

Abstract
Although apoptosis is a well-recognized phenomenon in chronic atherosclerotic disease, its role in sudden coronary death, in particular, acute plaque rupture is unknown. Culprit lesions from 40 cases of sudden coronary death were evaluated. Cases were divided into two mechanisms of death: ruptured plaques with acute thrombosis (n = 25) and stable plaques with and without healed myocardial infarction (n = 15). Apoptotic cells were identified by staining of fragmented DNA and confirmed in select cases by gold conjugate labeling combined with ultrastructural analysis. Additional studies were performed to examine the expression and activation of two inducers of apoptosis, caspases-1 and -3. Ruptured plaques showed extensive macrophage infiltration of the fibrous cap, in particular at rupture sites contrary to stable lesions, which contained fewer inflammatory cells. Among the culprit lesions, the overall incidence of apoptosis in fibrous caps was significantly greater in ruptured plaques (P < 0.001) and was predominantly localized to the CD68-positive macrophages. Furthermore, apoptosis at plaque rupture sites was more frequent than in areas of intact fibrous cap (P = 0.028). Plaque rupture sites demonstrated a strong immunoreactivity to caspase-1 within the apoptotic macrophages; staining for caspase-3 was weak. Immunoblot analysis of ruptured plaques demonstrated caspase-1 up-regulation and the presence of its active p20 subunit whereas stable lesions showed only the precursor; nonatherosclerotic control segments were negative for both precursor and active enzyme. These findings demonstrate extensive apoptosis of macrophages limited to the site of plaque rupture. The proteolytic cleavage of caspase-1 in ruptured plaques suggests activation of this apoptotic precursor. Whether macrophage apoptosis is essential to acute plaque rupture or is a response to the rupture itself remains to be determined.
It is well recognized that arterial thrombosis contributes to the onset of acute coronary syndromes. 1,2 The majority of sudden coronary deaths precipitated by acute thrombosis are associated with rupture of an advanced plaque. 1 Atherosclerotic plaques that are predisposed to rupture typically have a large lipid core, are covered by a thin fibrous cap, and are heavily infiltrated by macrophages and to a lesser extent by lymphocytes. 2-4 The rupture site is variable and is presumed to occur at the weakest point of the fibrous cap exposing the thrombogenic contents of the lipid core. 5 Most ruptures occur during normal diurnal activity without an obvious precipitating event although there is an increased association with exercise. 6 Among the many risk factors for coronary artery disease, an elevated ratio of total cholesterol to high-density lipoprotein cholesterol ratio is the strongest predictor of rupture. 4
Of the morphological features associated with rupture of an advanced plaque, fibrous cap thinning seems to be the most critical. 7,8 The precise nature of fibrous cap destruction is unknown although recent evidence suggests that smooth muscle cell (SMC) death by apoptosis may be partly responsible. 9 On the other hand, it has been proposed that ongoing apoptosis of macrophages in a stable plaque contributes to the enlargement of necrotic core (Bjorkrud) and vulnerability to rupture. The bulk of evidence of the occurrence of apoptosis in human arteries, however, is from lesions with chronic atherosclerotic disease. 10-14 The information in culprit lesions associated with plaque rupture and sudden coronary death is lacking.
This study was undertaken to determine the extent of apoptosis in varying cell populations in culprit lesions with acute thrombi attributed to plaque rupture. For comparison, culprit lesions with significant stenosis without evidence of rupture and luminal thrombi (stable plaques) were examined. 15 Additional studies were performed to assess the expression and activation of two mammalian death proteases, caspase-1 and -3, in association with apoptotic cells in plaque rupture.
Materials and Methods
Selection of Cases
Cases of sudden coronary death were identified as part of a consultative service provided to the Office of the Chief Medical Examiner for the State of Maryland. 16 Sudden coronary death was based on unexpected death, witnessed within 6 hours of the onset of symptoms, or death of a person known to be in stable condition <24-hours antemortem. A complete autopsy, including a toxicology screen, ruled out noncardiac causes of death.
Twenty-five cases of plaque rupture were collected and of these, 21 were used for DNA fragmentation staining and immunohistochemistry and four were processed for immunoblot analysis. Of the 15 stable plaques, 11 were used for DNA fragmentation staining and four were processed for immunoblot analysis; four nonatherosclerotic arteries served as controls for immunoblot studies.
Definitions
Culprit plaques were identified as those with an acute luminal thrombus and plaque rupture or in the absence of a thrombus, the artery with the greatest degree of luminal narrowing relative to the internal elastic lamina. 4,15 Acute plaque ruptures consisted of a luminal platelet-fibrin thrombus continuous with an underlying lipid-rich core. The connection between the thrombus and the lipid-rich core was through a disrupted thin fibrous cap infiltrated by macrophages. Stable plaque was defined as cross-sectional luminal narrowing of ≥75% with a fibrous cap >65 μm in thickness in the absence of plaque rupture or thrombus.
