

Abstract
Proteasomal dysfunction has been suggested to contribute to the degeneration of nigrostriatal dopamine neurons in Parkinson’s disease. A recent study reported that systemic treatment of rats with the proteasome inhibitor Z-lle-Glu(OtBu)-Ala-Leu-al (PSI) causes a slowly progressive degeneration of nigrostriatal dopamine neurons, the presence of inclusion bodies in dopamine neurons, and motor impairment. We examined in vitro and in vivo the effects of PSI on nigrostriatal dopamine neurons. Mass spectrometric analysis was employed to verify the authenticity of the PSI compound. PSI was non-specifically toxic to neurons in ventral mesencephalic organotypic slice cultures, indicating that impairment of proteasome function in vitro is toxic. Moreover, systemic administration of PSI transiently decreased brain proteasome activity. Systemic treatment of rats with PSI did not, however, result in any biochemical or anatomical evidence of lesions of nigrostriatal dopamine neurons, nor were any changes in locomotor activity observed. These data suggest that systemic administration of proteasome inhibitors to normal adult rats does not reliably cause an animal model of parkinsonism.
Keywords: Parkinson’s Disease, PSI, striatum, substantia nigra, tyrosine hydroxylase
1. Introduction
Idiopathic Parkinson’s disease (PD) is characterized pathologically by ubiquitin- and α-synuclein-containing intracytoplasmic inclusions (Lewy bodies) in the dopamine neurons of the substantia nigra (SN) (Spillantini et al., 1998). The formation of Lewy bodies suggests that the ubiquitin-proteasome system (UPS) does not adequately clear misfolded or damaged proteins, leading to protein aggregation and culminating in degeneration of SN dopamine neurons. Consistent with this idea, proteasome inhibitors have been reported to cause dopamine cell death in vitro (McNaught et al., 2002; Petrucelli et al., 2002), and intrastriatal infusion of proteasome inhibitors results in the presence of α-synuclein-positive inclusions and degeneration of SN dopamine neurons (Fornai et al., 2003; Miwa et al., 2005).
McNaught et al. (2004) reported that systemic administration of the proteasome inhibitor Z-lle-Glu(OtBu)-Ala-Leu-al (PSI) causes the progressive degeneration of nigrostriatal dopamine neurons and ubiquitinated, α-synuclein-immunoreactive (-ir) intracytoplasmic inclusions in surviving dopamine neurons, thus mimicking the pathology of PD. Moreover, decreased locomotor activity was reported to occur in response to PSI treatment. However, since this report three other papers have failed to replicate various aspects of the original findings (Bove et al., 2006; Kordower et al., 2006; Manning-Bog et al., 2006), while two other papers have replicated aspects of the orginal report (Schapira et al., 2006; Zeng et al., 2006).
Because of the importance in identifying an animal model of parkinsonism with progressive rather than acute dopamine cell loss and the significance of proteasomal inhibition to current models of cell death in PD, we examined the effects of PSI on nigrostriatal dopamine neurons in vitro and in vivo. Despite observing PSI-induced SN cell death in organotypic slice cultures and decreased proteasome activity in the brains of animals treated systemically with PSI, we did not observe any changes in pathological, biochemical, or behavioral indices of nigrostriatal dopamine function in animals treated with PSI.
2. Results
Identity of PSI
Mass spectrometric analysis of the PSI used in the experiments revealed a major 619 [M+H]+ ion corresponding to PSI, along with sodium [M+Na]+ and potassium [M+K]+ PSI adducts at 641 and 659, respectively. All ions except for one at m/z 510 were readily assigned to expected fragmentation pathways of the PSI molecule.
Effects of PSI in organotypic slice cultures of the ventral mesencephalon
Both freshly prepared PSI added to the culture media, and PSI solutions that were stored frozen for two months, caused concentration- and time-dependent increases in cell death (PI uptake) in SN slice cultures. Degenerating (PI-positive) neurons, including but not restricted to dopamine neurons, were seen in the SN (see Figure 1). Application of ≥200 nM PSI caused cell death, with more widespread toxicity seen in response to higher PSI concentrations. In cultures treated with 5 μM PSI for 40 hours and examined four days later, the number of PI-labeled cells was greater than in cultures examined two days after PSI treatment, suggesting some degree of ongoing neuronal loss (data not shown).
Figure 1.

Propidium iodide (PI) uptake by degenerating neurons in organotypic slice cultures of the ventral mesencephalon treated with vehicle or PSI. PSI caused a concentration-dependent increase in cell death compared to vehicle. (Bar = 10μM)
Brain Proteasome activity
We observed a non-significant trend toward nigral proteasome inhibition, including chymotrypsin- and trypsin-like as well as caspase-like activities, in animals sacrificed 2–3 hrs following cessation of PSI treatment as compared to vehicle-treated animals (data not shown). In animals sacrificed 2 weeks after cessation of treatment, whole brain proteasome activity was significantly decreased (Figure 2). A trend toward a decrease in caspase-like activity in the SN of animals surviving for two weeks was noted, but this did not reach statistical significance (see Figure 2). No change was seen in animals maintained for 12 weeks after cessation of PSI treatment. These findings suggest that SN proteasome activity was only transiently affected by PSI treatment. The significant decreases in chymotrypsin-, trypsin-, and caspase-like activities from whole brain samples (exclusive of the SN) in the two week survival group again suggest that PSI caused a transient decrease in proteasome activity.
Figure 2.

