

Abstract
Early on, intriguing biological activities were found associated with the EETs using in vitro systems. Although the EETs other than the 5,6-isomer, are quite stable chemically, they are quickly degraded enzymatically with the sEH accounting in many cases for much of the metabolism. This rapid degradation often made it difficult to associate biological effects with the administration of EETs and other lipid epoxides particularly in vivo. Thus, it is the power to inhibit the sEH that has facilitated the demonstration of many physiological processes associated with EETs and possibly other epoxy fatty acids. In the last few years it has become clear that major roles of the EETs include modulation of blood pressure and modulation of inflammatory cascades. There are a number of other physiological functions now associated with the EETs including angiogenesis, neurohormone release, cell proliferation, G protein signaling, modulation of ion channel activity, and a variety of effects associated with modulation of NFκB. More recently we observed a role of the EETs as modulated by sEHI in reducing non-neuropathic pain. The array of biological effects observed with sEHI illustrates the power of modulating the degradation of chemical mediators in addition to the modulation of their biosynthesis, receptor binding and signal transduction. Many of these biological effects can be modulated by sEHIs but also by the natural eicosanoids and their mimics all of which offer therapeutic potential.
Keywords: Arachidonic acid, Epoxyeicosatrienoic acid, sEH, Inhibitor, COX, Nociception, Hyperalgesia
1. Introduction
Over the last few years, the epoxides of arachidonic acid or epoxyeicosatrienoic acids (EETs) have been established as lipid mediators with important biological functions [1]. There are a variety of pathways involved in the degradation of these chemical mediators, but hydration of the epoxide to the corresponding 1,2-diols appears to be the major pathway [1,2]. Interestingly the enzyme that carries out this reaction was first found while studying the metabolism of a terpenoid epoxide that mimicked the insect juvenile hormone [3]. The enzyme was termed cytosolic and later soluble epoxide hydrolase (sEH) because of its localization in the soluble (and peroxisomal) fractions of the cell [4,5]. The human sEH is the product of EPXH-2, a single copy gene presents on chromosome 8 [6,7]. Although multiple epoxide hydrolase enzymes are present in all living organisms [8], the human sEH is the subject of intense research due to emerging roles of its substrates in inflammation and hypertension [9,10].
2. Structure and function of sEH
Following partial purification of the enzyme [11,12] its substrate selectivity was examined. Unlike the better studied microsomal epoxide hydrolase the sEH hydrolyzed epoxides on acyclic systems. Although the sEH hydrated trisubstituted terpenoid epoxides with a low Km, it showed the highest kCAT for cis-1,2 disubstituted compounds [13]. However, both saturated and unsaturated fatty acid epoxides are excellent sEH substrates, especially the EETs [2,12,14,15]. In the intervening 30 years the enzyme was characterized, its message and gene cloned, expressed and its catalytic mechanism determined [6,16–18]. Multiple crystal structures have been obtained for the murine and human enzymes [19,20]. The mammalian enzyme is a homodimer with the monomers arranged in an anti-parallel form. Each monomer is composed of two domains [19]. The N-terminal domain has a separate evolutionary history from the C-terminal domain and hydrolyzes phosphates on lipophilic backbones [21,22]. The biological role of this domain is not yet known. The C-terminal domain is an α/β-hydrolase fold structure and is responsible for the epoxide hydrolase activity [19]. Interestingly in many lower animals and in plants the N- and C-terminal domains are separate proteins from separate genes and messages. Based on the enzyme mechanism [18], and later on its structure [19] potent transition state inhibitors of the enzyme were developed [23,24]. These sEH inhibitors (sEHIs) stabilized endogenous lipid epoxides and thus facilitated investigations into numerous biological functions associated with EETs, many of which are discussed in this volume. These biological effects can be loosely grouped into two categories: cardiovascular and anti-inflammatory. The cardiovascular effects include a reduction in blood pressure in several hypertensive rodent models [10,25], a reduction in renal damage [26], and a reduction in ischemic injury in heart and brain [27,28]. The anti-inflammatory effects of EETS were first established by Node et al. [29] and Kozak et al. [30]. Node et al. observed that EETs prevented leukocyte adhesion to the vascular wall by a mechanism involving inhibition of transcription factor NFκB [29], while Kozak et al. demonstrated that arachidonic acid epoxides are anti-pyretic [30].