Evaluation of the Hearts and Preparation of Tissue Specimens
Hearts were studied under the supervision of the medical examiner. Coronary arteries were serial sectioned at 3-mm intervals. Segments showing narrowing of >50% were submitted for histological analysis. Collected arterial specimens were immediately immersed in ice-cold medium 199. Vessels were then snap-frozen in optimal cutting temperature tissue processing medium (OCT; Miles Diagnostics, Elkhart, IN).
In Situ End-Labeling (ISEL) for DNA Fragmentation
Serial cryostat sections (6 μm) were cut and air-dried onto Superfrost microslides (Columbia Diagnostics, Inc., Springfield, VA) and stored at −70°C before use. In situ labeling of DNA fragmentation was performed using terminal deoxyribonucleotide transferase (TdT)-mediated nick end labeling based on an in situ apoptosis detection kit (TACS; Trevigen, Gaithersburg, MD). Sections were fixed for 10 minutes in neutral-buffered formalin, rinsed in phosphate-buffered saline (PBS), and incubated for 5 minutes in 0.1% Triton X-100 in 0.1% sodium citrate. Treating the slides with 0.3% hydrogen peroxide for 10 minutes inactivated endogenous peroxidase. DNA fragments were labeled with biotinylated nucleotides (dNTPs) and TdT for 1 hour at 37°C. The incorporation of biotinylated nucleotides into DNA was detected with a streptavidin-conjugated horseradish peroxidase. The chromogenic substance used to visualize the reaction was TACS Blue Label (Trevigen) which produces a blue reaction product. Sections of rat mammary glands (weaning animals) were used a positive controls for apoptosis (Oncor, Gaithersburg, MD).
Detection of Fragmented DNA by Electron Microscopy
The method of tissue processing for ultrastructural analysis and immunogold labeling was adapted from the procedure described by Berryman and Rodewald, 17 and all processing steps were performed at 4°C. Semithin sections were cut, stained with toluidine blue, and examined by light microscopy for the site of rupture. Artifact-free areas showing rupture sites were selected for ultrathin section cutting.
The TdT immunogold assay was performed as above with slight modifications. 18 Sections were etched for 10 minutes in absolute ethanol, rinsed in PBS, and permeabilized in CytoPore (Trevigen, Inc.). Endogenous peroxidase was quenched by immersion in 3% hydrogen peroxide in 40% methanol. Sections were then incubated in the TdT-labeling mixture in a humidified chamber at 37°C for 1 hour. The reaction was stopped and sections were then incubated overnight at 4°C with colloidal gold-streptavidin (Zymed, San Francisco, CA) for direct visualization of biotinylated dNTPs. Immunogold labeling was examined using an electron microscope (Zeiss EM 10, Germany). Omitting TdT during the procedure as the negative control checked the validity of the method.
Immunohistochemistry
Cell Populations
Frozen sections were fixed in acetone for 10 minutes at −20°C and then air-dried. Treating the sections with 0.3% hydrogen peroxide for 20 minutes inactivated endogenous peroxides. Sections were incubated in protein-free block (DAKO, Carpinteria, CA) for 10 minutes to block nonspecific binding of primary antibody. Sections were incubated for 1 hour at room temperature with primary antibodies against human muscle-α-actin (HHF-35, dilution 1:50; DAKO), the macrophage marker CD68 (DAKO-CD68, KP1, dilution 1:600; DAKO) or the T-cell marker CD3 (DAKO-UCHT1, dilution 1:150; DAKO). Primary antibodies were labeled by a biotinylated link antibody directed against mouse using a peroxidase-based kit (LSAB; DAKO). Immunostains were visualized (reddish reaction product) by an aminoethylcarbazol substrate-chromogen system (DAKO) and counterstained with Gill’s hematoxylin.
Double-Immunohistochemistry
To identify cell types undergoing apoptosis, double-staining was performed by combining ISEL and immunohistochemistry with antibodies against actin, CD68, or CD3. Tissue sections were first stained for DNA fragmentation (as described above); however, labeling was detected using DAB as the chromogenic substrate followed by enhancement with nickel salts (brown-black reaction product). Immunostains were visualized with a red streptavidin-alkaline phosphatase substrate (Vector, Burlingame, CA); slides were then counterstained with methyl green.
Quantification of Cell Type and Apoptosis
Double-stained slides were used for quantification of cell type and apoptosis. For each arterial section, at least 300 total cells were counted in random high-power fields within the fibrous cap using an eyepiece reticle with a 0.04 mm 2 grid. Quantitatively the size of the rupture site varied between cases such that cell counts were performed in areas ranging from 0.12 to 0.20 mm 2 (Figure 1) ▶ . Remote areas of the fibrous cap were defined as those at least 0.4 mm away from the nearest field representing the rupture site. Care was taken to avoid counting inflammatory cells entrapped within the thrombus. The varying cell types are expressed as the percentage of total cells or apoptotic nuclei. Only nuclei with positive immunoreactivity and cell type were counted as some failed to stain positive with macrophage, SMC, or T cell-specific antibodies.
