

Abstract
Large and protracted elevations of intracellular [Ca2+] and [Na+] play a crucial role in neuronal injury in ischemic conditions. In addition to excessive glutamate receptor activation, other ion channels may contribute to disruption of intracellular ionic homeostasis. During episodes of ischemia, extracellular [Ca2+] falls significantly. Here we report the emergence of an inward current in hippocampal CA1 pyramidal neurons in acute brain slices from adult mice upon reduction/removal of [Ca2+]e. The magnitude of the current was 100–300 pA at −65 mV holding potential, depending on intracellular constituents. The current was accompanied by intense neuronal discharge, observed in both whole-cell and cell-attached patch configurations. Sustained currents and increased neuronal firing rates were both reversed by restoration of physiological levels of [Ca2+]e, or by application of spermine (1 mM). The amplitudes of the sustained currents were strongly reduced by raising intracellular [Mg2+], but not by extracellular [Mg2+] increases. Elevated intracellular ATP also reduced the current. This conductance is similar in several respects to the “calcium-sensing, non-selective cation current” (csNSC), previously described in cultured mouse hippocampal neurons of embryonic origin. The dependence on intracellular [ATP] and [Mg2+] shown here, suggests a possible role for this current in disruption of ionic homeostasis during metabolic stress that accompanies excessive neuronal stimulation.
Keywords: transient receptor potential, store-operated Ca2+ entry, spreading depression, stroke, TRPM7, Ca2+ paradox
An excessive increase in intracellular Ca2+ concentration has been repeatedly implicated as a key determinant of neuronal cell death pathways (Arundine and Tymianski 2003). Sustained cytosolic Ca2+ increases can be triggered by a variety of insults, and often involve persistent activation of the NMDA subtype of glutamate receptors. This leads to loading of mitochondria with large amounts of Ca2+ compromising oxidative phosphorylation, ultimately resulting in intracellular ATP depletion (Greene and Greenamyre 1996;Nicholls and Budd 2000).
Interstitial [Ca2+] levels drop during neurotransmission at central excitatory synapses, as a consequence of influx into both pre- and post-synaptic neurons (Nicholson et al. 1978). This is exacerbated in pathological states involving excessive glutamate receptor stimulation, such as during ischemia or traumatic brain injury, in which extracellular Ca2+ concentration can be reduced to approximately 0.1 mM (Harris et al. 1981;Silver and Erecinska 1990;Nilsson et al. 1996). In ischemia, [Ca2+]e reductions produced by neuronal Ca2+ influx may also be compounded by the accumulation of extracellular lactate, which is an effective chelator of divalent cations (Martell and Smith 1977;Immke and McCleskey 2001).
Fluctuations in [Ca2+]e are sensed by a range of plasma membrane Ca2+-sensing receptors and ion channels (Hofer 2005). Furthermore, the voltage dependence of many types of monovalent ion channels is shifted in the absence of divalent cations. For example, the voltage-dependence of a number of voltage-gated Ca2+ and Na+ channels may be shifted to more negative potentials by lowering [Ca2+]e, as a consequence of reduced shielding of negatively charged groups located at the membrane surface (Armstrong 1999;Armstrong and Cota 1999;Frankenhaeuser and Hodgkin 1957;Fukushima and Hagiwara 1985). Chloride channels are also sensitive to alterations in [Cl−]e (Liu and Stimers 1998). In extreme cases, selectivity of voltage-dependent channels can also be lost when [Ca2+]e is experimentally reduced to ultra-low (<1 μM) concentrations (Xiong and MacDonald 1999) and lead to significant additional cation influx.
Recently, a number of non-selective currents triggered by reductions in [Ca2+]e have been described (Formenti et al. 2001;Undem et al. 2003;Xiong et al. 1997;Smith et al. 2004;Mubagwa et al. 1997;Smith et al. 2004). These channels can produce significant inward current and their lack of inactivation may lead to Na+ overloading following extracellular Ca2+ decreases. While the channel identities carrying these currents are at present unknown, conductances attributed to several members of the “transient receptor potential” (TRP) family, such as TRPC1-3-6-7, TRPM7 and TRPV5-6 are known to be enhanced by [Ca2+]e reductions (Lintschinger et al. 2000;Shi et al. 2004;Aarts et al. 2003;Owsianik et al. 2005). Relevant to this, it has been shown that TRPM7 channels underlie currents measured in cultured neurons that contribute to cell death in an in vitro model of ischemic neuronal damage (Aarts et al. 2003).
Since previous work in cultured hippocampal neurons demonstrated a non-selective cation current activated by [Ca2+]e decreases (Xiong et al., 1997), the present study was undertaken to determine whether in situ mature neurons express an analogous current since it is widely recognized that there can be significant differences in channel expression and functioning between cell culture models and mature neurons. We show that the CA1 pyramidal neurons in acute hippocampal slices from adult mice exhibit a significant inward current upon reduction of [Ca2+]e. Furthermore, this current is regulated by intracellular [Mg2+] and [ATP], making it a potential contributor to increased Na+ influx during neuronal metabolic compromise.
EXPERIMENTAL PROCEDURES
Slice preparation
Male mice (FVB/N) were obtained from Harlan Laboratories (Indianapolis, IN) at 4–6 weeks of age and were housed in standard conditions (12 hr light/dark cycle) for at least 2 weeks before death. The currents triggered by extracellular Ca2+/Mg2+ removal (see Results) were not as readily observed in slices prepared from younger animals (<6 weeks), and therefore the study was restricted to the 6–8 week age group. All animal procedures were carried out in accordance with the National Institutes of Health and the University of New Mexico, Animal Care and Use Committee Guidelines. Mice were anesthetized with a mixture of ketamine and xylazine (85 and 15 mg/ml, respectively; 0.2 ml, subcutaneously) and subsequently decapitated. The use of ketamine during brain slice preparation improves the viability and maintains in situ morphology of CA1 neurons (Feig and Lipton 1990;Aitken et al. 1995;Lipton et al. 1995), and is washed from slices before recording. Brains were removed and placed in ice-cold cutting solution (see below for composition), equilibrated with 95%O2/5%CO2. Coronal sections (350 μm) were cut with a Vibratome (Technical Products International, St. Louis, MO), and slices were transferred into artificial cerebrospinal fluid (ACSF, see below for composition), equilibrated with 95%O2/5%CO2 and kept at 34°C. Cutting and recording solutions were both 300–310 mOsm. After being warmed to 34°C for 1 hr, ACSF was changed again, and the slices were maintained at room temperature until used for recording. Individual slices were transferred to the recording chamber and perfused with warmed (32±0.5 °C), oxygenated ACSF at 2 ml/min.