3. Novel aspects of sEH function: pain and inflammation
Because EETs and other lipid epoxides [15,31] appear to be major chemical mediators we developed GLC–MS methods for their analysis [32]. With the availability of LC–MS technology we began to add a number of other analytes in the arachidonic acid cascade to our method [9,33]. As expected, we observed that high level of sEH decreases EETs levels while increasing the corresponding diols, DHETs [25]. Analogously inhibiting sEH increases the epoxide to diol ratio in several animal models [9,10,23,25]. Using this analytical method we surprisingly observed that PGE2 levels in plasma were reduced following the administration of sEHI to mice treated with lipopolysaccharide (LPS) to induce inflammation [9]. PGE2 levels did not change significantly in control mice when administered sEHI alone. Based on these observations, and other eicosanoids involved in inflammation and pain, we developed the hypothesis that sEHI and EETs themselves should dramatically reduce pain. This proved to be the case in several classical rodent inflammatory pain models (Fig. 1).
Fig. 1.

The sEHI AUDA-be blocks carrageenan (CAR) induced local thermal hyperalgesia effectively. Male rats (n = 8) were administered 100 μl (2%) of CAR in saline by subplantar injections. CAR was injected immediately following baseline measurement of thermal hind paw withdrawal latency (PWL). AUDA-be was formulated in a neutral cream (Vanicream™) and applied topically to the treated paw (100 mg/kg) 20 h post-CAR injection. PWL for the contralateral and ipsilateral (treated) paws were measured at each time point. Therefore AUDA-be had not only a prophylactic effect in reversing thermal hyperalgesia (data not shown), but it was also effective therapeutically.
There are a multitude of modulating mechanisms and receptors involved in pain perception [34,35]. Peripherally, at the site of tissue injury, the nerve cell endings termed nociceptors are composed of small-diameter nerve fibers activated and sensitized by noxious stimuli (mechanical, electromagnetic, electrical, thermal, and chemical) or by substances released in response. Subsequently, allodynia, spontaneous pain in the absence of stimulus or in response to a previously non-painful stimulus such as gentle stroking, and hyperalgesia, a disproportionately severe pain produced by a mildly noxious stimulus, develops. The many neuroactive substances released in response to tissue injury also termed the ‘inflammatory soup’ stimulate nociceptors, thus playing a major role in the development of inflammatory pain. Components of the inflammatory soup include: protons, ATP, histamine, serotonin, kinins, cytokines and arachidonic acid metabolites such as prostanoids. These mediators released by damaged cells, immune cells, or by nociceptor terminals themselves via local axon reflexes can directly activate the primary afferent fibers by depolarizing their endings or by enhancing their responsiveness to other physical and chemical depolarizing agents. Beyond the periphery, several mediators including prostaglandins and nitric oxide send feedback signals to central presynaptic endings in the dorsal horn of the spinal cord eliciting response in the central nervous system leading to central sensitization. This highly sophisticated system, with many mediators and receptors, establishes a fine balance to monitor the health status of an organism and intervenes as it becomes necessary. One of the pivotal molecules in inflammation is arachidonic acid, which when released in response to tissue injury has three potential metabolic fates [1,36]. It can be metabolized by the COX, LOX and/or cytochrome P450 pathways resulting in the production of prostaglandins, monohydroxys, leukotrienes and epoxyeicosanoids, respectively. The cytochrome P450 oxidation products, well known as EETs, are among the major anti-inflammatory arachidonic acid metabolites with a variety of biological effects [29].
By way of increasing EET concentrations through either exogenous delivery or by stabilizing EETs via inhibition of sEH, inflammatory pain can be significantly reduced. In two rodent models of inflammatory pain, one elicited by LPS [37] and the other by carrageenan, we observed significant anti-hyperalgesic effect upon administration of two structurally dissimilar, but equally potent sEH inhibitors, with or without exogenous EETs. In the carrageenan induced pain model thermal hyperalgesia was restricted to the carrageenan treated limb (Fig. 1). Therapeutic topical administration of sEHI AUDA-be blocked hyperalgesia effectively for at least 8 h after which the hyperalgesia resumed. Notably, sEHIs attenuate both hyperalgesia and allodynia equally well in the LPS induced inflammatory pain model. Analogous to the action of non-steroidal anti-inflammatory drugs sEHIs did not have an impact on nociception in the absence of induced pain.