Figure 1.

Cartoon diagram illustrating where measurements of cell type and apoptosis were performed. The color key at the bottom describes the various plaque constituents. The black-hatched regions represent areas where measurements were taken. Quantitatively, the size of the rupture site varied between cases such that cell counts were performed in areas ranging from 0.12 to 0.20 mm2.
Localization of Caspases-1 and -3
Immunostaining for caspases-1 and -3 was performed as described above. To recognize caspase-1, serial cryosections were stained with a polyclonal rabbit anti-human antibody specific for the 10-kd subunit, (Clone C-20; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Caspase-3 was immunolocalized using a polyclonal rabbit anti-human antibody (DAKO) which is found to react primarily with the 32-kd proenzyme. Nonimmune rabbit serum was used as a negative control.
Electrophoresis and Immunoblot Analysis
Culprit plaques were identified by gross histological analysis using a dissection microscope and further confirmed by histological examination of adjacent sections. Atherosclerotic segments (3 to 5 mm) with and without plaque rupture and thrombi were removed and immediately placed in ice-cold M199; nonatherosclerotic vessels were collected as controls. After removal of the adventitia, samples were snap-frozen in liquid nitrogen and stored at −70°C until protein extraction.
For protein extraction, the tissue was weighed, minced with scissors, and extracted in ice-cold lysis buffer (pH 7.6) containing 50 mmol/L Tris-HCl, 15 mmol/L NaCl, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/ml aprotonin, 10 μg/ml leupeptin, 1% Nonidet P-40, and 1 mmol/L dithiothreitol.
Proteins (50 μg per lanes) from plaque homogenates were separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis mini gels and transferred to nitrocellulose membranes (Bio-Rad, Richmond, CA) using a semidry blotting system. Blocking of nonspecific binding was achieved with incubation of the membrane in 5% milk PBS, pH 7.2, containing 0.05% Tween 20. Polyclonal antibodies against caspase-1 (Upstate Biotechnology, Lake Placid, NY) or caspase-3 (Santa Cruz Biotechnology, Inc.) were used for staining. Antigen detection was performed with a chemiluminescent detection system (ECL; Amersham, Arlington Heights, IL).
Statistical Analysis
All values are expressed as the mean ± SEM. Comparisons of apoptotic index among cell types in the different regions of the plaque were performed by factorial analysis of variance (StatView 4.5; Abacus Concepts Inc., Berkeley, CA) and analyzed simultaneously with post hoc testing by the Scheffé’s procedure; statistical significance was established at P < 0.05.
Results
Evidence of Extensive Apoptosis at the Site of Plaque Rupture
Labeling of DNA fragmentation alone was undertaken for the detection of apoptosis by light microscopy in ruptured and stable plaques. Overall apoptosis within the fibrous cap was more prevalent in plaque ruptures than in stable plaques (P = <0.001). The bulk of this difference was accounted for by the high degree of apoptosis at plaque rupture sites (Figure 2) ▶ .
Figure 2.
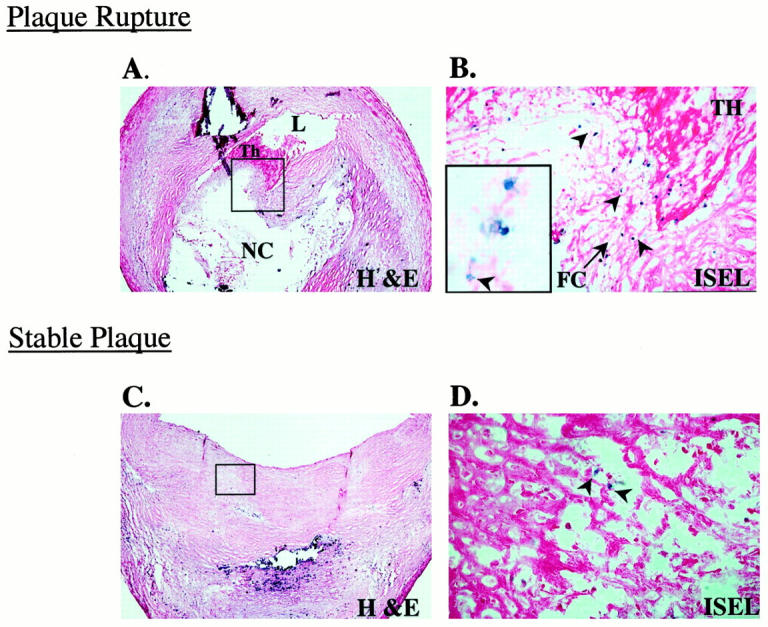
Apoptosis in culprit plaques. A: Micrograph of a cross-section of an epicardial coronary artery shows a plaque rupture with an acute luminal thrombus (Th); note the thin fibrous cap, boxed area (H&E stain; original magnification, ×30). B: Serial section of A after DNA fragmentation staining by ISEL (see Materials and Methods). Numerous apoptotic cells (blue nuclear staining, arrowheads) are identified at the plaque rupture site (eosin counterstain; original magnification, ×150). The inset shows a high-power view illustrating the nuclear detail; note the fragmented nucleus (arrowhead; original magnification, ×1000). C: Micrograph of a cross-section of an epicardial coronary artery shows an eccentric stable plaque. The lesion is characterized by dense fibrous cap, boxed area, overlying a calcified region (H&E; original magnification, ×30). D: Serial section of C after DNA fragmentation staining, there is a paucity of apoptotic cells (arrowheads) relative to rupture site in B (eosin counterstain; original magnification, ×150). Abbreviations: L, lumen; NC, necrotic core.