Electrophysiological recordings
Somatic whole-cell and cell-attached recordings were made from visualized CA1 pyramidal neurons, using a Multiclamp 700A amplifier (Axon Instruments, Union city, CA). Signals were digitized at 10–25 kHz (Digidata 1322A), and acquired using Clampex version 9 software (Axon Instruments). Patch pipettes intended for whole-cell recordings had resistances of 2–4 MOhm, and 2–2.5 MOhm for cell-attached recordings. Seal resistances were typically 2–7 GOhm before whole-cell access. Immediately after obtaining whole-cell access, cells were discarded if the access resistance was more than 20 MOhm, or leak currents exceeded ±100 pA at −65 mV holding potential. All cells were dialyzed for 3–4 min before initiation of experimental manipulations. If the access resistance varied more than 20% from the beginning to the end of the experiment, the recordings were excluded from analyses. For cell-attached recordings, pipettes were filled with ACSF (including 2 mM CaCl2 and 1 mM MgSO4) and the pipette potential was held at 0 mV. “Leak” currents were not subtracted for any of the recordings.
Materials
All chemicals used in this study were obtained from Sigma Chemical Co. (St. Louis, MO). Slice cutting solution comprised (in mM): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 6 MgSO4, 26 NaHCO3, 0.2 CaCl2, 10 glucose, and 0.43 ketamine and was equilibrated with 95%O2/5%CO2. The use of ketamine is based on observations described in (Aitken et al. 1995), and ketamine is thoroughly washed from slices (>2 hours, multiple solution exchanges) before beginning experiments. Artificial cerebrospinal fluid (ACSF) comprised (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 glucose was equilibrated with 95%O2/5%CO2. Cutting and recording solutions were both 300–310 mOsm. Nominally Ca2+-free solutions were prepared by omission of CaCl2 (no added chelators) and the free extracellular Ca2+ concentration of nominally-Ca2+-free ACSF was estimated at 3.8 μM, using Fura 6F ratio fluorescence measurements, and assuming a Kd value of 2.47 μM (Chinopoulos et al. 2003). In some experiments [Ca2+]e was buffered to very low concentrations (~1 nM) by the omission of extracellular CaCl2 and the inclusion of 0.5 mM EGTA. Table 1 lists the composition of intracellular recording solutions. In experiments to examine the effects of intracellular [Mg2+] and [ATP], estimated free [Mg2+] was calculated by using Winmaxc software (Bers et al., 1994). The ATP source contained less than 0.005% Mg2+ as a contaminant (Sigma, A7699, disodium salt), which was calculated to contribute less than 0.2 μM Mg2+ to pipette solutions when used at the highest concentration tested (4 mM).
Table 1.
Intracellular solutions used for electrophysiological recordings.
| CaCl2 | MgCl2 | NaCl | KCl | Glucose | EGTA | K gluconate | CsCl | Cs MES | ATP DiNa+ | |
|---|---|---|---|---|---|---|---|---|---|---|
| Int* 1 | 1 | - | 15 | 40 | 10 | 11 | 70 | - | - | - |
| Int 2 | 1 | 0.2 | 15 | 40 | 10 | 11 | 70 | - | - | - |
| Int 3 | 1 | 2 | 15 | 40 | 10 | 11 | 70 | - | - | - |
| Int 4 | 1 | 2 | 7 | 40 | 10 | 11 | 70 | - | - | 4 |
| Int 5 | 1 | - | 15 | - | 10 | 11 | - | 40 | 70 | - |
| Int 6 | 1 | 0.2 | 15 | - | 10 | 11 | - | 40 | 70 | - |
| Int 7 | 1 | 2 | 15 | - | 10 | 11 | - | 40 | 70 | - |
| Int 8 | 1 | 2 | 7 | - | 10 | 11 | - | 40 | 70 | 4 |
Int: Intracellular solution. All values are given in mM. pH was adjusted to 7.25 with KOH or CsOH (with buffer HEPES, 10 mM), depending whether the major cation was vK+ or Cs+, respectively. Intracellular solutions containing CsCl and Cs-MES were also supplemented with 2 mM TEA chloride. Cs-MES: Cs+ methanosulfonate. The free [Ca2+]i was estimated to be 10–30 nM, using the Winmaxc software (Bers et al. 1994) for all solutions.
Statistics
Data are presented as mean ± SEM; significant differences between group data were evaluated by using Mann-Whitney rank sum test, with p < 0.05 considered significant, unless otherwise indicated. For results showing representative recordings, each experiment was recorded from 4 to 17 neurons. Meta-analysis of electrophysiological recordings was performed using Clampfit software version 9.2 (Axon Instruments) and Mini Analysis version 5 (Synaptosoft Inc, Decatur, GA).
RESULTS
Non-inactivating inward current and neuronal discharge produced by reduction of extracellular divalent cation concentration
Figure 1A shows an example of the effects of reducing extracellular divalent cation concentration on currents measured from a CA1 pyramidal neuron. The neuron soma was voltage clamped at −65 mV, and the superfusion buffer was switched from normal ACSF (2 mM Ca2+, 1 mM Mg2+) to a modified ACSF containing no added Ca2+ or Mg2+ (nominally Ca2+/Mg2+-free). A K+-based intracellular solution was used (termed “Int 2”). Approximately 50 sec after onset of exposure, a slowly-developing inward current was observed. This sustained current was also associated with intense neuronal discharge, as evidenced by superimposed large transient inward currents, (see also figure 8). Both the sustained inward current and neuronal discharge were abolished by restoration of normal extracellular Ca2+/Mg2+ concentrations. Similar sustained currents were produced in 10 out of 12 cells examined under these conditions, and the mean peak amplitude of responders was −145±18 pA. In the remaining two neurons, no inward current was observed throughout at least 6 min of divalent cation removal. In responding neurons, the delay before demonstrable onset of the inward current was variable (range: 50–250 sec). The time lag is likely due to the relatively slow exchange of solutions in the slice superfusion system, and also the range of depths from which CA1 neurons were recorded (<60 μm from the top surface of the slice).
Figure 1. Removal of extracellular divalent cations produces a non-inactivating inward current accompanied by neuronal firing.