4. Synergistic interactions in the arachidonic acid cascade
Another surprising implication of the metabolic profiling was that the analgesic effect of inhibiting sEH correlated with decreased induction of COX-2 without affecting COX-1. Further work has shown that COX inhibitors can increase EETs concentrations and that a combination of these therapeutics can have an improved analgesic effect. COX inhibitors increase EETs levels dramatically enough that it is likely that at least some of the analgesic effects of non-steroidal anti-inflammatory drugs (NSAIDs) are due to this increase in EET levels. This can be clearly seen in Fig. 2, where a sEHI is used in conjunction with the selective COX-2 inhibitor celecoxib. The effect has been observed with all of the COX-1, COX-2 and mixed NSAIDs tested. Unlike various COX inhibitors, the sEHI did not cause a large increase in lipoxygenase 5 metabolites suggesting that LOX 5 was also down regulated and sEHIs are dampening down the effect of inflammation. It is feasible that the reduction in proinflammatory mediators and pain are the result of inhibiting sEH, which increases EETs and that leads to transcriptional down-regulation of COX-2 induction, while at the same time the NSAIDs directly reduce the enzyme activity of the remaining protein (Fig. 3).
Fig. 2.
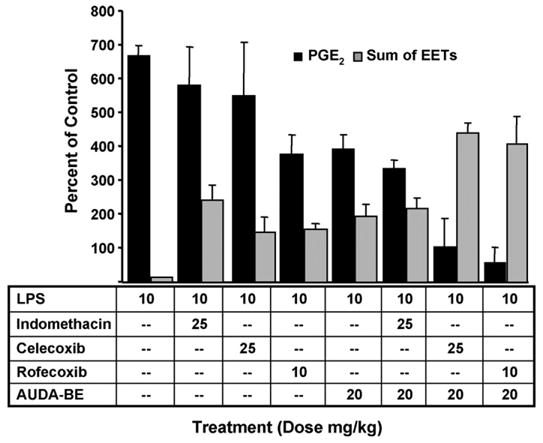
Synergistic reduction of PGE2 plasma levels by combined treatment of COX and sEHI. Co-administration AUDA-be and NSAIDs produce a synergistic decrease in prostaglandin PGE2 (black bars) and increase in EpETrEs (EETs grey bars), 6 h after LPS exposure. The data indicate that using a prophylactic dose of AUDA-be with a non-optimal therapeutic dose of COX inhibitor can further reduce PGE2. The data represent an average ± S.D. (n = 4) and are depicted as percent of control mice receiving vehicle without LPS. *Significantly different from NSAID alone (p < 0.05) as determined by analysis of variance followed by Tukey’s test.
Fig. 3.
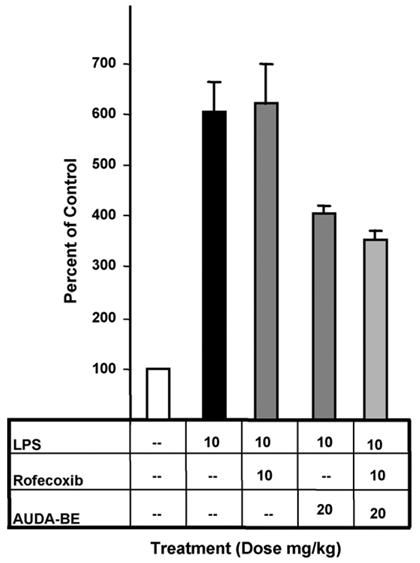
A prophylactic dose of AUDA-be reduces hepatic COX-2 protein expression 6 h after LPS exposure relative to mice receiving LPS only. Results from individual inhibitors at various doses are in dark grey bars. Co-administration of the sEHI and rofecoxib is depicted as a light grey bar, indicating that a prophylactic dose of AUDA-be used in conjunction with a non-optimal therapeutic dose of rofecoxib (10 mg/kg) can further decrease COX-2 induction. Data represent the COX-2 protein levels ± S.D. (n = 3) in murine liver after exposure to LPS as determined by independent western blots. *Significantly different from vehicle (p < 0.05) as determined by analysis of variance followed by Tukey’s test. #Significantly different from AUDA-be alone (p < 0.05) as determined by analysis of variance followed by Tukey’s test.