Ultrastructural examination was performed to confirm the occurrence of apoptosis in plaque rupture (Figure 3) ▶ . Many cells at the ruptured site demonstrated varying stages of apoptosis with characteristic changes of nuclear chromatin condensation and clumping, and loss of cell volume. DNA fragmentation was shown by combined ultrastructure and ISEL using a gold conjugate. The gold-labeled fragments were commonly found at the periphery of the nucleus at sites of chromatin condensation in cells undergoing apoptosis. Only occasional gold particles were noted in cells with normal ultrastructure and were found irrespective of nuclear chromatin material. Unlike ruptures, apoptotic cells were rarely observed in the fibrous caps of stable plaques.
Figure 3.

Immunoelectron microscopic characterization of apoptosis at the rupture sites. A: Ultrastructural examination of the macrophages (arrows) at the rupture site showed that the majority had nuclear changes suggestive of apoptosis. B: Example demonstrating varying stages of nuclear shrinkage with condensation of chromatin at the nuclear periphery. The presence of lipid globules and lack of contractile elements help identify the macrophages. ISEL was performed on ultra-thin sections with TdT and biotinylated nucleotides. Fragmented DNA was visualized using a 10-nm gold, streptavidin conjugate. C: High-power electron micrograph of the region indicated by the inset in B, note the gold particles are selectively localized on the condensed chromatin material.
Apoptosis at the Plaque Rupture Site Occurs Essentially in Macrophages
Immunohistochemical staining of serial sections for the identification of cell type showed that the majority of cells at the rupture site were macrophages. Of the total number of immunoreactive cells, 193 ± 10 per 0.1 mm 2 were macrophages; the number of smooth muscle and T cells were markedly less, 16 ± 2 and 15 ± 5 per 0.1 mm2, respectively. In contrast, stable plaques showed a decreased prevalence of macrophages (30 ± 9 per 0.1 mm2, P < 0.0001) and an increase in SMCs (80 ± 13, P = 0.0003); T cells comprised ∼3 ± 0.5 per 0.1 mm2.
Double-staining for the immunohistochemical typing of cells in combination with in situ labeling for apoptosis showed that apoptotic nuclei at rupture sites were essentially those of macrophages (Figures 4 and 5) ▶ ▶ . Apoptosis in macrophages was more frequent at the rupture site (93 ± 8 per 0.1 mm2) as opposed to areas of intact fibrous cap (10 ± 2 per 0.1 mm2, P = 0.03). Apoptotic smooth muscle and T lymphocytes at the rupture site were only 5 ± 0.9 per 0.1 mm 2 and 5 ± 1 per 0.1 mm2, respectively, contributing little to the total apoptotic cell population. Conversely, stable plaques showed a much lower frequency of apoptotic cells compared with ruptures (Figures 2 and 4) ▶ ▶ . The number of apoptotic nuclei expressed per 0.1 mm 2 in stable plaques were 7 ± 1.2 for macrophages, 3 ± 0.8 for SMCs, and 0.8 ± 0.4 for T lymphocytes.
Figure 4.

Identification of cells undergoing apoptosis in culprit plaques. Apoptotic cells were characterized using a combination of ISEL and specific antibodies for macrophages (KP-1/CD68; A and C) or SMCs (HHF-35; B and D). ISEL-positive nuclei were visualized with diaminobenzidine (dark-brown reaction product) and antibody staining was detected using an anti-mouse IgG conjugated with biotin and an avidin alkaline phosphatase-substrate system (red reaction product). The methyl green counterstain yields blue-green nuclei. A and B are photomicrographs of plaque rupture and C and D are stable plaques. A: CD68 and ISEL staining in plaque rupture, note the majority of apoptotic cells are macrophages (original magnification, ×150). Abbreviations: Th, thrombus; FC, fibrous cap. B: Serial section showing DNA fragmentation and HHF-35 staining in the fibrous cap in a ruptured plaque. Note that most of the apoptotic nuclei (upper left, rupture site) are negative for SMCs (original magnification, ×300). C: Stable plaque showing occasional apoptotic macrophages (arrowheads) in a region of the fibrous cap (original magnification, ×300). D: Serial section of C shows rare apoptotic SMCs (arrowheads) in the fibrous cap.
