB, Allis CD (2006) Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc Natl Acad Sci USA 103: 6428–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM (1991) Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 65: 753–763 [DOI] [PubMed] [Google Scholar]
- Hildebrandt T, Preiherr J, Tarbe N, Klostermann S, Van Muijen GN, Weidle UH (2000) Identification of THW, a putative new tumor suppressor gene. Anticancer Res 20: 2801–2809 [PubMed] [Google Scholar]
- Isono K, Fujimura Y, Shinga J, Yamaki M, Wang OJ, Takihara Y, Murahashi Y, Takada Y, Mizutani-Koseki Y, Koseki H (2005) Mammalian polyhomeotic homologues Phc2 and Phc1 act in synergy to mediate polycomb repression of Hox genes. Mol Cell Biol 25: 6694–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, Van Lohuizen M, Band V, Campisi J, Dimri GP (2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol 23: 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999) The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397: 164–168 [DOI] [PubMed] [Google Scholar]
- Jin J, Cai Y, Li B, Conaway RC, Workman JL, Conaway JW, Kusch T (2005) In and out: histone variant exchange in chromatin. Trends Biochem Sci 30: 680–687 [DOI] [PubMed] [Google Scholar]
- Kim H, Lapointe J, Kaygusuz G, Ong DE, Li C, van de Rijn M, Brooks JD, Pollack JR (2005) The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res 65: 8118–8124 [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100: 11606–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM, Zhang Y (2006) JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7: 715–727 [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev 16: 2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine SS, Weiss A, Erdjument-Bromage H, Shao Z, Tempst P, Kingston RE (2002) The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol 22: 6070–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Andrews LG, Tollefsbol TO (2006) Loss of the human polycomb group protein BMI1 promotes cancer-specific cell death. Oncogene 25: 4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg AS, Hahn WC, Gupta P, Weinberg RA (2000) Genes involved in senescence and immortalization. Curr Opin Cell Biol 12: 705–709 [DOI] [PubMed] [Google Scholar]
- Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML (2005) Defective gap junctional intercellular communication in the carcinogenic process. Biochim Biophys Acta 1719: 125–145 [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Fukutomi T, Akashi-Tanaka S, Hasegawa T, Asahara T, Sugimura T, Ushijima T (2005) Identification of 20 genes aberrantly methylated in human breast cancers. Int J Cancer 116: 407–414 [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ (2003) Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425: 962–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208 [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF (2003) Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423: 302–305 [DOI] [PubMed] [Google Scholar]
- Park YB, Park MJ, Kimura K, Shimizu K, Lee SH, Yokota J (2002) Alterations in the INK4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet Cytogenet 133: 105–111 [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Beato M, Sanchez E, Gonzalez-Carrero J, Morente M, Diez A, Sanchez-Verde L, Martin MC, Cigudosa JC, Vidal M, Piris MA (2006) Variability in the expression of polycomb proteins in different normal and tumoral tissues. A pilot study using tissue microarrays. Mod Pathol 19: 684–694 [DOI] [PubMed] [Google Scholar]
- Satijn DP, Olson DJ, van der Vlag J, Hamer KM, Lambrechts C, Masselink H, Gunster MJ, Sewalt RG, van Driel R, Otte AP (1997) Interference with the expression of a novel human polycomb protein, hPc2, results in cellular transformation and apoptosis. Mol Cell Biol 17: 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, Freemont PS (1998) The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol 142: 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold S, Rudroff C, Weber M, Galle J, Wanner C, Marx M (2003) Identification of a new tumor suppressor gene located at chromosome 8p21.3-22. FASEB J 17: 1180–1182 [DOI] [PubMed] [Google Scholar]
- Serrano M, Blasco MA (2001) Putting the stress on senescence. Curr Opin Cell Biol 13: 748–753 [DOI] [PubMed] [Google Scholar]
- Thiebault K, Mazelin L, Pays L, Llambi F, Joly MO, Scoazec JY, Saurin JC, Romeo G, Mehlen P (2003) The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc Natl Acad Sci USA 100: 4173–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro GJ, Green H (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M (2004) Stem cells and cancer; the polycomb connection. Cell 118: 409–418 [DOI] [PubMed] [Google Scholar]
- van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ (2001) Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood 97: 3896–3901 [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A (1991) Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65: 737–752 [DOI] [PubMed] [Google Scholar]
- Vanaja DK, Ballman KV, Morlan BW, Cheville JC, Neumann RM, Lieber MM, Tindall DJ, Young CY (2006) PDLIM4 repression by hypermethylation as a potential biomarker for prostate cancer. Clin Cancer Res 12: 1128–1136 [DOI] [PubMed] [Google Scholar]
- Voncken JW, Schweizer D, Aagaard L, Sattler L, Jantsch MF, van Lohuizen M (1999) Chromatin-association of the Polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J Cell Sci 112 (Part 24): 4627–4639 [DOI] [PubMed] [Google Scholar]
- Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM, van Lohuizen M, Betticher DC (2001) The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer 84: 1372–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Ambinder R, Tao Q (2005) The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clin Cancer Res 11: 6442–6449 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1