A direct prediction from the above work is that the EETs and thus the sEHI should show additive to synergistic interaction with NSAIDs in reducing inflammatory metabolites in the arachidonic acid cascade. The EETs transcriptionally down regulate COX-2 in an inflamed state resulting in a decrease in inflammatory metabolites. NSAIDs of course inhibit COX-1 and/or COX-2 also resulting in a dramatic reduction in inflammatory metabolites, and the combined effect is predictably dramatic. Co-administration of sEHI with a low dose of NSAIDs results in a large reduction in inflammatory metabolites. Thus, for example, if the deleterious effects associated with the use of high doses of rofecoxib are compound specific, one could use sEHI to maintain efficacy while reducing NSAID dose. Another hypothesis surrounding the side effects of COX-2 inhibitors is that they cause an imbalance in eicosanoids, specifically PGI2 and TXA2. The observation that the sEHI down regulate, but do not completely inhibit induced COX-2, suggests that the sEHI are less likely to demonstrate this toxicity. Therefore, co-administration of sEHI and NSAIDs may reduce the side effects associated with some NSAIDs.
Additionally, a low dose of NSAIDs increases the biological effects of sEHI, by shuttling the arachidonic acid precursors into the P450 branch of the arachidonate cascade and increasing EETs. Low doses of sEHI synergize NSAIDs and other drugs influencing the arachidonate cascade by transcriptionally down regulating key inflammatory enzymes. This would allow COX inhibitors, for example, to be used at dramatically reduced doses while retaining therapeutic effects and providing an alternative to combined COX/proton pump inhibitor cocktails which are currently under development by the pharmaceutical industry.
The effects of co-administration of NSAIDs and sEHIs can be extrapolated to other combination therapies within the arachidonic acid cascade. These data predict as well that low doses of NSAIDs and other drugs which shift eicosanoid flux should synergize the action of sEHI, and this synergism has been observed in rodent experiments. These observations are likely to lead to useful combinations of drugs. However, there is the caution that interaction of compounds acting on the arachidonate cascade or physiological processes downstream from the cascade could lead to undesirable interactions.
Our recent data indicate that EETs, like other oxylipins, not only regulate cytokines and chemokines but also regulate other key enzymes in the arachidonic acid cascade. An increase in EETs caused by sEHI seems to have little effect in constitutive COX-1 or even COX-2 as measured by plasma metabolite levels. This observation was confirmed by PCR and western analysis of the messages and proteins. However, an increase in EETs results in a dramatic down regulation of induced but not constitutive COX-2 and its associated metabolites leading to a reduction in symptoms associated with severe inflammation. At first glance an increase in EETs, which leads to the transcriptional down regulation of COX-2, might shift arachidonic acid into the LOX pathway, thus increasing proinflammatory eicosanoids. Our data show a decrease in 5-LOX metabolites. Simplistically the sEHI mediated increase in EETs can be seen as resulting in a shift in eicosanoids from a pattern generally initiating and propagating inflammation to a pattern of resolution of inflammation. When analyzing multiple metabolites, patterns of eicosanoid profiles are far more informative than individual biomarkers. We feel that this observation of cross talk among the pathways in the arachidonate and other lipid cascades is likely to be a general phenomenon.
The metabolomic approach to eicosanoid profiling has provided tremendous insight into the regulation of the arachidonic acid and other regulatory lipid cascades. By no means are these approaches easy, nor have we done more than demonstrate the concept of the approach of metabolomic profiling to the evaluation of the many known and unknown lipid mediators. However, the availability of a quantitative multianalyte method could prove valuable. From earlier work it is clear that modulation of enzymes in the arachidonic acid cascade shifts the flow of arachidonate among the pathways which can result in dramatic biological effects. Possibly the best known example of this effect is the observation that administration of aspirin can shift the arachidonate flow from the COX pathways to the LOX pathways including LOX 5 resulting in increased production of leukotrienes and thus asthmatic symptoms. The COX-2 inhibitor data demonstrates this type of transfer of arachidonic acid from COX to a cytochrome P450 pathway, supporting our previous speculation that some of the analgesic effects of COX inhibitors is be due to the increased concentrations of EETs.