Figure 5.

Comparision of apoptosis relative to specific cell types in culprit plaques. Bar graphs represent the different cell populations and apoptotic index in the fibrous cap of ruptured and stable plaques. The cells were quantified after double-labeling experiments using serial frozen sections as shown in Figure 4 ▶ . Not all cells undergoing apoptosis could be identified by antibodies. The results are expressed as the mean ± SE of 21 ruptures and 11 stable plaques. A: Overall, cell populations expressed as a percentage of the total number of nuclei; note the predominance of macrophages and paucity of SMCs at the rupture site. B: Apoptotic cells as represented as a percentage of cell type; ∼45% of macrophages were recognized as apoptotic. C: Macrophage apoptosis compared with a remote region of the fibrous cap. Abbreviations: MΦs, macrophages; SMC, smooth muscle cells.
The percentage of macrophage apoptosis was plotted against postmortem interval in 21 cases of plaque rupture to determine whether apoptosis of macrophages was dependent on the interval between death and autopsy. In our cases, the postmortem interval ranged between 10 and 53 hours with the majority of cases (n = 16) below 24 hours. Apoptosis of macrophages associated with plaque rupture sites showed no correlation with postmortem interval (R 2 = 6.2 × 10−5).
Apoptosis at the Rupture Site Is Associated with Interleukin-1β-Converting Enzyme Activation
Plaque rupture sites demonstrated strong immunoreactivity to caspase-1 corresponding to regions of apoptotic macrophages (Figure 6A) ▶ . In contrast, immunoreactivity for caspase-3 at rupture sites was weak (not shown). In stable plaques, areas positive for apoptotic cells, in particular regions bordering the necrotic core showed immunoreactivity to both caspases-1 and -3 (Figure 6B) ▶ ; staining in the fibrous cap was essentially negative for either protein.
Figure 6.

Immunolocalization and biochemical activation of caspase-1 in plaque rupture. A: Immunohistochemical staining demonstrating caspase-1 at the site of plaque rupture (reddish-brown reaction product; original magnification, ×150). Note the intense immunoreactivity at the rupture site (a serial section from the case as shown in Figure 4 ▶ ). B: Stable plaque showing the localization of caspase-1 in an area of the fibrous cap (reddish-brown reaction product; original magnification, ×150). C: Representative immunoblot analysis of coronary segments showing extensive cleavage of caspase-1 in rupture; the stable plaques showed minimal cleavage. The analyses were repeated at least four times, yielding similar results.
Immunoblot analysis showed the presence of the 45-kd precursor form of caspase-1 in both ruptured and stable plaques (Figure 6C) ▶ . In contrast, the cleaved p20 band of caspase-1 was noted only in the ruptured plaques. Stable plaques did not show the evidence of caspase-1 activation. Nonatherosclerotic control arterial segments failed to show either the caspase-1 precursor or cleavage products. Unlike interleukin-1β-converting enzyme in plaque ruptures, caspase-3 cleavage to p17 protein was not detected in any of the cases; only the uncleaved 32-kd band was present (data not shown).
Discussion
Several reports have described the occurrence of apoptosis in morphologically diverse human plaques from carotid endarterectomies, explanted hearts, atherectomies (de novo and restenotic), and aorta and coronary arteries at autopsy. 10-14 Most of these studies involve early or advanced lesions with chronic atherosclerotic disease; those resulting in an acute thrombus and death have not been studied. The present investigation provides new information regarding apoptosis of macrophages, within the fibrous cap at sites of plaque rupture. Ultrastructural studies corroborated the occurrence of extensive apoptosis at rupture sites and biochemical analysis showed the expression and selective activation of caspase-1.
SMC Apoptosis and the Vulnerable Plaque
There is a well-established association of rupture with thin fibrous caps 19 and cap thickness constitutes a major part of the definition of the vulnerable plaque. 4 Although apoptosis of SMCs, in part may lead to fibrous cap thinning, this was not appreciated in the present study perhaps because the lesions were already ruptured. The paucity of SMCs at plaque rupture sites; however, suggests that SMC death may have occurred at an earlier time point. One preliminary study suggests that human monocytes/macrophages induce vascular SMC apoptosis; 20 although in fatty streaks, both appear to co-exist. The finding of decreased SMCs at plaque rupture sites agrees with earlier studies involving ulcerated lesions of the aorta. 21,22 The overall loss of SMCs within the fibrous cap, most likely represents a chronic process before the actual rupture event.