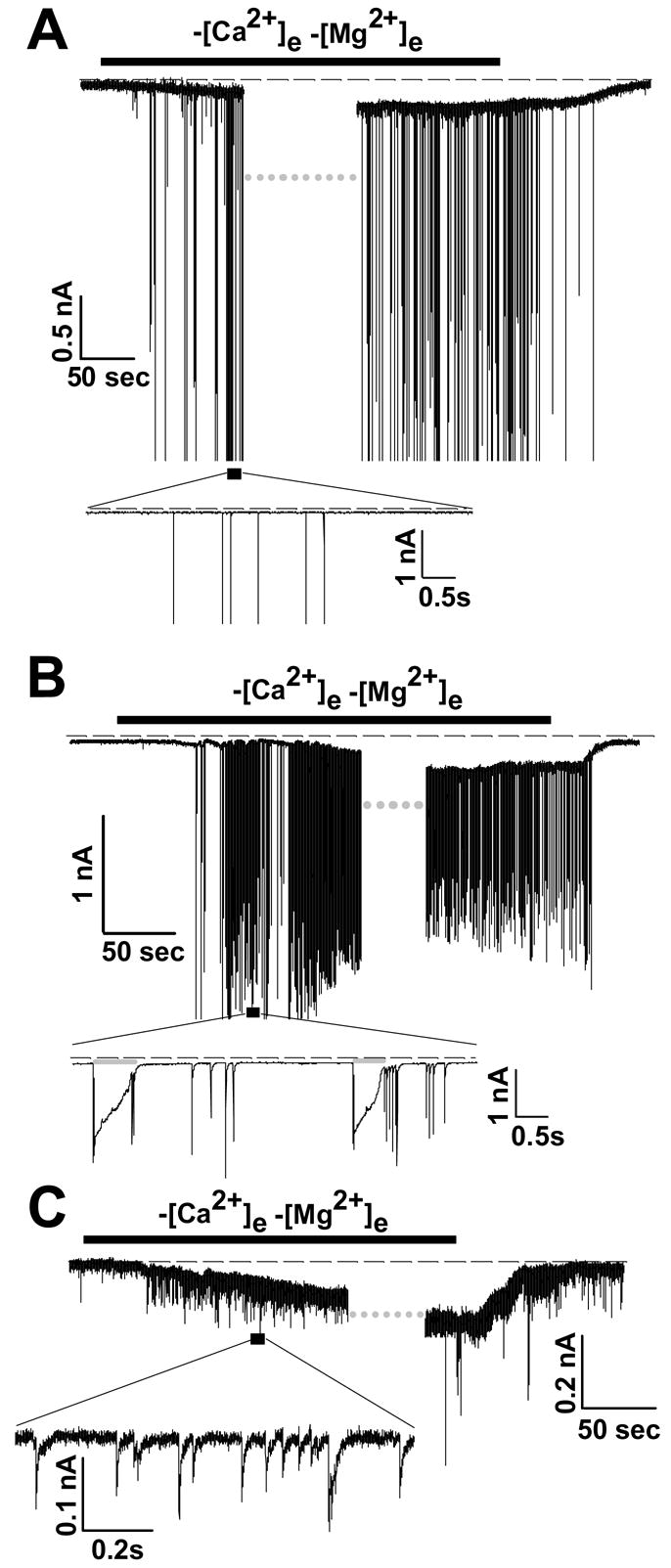
A: Whole cell recording, with K+-containing intracellular solution, in normal ACSF (2mM Ca2+, 1mM Mg2+). After onset of a sustained inward current triggered by Ca2+/Mg2+ removal (indicated by black bar), data acquisition was interrupted (at grey dotted bar) for application of voltage ramp protocols (see figure 6). Following resumption of continuous data acquisition, the current and spiking were terminated by restoration of Ca2+ and Mg2+. Dashed line indicates zero current in this and all figures. B: Conditions of this experiment are identical to those in A, except that the intracellular solution contained Cs+ as the major cation plus 2 mM TEA (“Int 6”), in lieu of K+. A similar inward current and spiking activity was recorded. C: The conditions of this experiment are identical as for “B”; however, no neuronal firing is observed in this neuron. The inset is an expanded region of the area indicated by the black box, to show small excitatory currents generated by Ca2+/Mg2+ -free solution in this neuron.
Figure 8. Characterization of neuronal firing accompanying the sustained inward current.

A: Left graph: Current clamp recording of a patched neuron in the whole cell mode, during superfusion with nominally [Ca2+]e-free ACSF; right graph: Interspike voltage (mV) of the recording depicted on the left graph. B: Left graph: Quantification of current spike frequency as a function of interspike Vm, for the same neuron as shown in A. B, Right graph: Quantification of current spike frequency over gradual injection of current on the patched neuron through the pipette. Top x-axis depicts the measured Vm during injection of current. C: Voltage clamp recording of a patched neuron in the whole cell mode, during superfusion with nominally [Ca2+]e-free ACSF, but in the presence of intracellular Mg2+ and ATP (“Int 4”). Current spikes are truncated.
A sustained inward current and neuronal discharge were also observed when K+ channels were blocked by substitution of intracellular K+ with Cs+ and TEA (solution “Int 6”). An example is shown in Figure 1B, and similar responses were observed in 7/9 neurons tested (12±3 Hz firing rate). K+ channel block also increased the duration of current spikes (grey bars Figure 1B, inset), that followed extracellular divalent removal (compare with Figure 1A inset). This increased duration implies that a portion of the transient current was generated intrinsically in unclamped regions of the recorded neuron, and was not entirely due to presynaptic input.
Although [Ca2+]e reduction triggered sustained inward currents in a high proportion of neurons (see above), a concomitant increase in current spike rate was not observed in every case. Some neurons (12 out of a total of 109 cells patched in the whole-cell mode under all intra- and extracellular conditions described throughout this study) showed no increase in spike rate, while 7 neurons showed an increase in rate of small excitatory currents (Figure 1C, inset) without the appearance of the large, spike-like events.
As a control for dialysis effects in the whole cell mode, figure 2 shows the effects of extracellular Ca2+ and Mg2+ reduction on recordings made in the cell-attached recording mode. While small sustained inward currents are not observable in the cell-attached mode, increases in the rate of large transient events (current spikes) should be detectable. As before, neurons were bathed in normal ACSF and then exposed to Ca2+/Mg2+-free solution. In most cases (~75%), tonic firing was observed following Ca2+/Mg2+ reduction (28±1 Hz). Figure 2A shows an example where a progressive increase in tonic discharge rate was followed by a progressive decrease in amplitude of the membrane current observed through the window of membrane capacitance; the decrease representing either a decrease of the spike upstroke slope, or increasing distance of spike generation from the soma. In a minority of cases (~15%) a bursting firing pattern was observed (figures 2C&D) following Ca2+/Mg2+ reduction, and the number of firing per cluster progressively increased over time in Ca2+/Mg2+-free media. Similar responses (both tonic and burst-like) were recorded in the cell-attached mode following removal of extracellular Ca2+ only (not shown). In the remaining population of patched neurons, no spike activity was observed.
Figure 2. Increased activity produced by removal of divalent cations, recorded in cell-attached mode.

A: Recording of a CA1 pyramidal neuron perfused in the absence of extracellular divalent cations, as indicated by the top black bar. B: Expanded regions corresponding to those depicted in the recording shown in A, showing a tonic firing pattern (seen in ~75 % of neurons tested). C&D: Identical experimental conditions as in A, but in this example, a burst firing pattern was seen following removal of extracellular divalent cations (seen in ~15% of neurons tested).