5. EETs and receptors
Despite the fact that no specific EET receptor has yet been characterized, several studies suggest there are both intracellular and membrane bound EET high affinity binding sites [1]. Interestingly, when EETs were exogenously applied (100–300 mg/kg) to male rats, the animals displayed a unique set of behaviors including a short period of increased activity, exploratory behavior, grooming and chewing. In pursuit of a better understanding of the observed analgesic effects and other behavior we investigated a set of cellular receptors on which EETs may have actions. A group of 47 potential receptors were selected, and screened for ability of EETs to displace high affinity radioligand binding (Table 1). Among these we found that respective high affinity radioligands of the peripheral benzodiazepine receptor (PBR), cannabinoid CB2 receptor, neurokinin NK2 receptor and dopamine D3 receptor were displaced by low micromolar concentrations of EETs whereas other subtypes of these receptors including the central benzodiazepine, cannabinoid CB1, neurokinin NK1 and NK2 and dopamine D1 and D2 receptors were not effected. These effects were further investigated by screening the activity of individual EET regioisomers on each of these receptors (Table 1).
Table 1.
Interaction of EETs with selected cellular receptors
| CB1 | CB2 | NK1 | NK2 | NK3 | Peripheral benzodiazepine |
Central benzodiazepine |
D3 | |
|---|---|---|---|---|---|---|---|---|
| EET-me mixture (μM) | >100 | 19 | >100 | 14 | >100 | 4.6 | >100 | 30 |
| 5,6 EET-me (μM) | NT | 20 | NT | 36 | NT | 12 | NT | >100 |
| 8,9 EET-me (μM) | NT | >100 | NT | >100 | NT | >100 | NT | >100 |
| 11,12 EET-me (μM) | NT | >100 | NT | >100 | NT | 140 | NT | >100 |
| 14,15 EET-me (μM) | NT | >100 | NT | >100 | NT | 12 | NT | >100 |
Binding assays were conducted by CEREP according to standardized procedures. A mixture of regioisomers of EETs were initially screened broadly for displacing ability of high affinity ligands. In a second round the IC50 of the mixture and the individual isomers were determined. Reference compounds and their affinities (M) for respective receptors from left to right were CP 55940 (1.00E–09), WIN 55212-2 (7.60E–09), [Sar9,Met(O2)11]-SP (2.20E–10), [Nle10]-NKA(4–10) (9.30E–09), SB 222200 (1.00E–08), PK 11195 (2.70E–09), Diazepam (1.0E–08), (+) butaclamol (8.90E–09). NT: not tested.
Although these binding studies resulted with weak effects and the functional consequences of these reported activities are largely unknown these receptors may have links to nociception, the most notable being the CB2 receptor. CB2 receptor is expressed in the periphery, in particular on immune cells and its activation is independent from the CB1 receptor which mediates central cannabionoid effects such as hypothermia, catalepsy, and hypoactivity [38]. Activation of CB2 on non-neuronal cells in inflamed tissue is assumed to suppress the release of inflammatory mediators implicated in nociceptor sensitization [39]. Accordingly, CB2-selective agonists have recently been shown to induce antinociception in models of acute, inflammatory, and nerve injury-induced nociception [40–43]. These data lead us to predict that inhibition of sEH, thereby increasing epoxide fatty acids concentrations, may cause analgesia through multiple pathways including reduction in the levels of prostaglandins such as PGE2 and by directly acting on cellular receptors. Compounds that mimic EET structure can potentially be exploited to target these receptors.