Apoptosis versus Oncosis
There is at least one paper on carotid plaques suggesting that oncosis contributes to cell death. 23 Recognition between apoptosis and oncosis is best appreciated at the ultrastructural level, because the terminal dUTP nick-end labeling assay by light microscopy lacks specificity. These pathways of cell death are best distinguished early because the phase of necrosis is similar for both. 24 In our study, early changes of cell death showed morphological features consistent with the nuclear changes associated with apoptosis. Because of the variable uptake of lipids and differences in cell size, cell shrinkage (another feature of apoptosis), was not always apparent. There was also some degree of uncertainty of cell type, because lipid-uptake was not exclusive to macrophages. As the morphological changes of cell death advanced the distinction between apoptosis and oncosis became difficult, thus, death by oncosis could not be excluded.
Dissolution of Matrix and Cell Apoptosis Anoikis
The precise etiology of macrophage apoptosis at the plaque rupture site is speculative. There is experimental evidence that the dissolution of extracellular matrix may threaten cell survival. In epithelial and vascular endothelium, inhibition of integrin function has been shown to produce a loss of cell-cell adhesion and apoptosis. 25,26 This type of detachment-induced cell death, a form of apoptosis, is referred to as “anoikis.” 27 In another study, basement membrane matrix was shown to suppress apoptosis of mammary epithelial cells in tissue culture and in vivo. In these studies, antibodies to α-integrins or overexpression of stromelysin-1 stimulated apoptosis, whereas the loss of extracellular matrix correlated with the expression of caspase-1 such that specific inhibitors of its activity rescued cells from apoptosis. Although data on cell attachment and death of human macrophages is lacking, an integrin dependence of cell survival has been shown in the murine macrophage-like cell line RAW264.7. 28
TIMP-3 and Cell Survival in the Fibrous Cap
There is emerging evidence that inhibitors of metalloproteinases, specifically TIMP-3, directly affect cell survival. 29 For example, adenoviral-induced overexpression of TIMP-3 promotes apoptosis of vascular SMCs both in vivo and in vitro. Although little TIMP-3 protein is found in normal arteries, it is markedly elevated in advanced plaques and is primarily associated with intimal macrophages at rupture-prone sites. 30 It remains to be demonstrated whether macrophages themselves commit cellular suicide through the expression of TIMP-3.
Apoptosis of Macrophages and Thrombogenicity
Macrophage apoptosis may also facilitate the acute thrombotic event arising from the rupture itself. In a recent study, Mallat et al 31 examined the role of apoptotic cell death in relation to plaque thrombogenicity. Shed membrane microparticles, products of apoptotic cells from carotid plaques were captured by annexin V and analyzed for their procoagulant activity. These microparticles were primarily monocytic and lymphocytic in origin, and very rich in tissue factor activity. Thus, it is conceivable that microparticles from macrophage apoptosis at the rupture site may increase the procoagulant potential of the plaque.
Caspases and Cell Death in Plaques
Apoptosis in mammalian cells is essentially regulated by caspases that are homologous to the C. elegans (worm) death genes. 32,33 The prototypic enzyme caspase-1 is synthesized as a 45-kd proenzyme that is autocatalytically cleaved to generate an active homodimeric enzyme of 20- and 10-kd subunits. 34 Geng and Libby 10 first reported on the co-localization of caspase-1 with apoptosis in advanced human atherosclerotic plaques. This data is corroborated by our study in which extensive expression of caspase-1 was detected at rupture sites rich in apoptotic macrophages. In comparison, caspase-1 immunolocalization in stable plaques was mostly confined to areas surrounding the necrotic core with minimal staining within the fibrous cap.
Although caspase-3 was also detected in ruptured plaques, its expression was not localized to plaque rupture sites and was more associated with cells surrounding the necrotic core. In the apoptotic cascade, caspase-3 has been described as an important mediator, especially with DNA damage and non-DNA damage stress. Cytokine-mediated stress, however, routes through activation of caspase-8 and caspase-1. 35 This raises the possibility of differential caspase activation depending on the environment within the plaque.
The present study demonstrates significant caspase-1 cleavage with plaque rupture when compared with stable plaque. Although cell types are indistinguishable in plaque homogenates, the strong expression of the caspase-1 precursor and its cleavage fragment complements the immunohistochemical localization of caspase-1 in macrophages at plaque rupture sites. The strong evidence of caspase-1 cleavage in the ruptured plaque may stem from the focal and concentrated presence of macrophages undergoing apoptosis. However, the absence of activation of caspase-1 in stable plaques does not rule out the possibility of apoptosis in these lesions but only suggests that the degree of apoptosis is too infrequent to be detected in whole plaque homogenates.