The non-inactivating current cannot be accounted for by seal deterioration upon reduction of extracellular divalent cations
Formenti and De Simoni (Formenti and De 2000) previously demonstrated that reduction of extracellular Ca2+ leads to a decrease in seal resistance and therefore “currents” may appear due to leak conductance from the pipette to the bath solution. The extent of this decrease depends on the value of the initial resistance. To address this possibility, and to quantify how much “leak-seal” current potentially contaminates our recordings, we examined the effects of extracellular divalent cation removal on seal resistance, when recording in the cell-attached mode (figure 3). Seal resistances were calculated from current-voltage (I–V) curves (r2>0.95 in all cases). When the initial seal resistance was 1.96±0.11 GOhm, (n=5, initial pipette resistance ~2 MOhm), removal of extracellular Ca2+ and Mg2+ produced a small decrease in seal resistance (to 1.72±0.25 GOhm, p=0.41, not statistically significant, Student t-test, p=0.411, figure 3A). This translates to a difference in conductance from 510 to 581 pS. Assuming a resting membrane potential of −65 mV, and that the reversal potential of the “current” appearing due to deterioration of seal resistance is 0 mV, we estimate that the “leak-seal” conductance is (581 pS - 510 pS)*(−65mV - 0mV) = 4.6 pA. This is too small to alter the values obtained from whole-cell recordings, upon removal of extracellular Ca2+, which is on the order of hundreds of pA (see figure 7). Note also that the extrapolated value for the holding current is approximately −35 pA at −65 mV, when the seal is ~2 GOhm, a value which is close to the recorded one in control ACSF (see figure 7). However, as pointed out previously (Chu et al. 2003), it can be argued that during the formation of the whole-cell patch configuration, the seal resistance is reduced due to a physical disruption of the patch membrane, rendering it more sensitive to the changes in [Ca2+]e. We tested this by forming seals with an initial resistance of ~500 MOhm. Removal of extracellular Ca2+/Mg2+ then led to a larger, and statistically significant decrease in seal resistance from 0.569±16 to 0.411±17 MOhm (Figure 3B, n=5, Student t-test, p <0.001), as calculated from fitting a line on the slope of the I–V curve (r2= 0.96, both fittings). This translates to a difference in conductance from 1.76 to 2.43 nS. Applying the same calculations as above, the expected increase in “current” due to diminished seal formation, is ~ 44 pA, much larger than when the seal resistance was ~2 GOhm, and closer to the values obtained during our whole-cell recordings, comparing the holding current in the presence and absence of extracellular divalent cations. However, the values obtained from our whole-cell recordings are still significantly larger than 44 pA (figure 7). Moreover, in our hands immediately after whole-cell access, the holding current at −65 mV is significantly lower than 44 pA, therefore the extrapolated value of the seal resistance should be well above 500 MOhm. Hence, it seems reasonable to conclude that our measured conductances upon removal of extracellular divalent cations are due to biological phenomena, though most conservatively overestimated by ~40–50 pA.
Figure 3. Effects of reduction of extracellular divalent cations on seal resistance.

A: Current-voltage relationships from cell-attached recordings in the presence (black circles) or absence (grey triangles) of extracellular divalent cations (mean ±SEM of 5 cells each). Seal resistance (Rs) was adjusted by mouth suction to ~ 2 GOhm and divalent cations removal had a minor effect on seal resistance. B: Recordings performed as for A, except that Rs was adjusted by mouth suction to ~0.5 GOhm, and resulted in significantly greater decreases in seal resistance following divalent cations removal.
Figure 7. Estimates of currents induced by removal of extracellular Ca2+ at −65 mV holding potential in whole cell recordings, and effect of spermine.

A: The intracellular solution contains K+ as the major cation (Int “1–4”). B: The intracellular solution contains Cs+ as the major cation (Int “5–8”). Hatched bars signify reintroduction of Ca2+ (and Mg2+ if it was removed previously) to the perfusate. *, significant, (p<0.05), Mann-Whitney rank sum test. Each bar represents the mean value of 5–9 cells. Non-responding cells were not included in the calculations of the mean currents. Free [Mg2+]i and [ATP] concentration are shown on the corresponding group of bars. The total [Mg2+]i and [ATP] are identical to that shown for figure 6. The currents measured with 0.08 mM free [Mg2+]i is statistically significantly higher than in the presence of 0.07 mM free [Mg2+]i plus 2.2 mM ATP, irrespective if the major intracellular cation is K+ or Cs+ (p<0.005). C: Whole cell recordings of neurons perfused with either nominally Ca2+-free ACSF (grey bar) as compared to the presence of 0.1 mM spermine in the perfusate (grey-white hatched bar), or 1 mM spermine (white-black hatched bar). The intracellular solution for these experiments is “Int 6”. In panel D, a representative recording of a voltage clamped neuron in the whole cell mode is shown, where 1 mM spermine ameliorates against the –low-[Ca2+]e–induced current; current spikes are truncated.
Reduction of extracellular Ca2+ alone produces a sustained inward current and neuronal firing
Figure 4 shows the effects of removal of [Ca2+]e alone, while maintaining [Mg2+]e constant at 1 mM. This had an effect similar to removal of both Ca2+ and Mg2+ (compare with figures 1&2); current spikes have been thresholded out (by removing data points that overshot below −0.35 nA) for visual clarity of the sustained current. This was reversed by re-introduction of 2 mM Ca2+ to the perfusate. Removal of Ca2+e accompanied by equimolar replacement of extracellular Mg2+, in order to maintain a constant concentration of extracellular divalent cations and minimize possible changes in charge screening didn’t affect the sustained inward current (data not shown).
Figure 4. Inward currents produced by removal of Ca2+ alone.

Whole cell recording (holding potential =−65 mV), showing inward current produced by Ca2+ removal, retaining extracellular Mg2+ constant at 1 mM. Current spikes are truncated.
Figure 5A compares the mean inward currents obtained by reduction of extracellular Ca2+ from 2 mM to either nominally Ca2+-free or to 100 μM Ca2+, in whole-cell recordings. In cell-attached recordings, reduction of [Ca2+]e to 100 μM caused a trivial decrease in seal resistance, that would account to no more than 1–2 pA increase in “leak-seal” currents (not shown) and produced a demonstrable net inward current (white bar), though significantly less than that produced by reduction to nominally Ca2+-free solution (~3.8 μM, grey bar). An example of the effects of reduction to 100 μM extracellular Ca2+ is shown in Figure 5B (representative of 5 out of 5 neurons tested). The neuron was voltage clamped at −65 mV in the whole cell configuration and superfusion with 100 μM [Ca2+]e produced a small but discernible inward current and neuronal discharge. Reduction of [Ca2+]e to very low concentrations (~1 nM) by addition of 0.5 mM EGTA produced much larger currents (~ −700 pA, not shown). Although we recorded the greatest responses in the presence of 1 nM [Ca2+]e, we chose to characterize further the sustained inward current by reducing extracellular Ca2+ to 3.8 μM. This is because many types of channels are influenced by reduction of [Ca2+]e to nanomolar levels, reviewed by (Xiong and MacDonald 1999). Working with ACSF in which [Ca2+]e would be adjusted to 0.1 mM is potentially more relevant to in vivo conditions, however the currents that it produces are much smaller, making it more very difficult to evaluate the significance of changes due to intracellular Mg2+ and ATP manipulation described below.