6. Conclusion
The concepts presented here may have broader implication, with regard to both maintaining health and treating disease. Currently, over 15% of the world’s pharmaceuticals influence the arachidonic acid cascade, and many of the key enzymes in the biosynthesis and degradation of lipid chemical mediators are major targets of the pharmaceutical industry. Thus any discussion of the arachidonic acid cascade is quite general. This cascade has been exploited for therapy for many years in terms of single concept medications as exemplified by taking an aspirin for a headache. More modern therapies in some ways continue this simplistic approach exemplified by using celecoxib to treat joint pain. It is clear from data presented here that sEHI or for that matter EETs can be used successfully in combination with five lipoxygenases activating protein inhibitor and COX inhibitors to reduce pain and inflammation. As we begin to think in more sophisticated terms regarding the exploitation of the arachidonic acid cascade to preserve health and treat disease, we certainly should begin to consider logical combinations of two or more medications to alter the cascade in desirable ways. We know that regulatory lipids are altered by diet, nutraceuticals and lifestyle. The scientific community should be evaluating the effects of all of these perturbations on the arachidonic acid cascade and integrating this information with changes caused by pharmaceuticals. By no means are these improvements in health delivery simple to develop either from a scientific or sociological perspective, but the technologies to implement them seem clear. For example a near term goal will be the integration of analytical knowledge of the arachidonate cascade with information on the genome, transcriptome and proteome. We can plan toward a goal of individualized diagnostics in general and of the arachidonic cascade in particular and development of therapy geared to the individual rather than the “average person”. In this vision we would adjust lifestyle, nutraceuticals, diet, and drugs to modulate the arachidonate cascade to maintain health and if needed treat disease. Progress toward realizing such a vision would certainly be a tribute to eicosanoid pioneers such as Jack McGiff.
Acknowledgments
This work was supported by NIEHS Grant R37 ES02710, NIEHS Superfund Basic Research Program Grant P42 ES04699, NIEHS Center, P30 ES05707, NIEHS Center for Children’s Environmental Health and Disease Prevention Grant P01 ES11269, and UCDMC Translational Technology Research Grant.
Abbreviations
- EET
epoxyeicosatrienoic acid
- sEH
soluble epoxide hydrolase
- sEHI
sEH inhibitors
References
- 1.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Progress Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 2.Chacos N, Capdevila J, Falck JR, et al. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;233:639–48. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- 3.Gill SS, Hammock BD, Casida JE. Mammalian metabolism and environmental degradation of the juvenoid 1-(4′-ethylphenoxy)-3,7-dimethyl-6,7-epoxy-trans-2-octene and related compounds. J Agric Food Chem. 1974;22:386–95. doi: 10.1021/jf60193a058. [DOI] [PubMed] [Google Scholar]
- 4.Gill SS, Hammock BD. Distribution and properties of a mammalian soluble epoxide hydrase. Biochem Pharmacol. 1980;29:389–95. doi: 10.1016/0006-2952(80)90518-3. [DOI] [PubMed] [Google Scholar]
- 5.Arand M, Knehr M, Thomas H, Zeller HD, Oesch F. An impaired peroxisomal targeting sequence leading to an unusual bicompartmental distribution of cytosolic epoxide hydrolase. FEBS Lett. 1991;294:19–22. doi: 10.1016/0014-5793(91)81333-4. [DOI] [PubMed] [Google Scholar]
- 6.Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 7.Sandberg M, Meijer J. Structural characterization of the human soluble epoxide hydrolase gene (EPHX2) Biochem Biophys Res Commun. 1996;221:333–9. doi: 10.1006/bbrc.1996.0596. [DOI] [PubMed] [Google Scholar]
- 8.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Schmelzer KR, Kubala L, Newman JW, Kim I-H, Eiserich JP, Hammock BD. Soluble epoxide hyrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–7. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–4. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 11.Hammock BD, Gill SS, Stamoudis V, Gilbert LI. Soluble mammalian epoxide hydratase: action on juvenile hormone and other terpenoid epoxides. Comp Biochem Physiol. 1976;53B:263–5. doi: 10.1016/0305-0491(76)90045-6. [DOI] [PubMed] [Google Scholar]
- 12.Gill SS, Hammock BD. Hydration of cis- and trans-epoxymethyl stearates by the cytosolic epoxide hydrase of mouse liver. Biochem Biophys Res Commun. 1979;89:965–71. doi: 10.1016/0006-291x(79)91872-2. [DOI] [PubMed] [Google Scholar]
- 13.Wixtrom RN, Hammock BD. Membrane-bound and soluble-fraction epoxide hydrolases: methodological aspects. In: Zakim D, Vessey DA, editors. Biochemical pharmacology and toxicology, vol. 1: methodological aspects of drug metabolizing enzymes. New York: John Wiley & Sons Inc; 1985. pp. 1–93. [Google Scholar]