Study Limitations
Plaque rupture is a dynamic process and it is conceivable that an active cell such as macrophage plays an important role in the process. It must be emphasized that acute fatal ruptures are at their morphological endpoint and many of the mechanistic processes leading to the rupture event likely have occurred. Because autopsy material only provides an end-stage specimen, the physiological relevance of the present observations cannot be identified. Furthermore, limitations of autopsy material, including postmortem interval, must also be taken into account. In our study, specimens were often collected more than 8 hours after death, possibly allowing sufficient time for macrophages to undergo apoptosis after rupture was triggered rather than before. The lack of correlation of macrophage apoptosis at rupture sites with postmortem interval, however, argues against apoptosis occurring after the patient’s death. Other published studies in which simulated postmortem delay (up to 15 hours) failed to show increased apoptosis in myocardial and brain tissue corroborate our findings. 36,37 Finally, extensive macrophage apoptosis was primarily localized to the rupture site, whereas DNA fragmentation in macrophages in the fibrous cap away from the rupture site and in stable plaques with similar postmortem intervals was significantly less.
Conclusions
Our findings, restricted to the plaque rupture site, demonstrate extensive focal apoptosis of macrophages. The physiological relevance of this finding is unknown. Because extensive macrophage apoptosis was found predominantly at the rupture site, it is tempting to postulate a direct relationship between the two processes. In contrast, rather than precipitating plaque rupture macrophage apoptosis may also occur secondarily to the rupture itself. Furthermore, it is also possible that macrophage apoptosis contributes to plaque thrombogenicity or plaque healing and may have nothing to do with the rupture itself. Because of the complex dynamics of acute plaque rupture, autopsy material cannot reveal direct cause-and-effect relationships. Such questions can more readily be answered in an animal model of plaque rupture, which currently does not exist.
Acknowledgments
We thank Lila Adams, Russ Jones, and Marie Jenkins for technical support.
Footnotes
Address reprint requests to Renu Virmani, M.D., Armed Forces Institute of Pathology, Department of Cardiovascular Pathology, Building 54, Room 2005, 6825 16th St., NW, Washington, DC 20306-0004. E-mail: virmani@afip.osd.mil.
Supported in part by research grants from the American Registry of Pathology and the National Institutes of Health (RO1 HL61799–02).
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting views of the United States Army, Navy, Air Force, or the Department of Defense.
References
- 1.Farb A, Burke AP, Tang AL, Liang Y, Mannan P, Smialek J, Virmani R: Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation 1996, 93:1354-1363 [DOI] [PubMed] [Google Scholar]
- 2.Davies MJ: Anatomic features in victims of sudden coronary death. Coronary artery pathology. Circulation 1992, 85:19-24 [PubMed] [Google Scholar]
- 3.Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GR, Lee RT: Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol 1996, 7:330-335 [DOI] [PubMed] [Google Scholar]
- 4.Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R: Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med 1997, 336:1276-1282 [DOI] [PubMed] [Google Scholar]
- 5.Richardson PD, Davies MJ, Born GV: Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet 1989, 2:941-944 [DOI] [PubMed] [Google Scholar]
- 6.Burke AP, Farb A, Malcom GT, Liang Y, Smialek JE, Virmani R: Plaque rupture and sudden death related to exertion in men with coronary artery disease. JAMA 1999, 281:921-926 [DOI] [PubMed] [Google Scholar]
- 7.Davies MJ, Woolf N, Rowles P, Richardson PD: Lipid and cellular constituents of unstable human aortic plaques. Basic Res Cardiol 1994, 89(Suppl 1):33-39 [DOI] [PubMed] [Google Scholar]
- 8.Shah PK, Falk E, Badimon JJ, Fernandez-Ortiz A, Maihac A, Villareal-Levy G, Fallon JJ, Regnstrom J, Fuster V: Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 1995, 92:1565-1569 [PubMed] [Google Scholar]
- 9.Geng YJ, Henderson LE, Levesque EB, Muszynski M, Libby P: Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1997, 17:2200-2208 [DOI] [PubMed] [Google Scholar]
- 10.Geng YJ, Libby P: Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1 beta-converting enzyme. Am J Pathol 1995, 147:251-266 [PMC free article] [PubMed] [Google Scholar]
- 11.Isner JM, Kearney M, Bortman S, Passeri J: Apoptosis in human atherosclerosis and restenosis. Circulation 1995, 91:2703-2711 [DOI] [PubMed] [Google Scholar]
- 12.Mallat Z, Ohan J, Leseche G, Tedgui A: Colocalization of CPP-32 with apoptotic cells in human atherosclerotic plaques. Circulation 1997, 96:424-428 [DOI] [PubMed] [Google Scholar]