Figure 5. Effects of moderate decreases in extracellular Ca2+.

A: Mean differences of peak minus the base leak amplitudes of sustained inward currents, generated by nominally Ca2+-free solution (grey bar) or 0.1mM Ca2+e (white bar). [Mg2+]e was constant at 1 mM. B: Representative recording of a neuron perfused with ACSF containing 100 μM [Ca2+]e. The intracellular solution contains K+ as the major cation (Int “2”). Neuronal discharge also accompanies the sustained inward current.
Characterization of the non-inactivating inward current, as a function of intracellular Mg2+ and ATP
In Figure 6, representative experiments are shown in which somata were voltage-clamped in the whole cell configuration (Cs+ being the major intracellular cation in all cases), and voltage ramps (−40 to +40 mV) were applied starting from a holding potential of 0 mV (5–10 cells per condition, see figure 6 inset for voltage ramp waveform). This protocol avoids initiation of neuronal discharge, as Na+ channels remain inactivated by holding at 0 mV, and by not exceeding −40 mV during application of the waveform. All panels show currents recorded in 2 mM extracellular Ca2+ (thin black curves), and currents following exposure to nominally Ca2+-free solution (grey curves). [Mg2+]e was held constant at 1 mM, but intracellular Mg2+ was progressively increased in separate sets of recordings in different neurons. When Mg2+ was omitted from the patch pipette (Panel A), removal of extracellular Ca2+ produced a robust current activation. This current was still present at very low Mg2+i levels in the patch pipette (calculated 0.08 mM free Mg2+i, Panel B), but was absent at 0.9 mM Mg2+i (Panel C). In addition, the current observed in low free Mg2+i was prevented if the intracellular solution contained 4 mM ATP (Panel D). The sustained current appearing upon removal of extracellular Ca2+ was reversed following reintroduction of Ca2+ to the bathing medium (panel A, dotted curve, and figure 7). The emergence of this current (when [Mg2+]i is 0) shifted the reversal potential of the overall conductances of the cell by approximately 28 mV towards positive with Cs+ as the major intracellular cation, and by approximately 31 mV when K+ was the major cationic charge carrier (not shown). Subtraction of the I–V curve performed in the absence of extracellular [Ca2+] from that performed in the presence of [Ca2+]e, resulted in an I–V curve that reversed around −5 mV (panel A, thick black curve “d”). In conditions in which the conductance is not activated by removing [Ca2+] (i.e. in the presence of 0.9 mM [Mg2+]i) the I–V curves in the presence or absence of [Ca2+]e are virtually identical. Therefore, the subtraction I–V curve appears to be due to the emergence of a conductance induced by removal of extracellular Ca2+, rather than modification of existing voltage-dependent currents by cation removal. Figure 7 shows mean values of peak inward currents recorded from a population of neurons. When Mg2+ was absent or very low (0.08 mM) in intracellular solutions, significant increases in peak current were observed with removal of Ca2+ alone, or removal of both Ca2+ and Mg2+. This was the case in experiments where K+ was the charge carrier (Figure 7A), and also when Cs2+ was the charge carrier (Figure 7B), and in all cases the effects of extracellular cation removal was reversible (hatched bars). With a higher intracellular Mg2+ concentration (0.9 mM), there was no significant increase in peak inward current following extracellular cation removal, compared with control values (black bars). No significant increase in peak inward current was observed following cation removal if intracellular ATP levels were elevated to 2.2 mM while maintaining low intracellular Mg2+ (0.07 mM).
Figure 6. Inhibition of currents evoked by Ca2+ removal by intracellular [Mg2+] and [ATP].
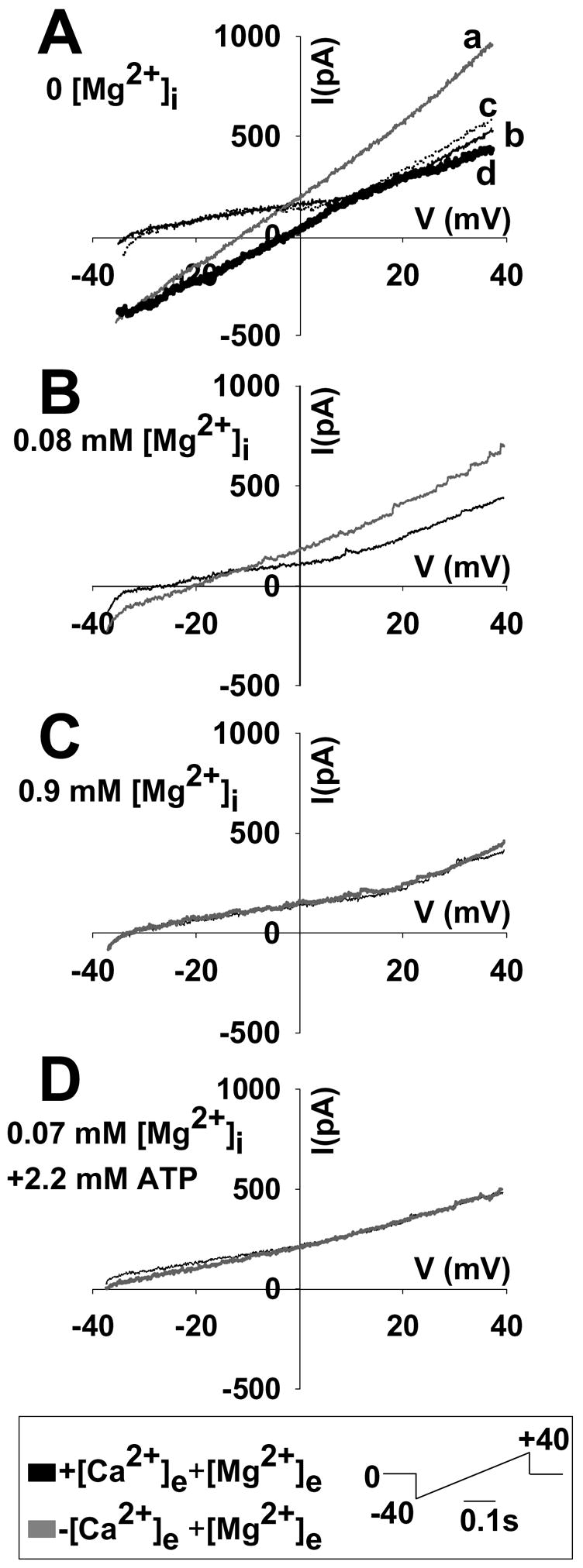
Currents were evoked by voltage ramps (−40 to +40 mV, see inset for waveform) from a holding potential of 0 mV. The intracellular solution contained Cs2+ as the major cation. Thin black curves indicate superfusion with normal ACSF, and grey curves indicate superfusion in nominally Ca2+-free ACSF (1mM Mg2+). Panels A–C show the effects of increasing Mg2+i concentrations. D shows the effects of addition of ATP to the intracellular solution. The calculated free [Mg2+]i and [ATP] are shown for each panel (calculated with Winmaxc, see Methods). The dotted curve on panel “A” indicates reintroduction of [Ca2+]e to the perfusate (curve “c”). The thick black curve on panel A is the result of subtracting curve “b” from curve “a”.