- 14.Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, Hammock BD. Improved radiolabeled substrates for soluble epoxide hydrolase. Anal Biochem. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- 15.Greene JF, Williamson KC, Newman JW, Morisseau C, Hammock BD. Metabolism of monoepoxides of methyl linoleate: bioactivation and detoxification. Arch Biochem Biophys. 2000;376:420–32. doi: 10.1006/abbi.2000.1753. [DOI] [PubMed] [Google Scholar]
- 16.Borhan B, Jones AD, Pinot F, Grant DF, Kurth MJ, Hammock BD. Mechanism of soluble epoxide hydrolase: formation of an (-hydroxy ester-enzyme intermediate through Asp-333. J Biol Chem. 1995;270:26923–30. doi: 10.1074/jbc.270.45.26923. [DOI] [PubMed] [Google Scholar]
- 17.Pinot F, Grant DF, Beetham JK, et al. Molecular and biochemical evidence for the involvement of the Asp333-His523 pair in the catalytic mechanism of soluble epoxide hydrolase. J Biol Chem. 1995;270:7968–74. doi: 10.1074/jbc.270.14.7968. [DOI] [PubMed] [Google Scholar]
- 18.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–33. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 19.Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Detoxification of environmental mutagens and carcinogiens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc Natl Acad Sci USA. 1999;96:10637–42. doi: 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716–23. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 21.Cronin A, Mowbray S, Durk H, et al. The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc Natl Acad Sci USA. 2003;100:1552–7. doi: 10.1073/pnas.0437829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc Natl Acad Sci USA. 2003;100:1558–63. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morisseau C, Goodrow MH, Dowdy D, et al. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA. 1999;96:8849–54. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW. Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J Biol Chem. 2000;275:15265–70. doi: 10.1074/jbc.M000278200. [DOI] [PubMed] [Google Scholar]
- 25.Yu Z, Xu F, Huse LM, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–8. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 26.Imig JD, Zhao X, Zaharis CZ, et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–81. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fornage M, Lee CR, Doris PA, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–37. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otsuka T, Sugo N, Koehler RC, Hurn PD, Traystman RJ, Alkayed NJ. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2003;34:301. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozak W, Kluger MJ, Kozak A, Wachulec M, Dokladny K. Role of cytochrome P-450 in endogenous antipyresis. Am J Physiol. 2000;279:R455–60. doi: 10.1152/ajpregu.2000.279.2.R455. [DOI] [PubMed] [Google Scholar]
- 31.Greene JF, Newman JW, Williamson KC, Hammock BD. Toxicity of epoxy fatty acids and related compounds to cells expressing human soluble epoxide hydrolase. Chem Res Toxicol. 2000;13:217–26. doi: 10.1021/tx990162c. [DOI] [PubMed] [Google Scholar]
- 32.Newman JW, Hammock BD. Optimized thiol derivatizing reagent for the mass spectral analysis of disubstituted epoxy fatty acids. J Chromatogr A. 2001;925:223–40. doi: 10.1016/s0021-9673(01)00998-0. [DOI] [PubMed] [Google Scholar]
- 33.Newman JW, Watanabe T, Hammock BD. The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J Lipid Res. 2002;43:1563–78. doi: 10.1194/jlr.d200018-jlr200. [DOI] [PubMed] [Google Scholar]
- 34.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 35.Coutaux A, Adam F, Willer JC, Le BD. Hyperalgesia and allodynia: peripheral mechanisms. Joint Bone Spine. 2005;72:359–71. doi: 10.1016/j.jbspin.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 36.McGiff JC. Cytochrome P-450 metabolism of arachidonic acid. Annu Rev Pharmacol Toxicol. 1991;31:339–69. doi: 10.1146/annurev.pa.31.040191.002011. [DOI] [PubMed] [Google Scholar]
- 37.Inceoglu AB, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. doi: 10.1016/j.lfs.2006.07.031. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549–63. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 39.Malan TP, Ibrahim MM, Deng H, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–45. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 40.Nackley AG, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB2 receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;119:747–57. doi: 10.1016/s0306-4522(03)00126-x. [DOI] [PubMed] [Google Scholar]
- 41.Quartilho A, Mata HP, Ibrahim MM, et al. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:95–960. doi: 10.1097/00000542-200310000-00031. [DOI] [PubMed] [Google Scholar]
- 42.Ibrahim MM, Deng H, Zvonok AM, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100:10529–33. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–53. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]