- 13.Han DK, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G: Evidence for apoptosis in human atherogenesis and in a rat vascular injury model. Am J Pathol 1995, 147:267-277 [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorkerud S, Bjorkerud B: Apoptosis is abundant in human atherosclerotic lesions, especially in inflammatory cells (macrophages and T cells), and may contribute to the accumulation of gruel and plaque instability. Am J Pathol 1996, 149:367-380 [PMC free article] [PubMed] [Google Scholar]
- 15.Kragel AH, Reddy SG, Wittes JT, Roberts WC: Morphometric analysis of the composition of atherosclerotic plaques in the four major epicardial coronary arteries in acute myocardial infarction and in sudden coronary death. Circulation 1989, 80:1747-1756 [DOI] [PubMed] [Google Scholar]
- 16.Farb A, Tang AL, Burke AP, Sessums L, Liang Y, Virmani R: Sudden coronary death. Frequency of active coronary lesions, inactive coronary lesions, and myocardial infarction. Circulation 1995, 92:1701-1709 [DOI] [PubMed] [Google Scholar]
- 17.Berryman MA, Rodewald RD: An enhanced method for post-embedding immunocytochemical staining which preserves cell membranes. J Histochem Cytochem 1990, 38:159-170 [DOI] [PubMed] [Google Scholar]
- 18.Goping G, Wood KA, Sei Y, Pollard HB: Detection of fragmented DNA in apoptotic cells embedded in LR white: a combined histochemical (LM) and ultrastructural (EM) study. J Histochem Cytochem 1999, 47:561-568 [DOI] [PubMed] [Google Scholar]
- 19.Lee RT, Libby P: The unstable atheroma. Arterioscler Thromb Vasc Biol 1997, 17:1859-1867 [DOI] [PubMed] [Google Scholar]
- 20.Boyle JJ, Bowyer DE, Proudfoot D, Weissberg PL, Bennett MR: Human monocyte/macrophages induce human vascular smooth muscle cell apoptosis in culture. Circulation 1998, 98(Suppl I):I-598(abstract) [Google Scholar]
- 21.Lendon CL, Davies MJ, Born GV, Richardson PD: Atherosclerotic plaque caps are locally weakened when macrophages density is increased. Atherosclerosis 1991, 87:87-90 [DOI] [PubMed] [Google Scholar]
- 22.Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J: Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J 1993, 69:377-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crisby M, Kallin B, Thyberg J, Zhivotovsky B, Orrenius S, Kostulas V, Nilsson J: Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis 1997, 130:17-27 [DOI] [PubMed] [Google Scholar]
- 24.Trump BF, Berezesky IK, Chang SH, Phelps PC: The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol 1997, 25:82-88 [DOI] [PubMed] [Google Scholar]
- 25.Meredith JE, Jr, Fazeli B, Schwartz MA: The extracellular matrix as a cell survival factor. Mol Biol Cell 1993, 4:953-961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch SM, Francis H: Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994, 124:619-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frisch SM, Ruoslahti E: Integrins and anoikis. Curr Opin Cell Biol 1997, 9:701-706 [DOI] [PubMed] [Google Scholar]
- 28.Judware R, McCormick TS, Mohr S, Yun JK, Lapetina EG: Propensity for macrophage apoptosis is related to the pattern of expression and function of integrin extracellular matrix receptors. Biochem Biophys Res Commun 1998, 246:507-512 [DOI] [PubMed] [Google Scholar]
- 29.Baker AH, Zaltsman AB, George SJ, Newby AC: Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest 1998, 101:1478-1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fabunmi RP, Sukhova GK, Sugiyama S, Libby P: Expression of tissue inhibitor of metalloproteinases-3 in human atheroma and regulation in lesion-associated cells: a potential protective mechanism in plaque stability. Circ Res 1998, 83:270-278 [DOI] [PubMed] [Google Scholar]
- 31.Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A: Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 1999, 99:348-353 [DOI] [PubMed] [Google Scholar]
- 32.Miller DK, Ayala JM, Egger LA, Raju SM, Yamin TT, Ding GJ, Gaffney EP, Howard AD, Palyha OC, Rolando AM: Purification and characterization of active human interleukin-1 beta-converting enzyme from THP. 1 monocytic cells. 1806, :2-18069 268: J Biol Chem 1993 [PubMed] [Google Scholar]
- 33.Miller DK, Myerson J, Becker JW: The interleukin-1 beta converting enzyme family of cysteine proteases. J Cell Biochem 1997, 64:2-10 [DOI] [PubMed] [Google Scholar]
- 34.Yamin TT, Ayala JM, Miller DK: Activation of the native 45-kDa precursor form of interleukin-1-converting enzyme. J Biol Chem 1996, 271:13273-13282 [DOI] [PubMed] [Google Scholar]
- 35.Nagata S: Biddable death. Nat Cell Biol 1999, 1:E143-E145 [DOI] [PubMed] [Google Scholar]
- 36.Schallock K, Schulz-Schaeffer WJ, Giese A, Kretzschmar HA: Postmortem delay and temperature conditions affect the in situ end-labeling (ISEL) assay in brain tissue of mice. Clin Neuropathol 1997, 16:133-136 [PubMed] [Google Scholar]
- 37.Veinot JP, Gattinger DA, Fliss H: Early apoptosis in human myocardial infarcts. Hum Pathol 1997, 28:485-492 [DOI] [PubMed] [Google Scholar]