Effect of extracellularly applied spermine
Currents measured in other type of neurons upon removal of extracellular Ca2+ were sensitive to inhibition by a high concentration of extracellular spermine (Formenti et al. 2001;Xiong et al. 1997). Figure 7 (C&D) shows the effects of extracellular spermine on currents produced by superfusion with nominally Ca2+-free solution. The fact that substitution of [K+]i with Cs2+ and TEA does not affect the appearance of the inward current supports the hypothesis that the conductance is non-selective for K+/Cs+. In the experiments where spermine was applied, K+-based intracellular solution (Int 2) was used. As shown, 0.1 mM spermine had no significant effect on the sustained current (n=4), but 1 mM spermine virtually abolished the current (n=4, current spikes have been thresholded out for visual clarity).
Characterization and origin of the current spikes accompanying the sustained inward current
To investigate the basis of current spikes observed during Ca2+ removal, we examined spikes in current clamp recordings. As shown in figure 8A (left panel), reducing extracellular Ca2+ resulted in high frequency action potential firing and a significant depolarizing shift in the interspike voltage (fig. 8A, right panel, action potentials have been removed by threshold-exclusion of data more positive than 0 mV, for visual clarity). The decrease in the interspike voltage was inversely related to the [Mg2+]i. With 0.2 mM [Mg2+] in the recording pipette, membrane potential decreased to −57±2 mV, and with 0 [Mg2+], it diminished further to −50±3 mV (n=5, both conditions).
To test whether the currents measured under soma voltage clamp conditions were of an appropriate magnitude to produce such firing and depolarization, we applied a range of depolarizing currents to neurons bathed in normal ACSF. The range included the values determined for the population of neurons tested under soma voltage clamp (see Fig 7A). The firing rate/interspike depolarization relationships shown in Fig. 8B (left panel) were obtained by manually increasing the injected current from 0 to 150 pA (~ 20 pA increment steps/5 sec) and determining the parameters within a sampling interval of 1 sec. For comparison, the firing rate and depolarization profile of this same neuron during low-[Ca2+]e exposure are given in Fig. 8B (right panel), and is representative of 5 independent experiments. The order of the protocol applied (injection of current versus superfusion with Ca2+-free ACSF) did not influence the outcome. The upward slope of firing rate reflects the development of inward current under low Ca2+ exposure mimicked by gradual increase of applied current. The falloff in firing rate at high stimulus current is likely due to Na+ conductance inactivation, as the interspike potential is quite depolarized (fig. 8B left panel, measured Vm marked above the plot). This is not the cause in the low Ca2+ condition judging from the interspike potential. Removal of extracellular Ca2+ caused a greater increase in firing frequency (25±1 Hz, n=5), as compared to the frequency of current spikes appearing by injection of current through the pipette (21±1 Hz, n=5, p=0.009 statistically significant). This difference could be due to additional synaptic inputs to the patched neurons from projecting neurons in the slice that would be excited by the nominally Ca2+-free ACSF but not by injection of current into the postsynaptic neuron. Also the magnitude of the nominally Ca2+-free induced current is measured at a voltage-clamped soma. If the channels carrying the current are distributed over the neuron, this will be only a fraction of the total cellular current acting to initiate action potentials.
Inclusion of high [ATP] in the recording pipette abolished the sustained current observed in nominally Ca2+-free solution (figure 8C, current spikes have been thresholded out for visual clarity). Firing frequency in the presence of ATP in the pipette upon superfusion with nominally Ca2+ free ACSF was 26±1 Hz (n=4, not significantly different compared to the absence of ATP in the pipette). This reflects the possibility that other sources of excitability participate in the high frequency firing. This hypothesis is also supported by the finding that the frequency of the spikes is diminished by blocking glutamatergic transmission with 50 μM CNQX + 100 μM APV in some, but not all cases (data not shown).
DISCUSSION
The results of this study demonstrate that extracellular Ca2+ reductions produce a sustained inward current in hippocampal CA1 pyramidal neurons in brain slices from adult mice, associated with an increased frequency in action potential generation. This current appears similar to Ca2+e-sensitive non-selective currents reported in cultured neuronal preparations or freshly isolated neurons (Formenti and De 2000;Undem et al. 2003;Xiong et al. 1997). A novel finding of the present study is the demonstration of a strong negative dependence on intracellular Mg2+ and ATP concentrations not examined in other preparations, suggesting that increasing metabolic stress could be an important trigger (or at least play a permissive role) for this influx pathway in mature neurons.
The onset rate and amplitude of sustained inward currents in the present study were significantly lower that those measured in cultured hippocampal neurons of embryonic origin (Xiong et al. 1997). On several occasions we observed that prolonged bathing of the slice to nominally Ca2+-free solution (with or without extracellular Mg2+) led to large and precipitous decreases in the measured resistance with a zero reversal potential of the associated currents. Reintroduction of extracellular Ca2+ abolished this phenomenon in about half of the cases. We have conservatively attributed such large responses to loss of seal formation, however, we do not exclude the possibility that initially, activation of major currents led to the large decrease in membrane resistance that itself quickly resulted in a loss of seal. As described in Results, changes in seal quality were considered, and do not appear to explain the currents observed with nominally Ca2+-free solutions.
A small current activation was observed with Ca2+ reductions to 0.1 mM but for most characterization of the current described here, studies were performed with nominally Ca2+-free solutions (containing ~ 3.8 μM Ca2+) that provided larger amplitude currents (Figure 5). If the current described here contributes to neuronal demise in injuries such as stroke, it is possible that adjuvant circumstances (e.g. an oxidative stress) create conditions by which channel activation could be significantly increased during the more modest Ca2+ reductions (see below).
Dependence on intracellular Mg2+ and ATP
The observation that currents were much larger without ATP in the recording pipette raises the possibility that metabolic state is a regulator of this inward current. Thus, this conduit may be a critical determinant in ischemic or other conditions involving excitotoxicity, in which intracellular ATP levels are significantly decreased (Greene and Greenamyre 1996;Nicholls and Budd 2000;Nicotera and Lipton 1999). The mechanism(s) underlying ATP effects here are currently unknown. A caveat here is that ATP is a weak chelator of Mg2+, and therefore the equilibrium of the free Mg2+ ion to the Mg-ATP complex may be such that there is considerably more free Mg2+ in the dialysed cytosol (Kozak and Cahalan 2003). Alternatively, the configuration of the Mg-ATP complex may be such that, the Mg2+ moiety is still able to act on its target.
Current spikes and action potentials
Intense neuronal firing upon reduction of extracellular Ca2+ has been previously observed in rat hippocampal slices (Konnerth et al. 1986). Robust increases in firing were observed in the present study even in the somata voltage-clamp mode, implying origin in distal axon and/or dendrites that may include synaptic responses from other neurons in the slice. Synaptic origin of some of the firing is suggested by the observation that increasing the extracellular Mg2+ to 3 mM upon removal of extracellular Ca2+, substantially diminished spiking, while the sustained current was not affected. However, there appear to be additional mechanisms contributing to this intense neuronal discharge, since blocking excitatory neurotransmission with CNQX+APV did not always abolish the current spikes. The reason for this variance is beyond the scope of the present study and was not further investigated. However it was previously reported that NMDA receptors mediate epileptiform activity induced by the absence of extracellular Mg2+ in some conditions but not others, depending on the individual morphology of the dendritic projections (Thomson and West 1986). In addition, it is not obvious if the projecting non-dialysed neurons meet the intracellular requirements (reduction of intracellular Mg2+ and ATP) during reduction of extracellular Ca2+, in order to exhibit the sustained current – or conditions might be different in nerve terminals – i.e. there may not be the same Mg2+/ATP dependence there. For a comprehensive discussion on the relation of extracellular Ca2+ to seizure-like phenomena in slice, the reader is referred to (Heinemann et al. 1990).
Relationship to known currents and channels
At this juncture, it is not known how the other Ca2+ sensing mechanisms that are found in the hippocampus, such as voltage-dependent Ca2+ channels, mGluRs or hemi-gap channels (Hofer 2005) may contribute to the observations recorded here. One interesting possibility is that the sustained conductance that we record on the pyramidal neurons is due to one or more members of the transient receptor potential (TRP) family (Clapham et al. 2001). TRP channels are widely expressed in mammalian tissues, especially in neurons of the central nervous system including the hippocampus (Moran et al. 2004). Not infrequently, it has been observed that several TRP channels conduct better in the absence of divalent cations. Among them, TRPM7 deserves particular attention as a candidate for the conductance that we present here, due to 1) the strong negative feedback it receives by intracellular Mg2+ (irrespective of whether it is weakly chelated by ATP (Nadler et al. 2001;Kozak and Cahalan 2003), 2) its activation upon removal of external divalent ions (Runnels et al. 2001;Nadler et al. 2001), and 3) its sensitivity to inhibition by spermine (Kozak et al. 2002;Kerschbaum et al. 2003). However, in our case, 100 μM spermine failed to inhibit the sustained current upon removal of extracellular Ca2+, a concentration that should fully eradicate the TRPM7-mediated current (Kerschbaum et al. 2003), plus the current was insensitive to inhibition even by high extracellular [Mg2+]. 1 mM spermine completely abolished the low-[Ca2+]e-induced sustained current in adult pyramidal neurons in the slice preparation, consistent with a previous observation in cultured hippocampal neurons (Xiong et al. 1997). Nonetheless, possible candidates may also include many voltage-gated channels (see Introduction) as well as ligand gated ion channels, since low, but not zero [Ca2+]e is known to elicit additional transmitter release, via modification of surface potential and subsequent Ca2+ influx (Piccolino and Pignatelli 1996). Icrac-conducting channels can also be regulated by extracellular divalent ions (Hoth and Penner 1993).
Consequences of current activation
[Na+]i increases due to non-inactivating current may be appreciable. Relevant to this, an increase of greater than 10 mM [Na+]i may up-regulate NMDA receptor activity (Yu and Salter 1998), and it was recently shown that extracellular Ca2+ depletion leads to [Na+]i elevation, that in turns increases NMDA channel activity (Xin et al. 2005). Large [Ca2+]i increases are known to be triggered by reintroduction of physiological Ca2+ concentrations to the extracellular milieu (reperfusion) after cardiac muscle tissue has experienced a prior [Ca2+]e-free challenge, or at least a severe reduction in Ca2+e concentration (ischemia), (Zimmerman and Hulsmann 1966) termed the “Ca2+ Paradox”. We have recently reported that cultured cortical neurons exhibit paradoxical Ca2+ entry (Chinopoulos et al. 2004) and it is possible that this entry pathway may be a significant contributor to excitotoxic neuronal death. Evidence for the existence of the Ca2+ Paradox in brain has been summarized previously (Young 1986), and it was proposed recently that the phenomenon may be substantiated by one or more members of the TRP family of channels (Chinopoulos and Adam-Vizi 2006;MacDonald et al. 2006). Activation of NMDAR/non-NMDARs during hypoxia is associated with a severe Ca2+ influx, imposing downstream injurious effects on cellular functions. Notably though, the conductance mechanism presented here is not [Ca2+]i activated, as there was 10 mM EGTA (free) at all times in the pipette solution available to chelate any incoming Ca2+, verified by Fura 2 imaging (not shown). Overall, we demonstrate the presence of a sustained inward conductance in adult hippocampal CA1 pyramidal neurons upon reduction of [Ca2+]e, provided that intracellular Mg2+/ATP concentrations are reduced. This could be an important causative mechanism in neuronal pathology encompassing diminished metabolic capacity in the presence of reduced levels of extracellular calcium, conditions that are both met in various disease states.
Acknowledgments
We thank Drs. Wolfgang Müller, Volodymyr Gerzanich and J. Marc Simard for critical comments during the preparation of the manuscript and Dr. C. Fernando Valenzuela for help with the Mini Analysis software. This work was supported by NIH grants NS43458 & RR15636 to C.W.S.
Abbreviations
- APV
2-Amino-5-phosphonopentanoic acid
- csNSC
calcium-sensing non-selective channel
- CNQX
6-Cyano-7-nitroquinoxaline-2,3-dione
- mGluR
metabotropic glutamate receptor
- NMDAR
NMDA receptor
- TEA
tetraethylammonium chloride
- TRP
Transient Receptor Potential
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Aitken PG, Breese GR, Dudek FF, Edwards F, Espanol MT, Larkman PM, Lipton P, Newman GC, Nowak TS, Jr, Panizzon KL. Preparative methods for brain slices: a discussion. J Neurosci Methods. 1995;59:139–149. doi: 10.1016/0165-0270(94)00204-t. [DOI] [PubMed] [Google Scholar]
- Armstrong CM. Distinguishing surface effects of calcium ion from pore-occupancy effects in Na+ channels. Proc Natl Acad Sci U S A. 1999;96:4158–4163. doi: 10.1073/pnas.96.7.4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Cota G. Calcium block of Na+ channels and its effect on closing rate. Proc Natl Acad Sci U S A. 1999;96:4154–4157. doi: 10.1073/pnas.96.7.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Gerencser AA, Doczi J, Fiskum G, Adam-Vizi V. Inhibition of glutamate-induced delayed calcium deregulation by 2-APB and La3+ in cultured cortical neurones. J Neurochem. 2004;91:471–483. doi: 10.1111/j.1471-4159.2004.02732.x. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G. Cyclosporin A-insensitive permeability transition in brain mitochondria: inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem. 2003;278:27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- Chu XP, Zhu XM, Wei WL, Li GH, Simon RP, MacDonald JF, Xiong ZG. Acidosis decreases low Ca(2+)-induced neuronal excitation by inhibiting the activity of calcium-sensing cation channels in cultured mouse hippocampal neurons. J Physiol. 2003;550:385–399. doi: 10.1113/jphysiol.2003.043091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strubing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Feig S, Lipton P. N-methyl-D-aspartate receptor activation and Ca2+ account for poor pyramidal cell structure in hippocampal slices. J Neurochem. 1990;55:473–483. doi: 10.1111/j.1471-4159.1990.tb04160.x. [DOI] [PubMed] [Google Scholar]
- Formenti A, De SA. Effects of extracellular Ca2+ on membrane and seal resistance in patch-clamped rat thalamic and sensory ganglion neurons. Neurosci Lett. 2000;279:49–52. doi: 10.1016/s0304-3940(99)00951-9. [DOI] [PubMed] [Google Scholar]
- Formenti A, De SA, Arrigoni E, Martina M. Changes in extracellular Ca2+ can affect the pattern of discharge in rat thalamic neurons. J Physiol. 2001;535:33–45. doi: 10.1111/j.1469-7793.2001.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. The Journal Of Physiology. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima Y, Hagiwara S. Currents carried by monovalent cations through calcium channels in mouse neoplastic B lymphocytes. J Physiol. 1985;358:255–284. doi: 10.1113/jphysiol.1985.sp015550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- Harris RJ, Symon L, Branston NM, Bayhan M. Changes in extracellular calcium activity in cerebral ischaemia. J Cereb Blood Flow Metab. 1981;1:203–209. doi: 10.1038/jcbfm.1981.21. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Stabel J, Rausche G. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res. 1990;83:197–214. doi: 10.1016/s0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- Hofer AM. Another dimension to calcium signaling: a look at extracellular calcium. J Cell Sci. 2005;118:855–862. doi: 10.1242/jcs.01705. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001;4:869–870. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J. 2003;84:2293–2305. doi: 10.1016/S0006-3495(03)75035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Heinemann U, Yaari Y. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I Development of seizurelike activity in low extracellular calcium. J Neurophysiol. 1986;56:409–423. doi: 10.1152/jn.1986.56.2.409. [DOI] [PubMed] [Google Scholar]
- Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalent cations but not ATP. Biophys J. 2003;84:922–927. doi: 10.1016/S0006-3495(03)74909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak JA, Kerschbaum HH, Cahalan MD. Distinct Properties of CRAC and MIC Channels in RBL Cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Lipton P, Aitken PG, Dudek FE, Eskessen K, Espanol MT, Ferchmin PA, Kelly JB, Kreisman NR, Landfield PW, Larkman PM. Making the best of brain slices: comparing preparative methods. J Neurosci Methods. 1995;59:151–156. doi: 10.1016/0165-0270(94)00205-u. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Stimers JR. Calcium-inhibitable current in cultured embryonic chick cardiac myocytes: possibly via a novel chloride channel. Exp Physiol. 1998;83:323–336. doi: 10.1113/expphysiol.1998.sp004116. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca(2+) signaling, cell death and stroke. Trends Neurosci. 2006;29:75–81. doi: 10.1016/j.tins.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Martell A, Smith R. Critical Stability Constants. Plenum; New York: 1977. [Google Scholar]
- Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Curr Opin Neurobiol. 2004;14:362–369. doi: 10.1016/j.conb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Stengl M, Flameng W. Extracellular divalent cations block a cation non-selective conductance unrelated to calcium channels in rat cardiac muscle. J Physiol. 1997;502 ( Pt 2):235–247. doi: 10.1111/j.1469-7793.1997.235bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg. ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Ten BG, Stockle H, Steinberg R. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J Neurophysiol. 1978;41:1026–1039. doi: 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Lipton SA. Excitotoxins in neuronal apoptosis and necrosis. J Cereb Blood Flow Metab. 1999;19:583–591. doi: 10.1097/00004647-199906000-00001. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Laursen H, Hillered L, Hansen AJ. Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J Cereb Blood Flow Metab. 1996;16:262–270. doi: 10.1097/00004647-199603000-00011. [DOI] [PubMed] [Google Scholar]
- Owsianik G, Talavera K, Voets T, Nilius B. Permeation and Selectivity of TRP Channels. Annu Rev Physiol. 2005 doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- Piccolino M, Pignatelli A. Calcium-independent synaptic transmission: artifact or fact? Trends Neurosci. 1996;19:120–125. doi: 10.1016/s0166-2236(96)80017-8. [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–432. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Intracellular and extracellular changes of [Ca2+] in hypoxia and ischemia in rat brain in vivo. J Gen Physiol. 1990;95:837–866. doi: 10.1085/jgp.95.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron. 2004;41:243–256. doi: 10.1016/s0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC. N-methylaspartate receptors mediate epileptiform activity evoked in some, but not all, conditions in rat neocortical slices. Neuroscience. 1986;19:1161–1177. doi: 10.1016/0306-4522(86)90130-2. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Oh EJ, Lancaster E, Weinreich D. Effect of extracellular calcium on excitability of guinea pig airway vagal afferent nerves. J Neurophysiol. 2003;89:1196–1204. doi: 10.1152/jn.00553.2002. [DOI] [PubMed] [Google Scholar]
- Xin WK, Zhao XH, Xu J, Lei G, Kwan CL, Zhu KM, Cho JS, Duff M, Ellen RP, McCulloch CA, Yu XM. The removal of extracellular calcium: a novel mechanism underlying the recruitment of N-methyl-D-aspartate (NMDA) receptors in neurotoxicity. Eur J Neurosci. 2005;21:622–636. doi: 10.1111/j.1460-9568.2005.03888.x. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Lu W, MacDonald JF. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc Natl Acad Sci U S A. 1997;94:7012–7017. doi: 10.1073/pnas.94.13.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, MacDonald JF. Sensing of extracellular calcium by neurones. Can J Physiol Pharmacol. 1999;77:715–721. [PubMed] [Google Scholar]
- Young W. Ca paradox in neural injury: a hypothesis. Cent Nerv Syst Trauma. 1986;3:235–251. doi: 10.1089/cns.1986.3.235. [DOI] [PubMed] [Google Scholar]
- Yu XM, Salter MW. Gain control of NMDA-receptor currents by intracellular sodium. Nature. 1998;396:469–474. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]
- Zimmerman AN, Hulsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature. 1966;211:646–647. doi: 10.1038/211646a0. [DOI] [PubMed] [Google Scholar]