The effects of PSI treatment on proteasome activity. Chymotrypsin (Suc-LLV-AMC)-, trypsin (BOC-LLR-AMC), and caspase (Z-LLE-AMC)-like activities were significantly decreased in whole brain of PSI-treated rats sacrificed two weeks after the last PSI injection. In contrast, proteasome activities were not significantly decreased in the SN, although a trend toward a decrease in caspase-like activity was noted. *p ≤ .05, **p ≤ .01
Biochemical and immunohistochemical assessment of nigrostriatal dopamine neurons
PSI treatment did not significantly decrease striatal concentrations of dopamine, nor did it change the concentrations of the dopamine acidic metabolites DOPAC and HVA (see Table 1). Neither concentrations of norepinephrine nor serotonin differed significantly between control and PSI-treated groups (Table 1). Consistent with these biochemical observations, PSI did not subjectively appear to decrease the number of midbrain dopamine neurons or the morphology of dopamine (tyrosine hydroxylase-immunoreactive) neurons, and the density of striatal dopaminergic axons in PSI-treated animals appeared to be comparable to that observed in vehicle-treated animals (see Figure 3). Ubiquitin immunohistochemistry did not reveal the presence of ubiquitin-immunoreactive aggregates within the SN or other brain areas of PSI-treated animals (data not shown).
Table 1.
Concentrations of striatal monoamines and metabolites.
| DA | NE | 5-HT | DOPAC | HVA | |
|---|---|---|---|---|---|
| Vehicle | 179.20 ± 5.99 | 1.24 ± 0.47 | 5.56 ± 0.45 | 10.99 ± 0.78 | 6.79 ± 0.35 |
| PSI | 209.30 ± 11.95 | 0.78 ± 0.08 | 6.37 ± 0.40 | 13.62 ± 1.15 | 7.81 ± 0.67 |
The effects of systemic PSI treatment (N=7/gp) on striatal concentrations (ng/mg protein) of dopamine (DA), norepinephrine (NE), serotonin (5-HT), 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA). PSI treatment did not result in any significant differences in monoamine or metabolite concentrations relative to vehicle-injected rats.
Figure 3.
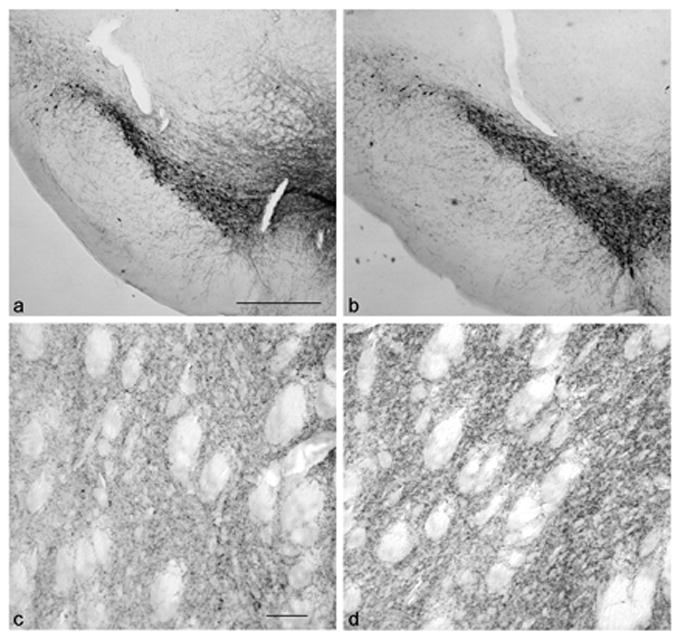
Tyrosine hydroxylase immunoreactive cells and axons in the SN and striatum of vehicle (a, c)- and PSI (b, d)-treated subjects. There is no apparent difference in the numbers or distribution of nigral dopamine neurons or density of striatal dopamine axons. (a, bar = 500μM; c, bar = 50 μM).
Behavioral effects of systemic PSI
Both vehicle-treated and PSI-treated animals developed granulomas over the course of the administration schedule. Because both treatment groups were affected, the presence of the granulomas was attributed to the vehicle (70% ethanol).
No overt behavioral changes were seen during the 12 weeks after PSI treatment. PSI-treated rats appeared well groomed and healthy, and gained weight at a rate that was essentially identical to that of vehicle-treated control subjects. At no point did animals exhibit overt impoverishment of movement, rigidity, or tremors. No significant differences in locomotor or total activity between experimental and control groups were seen at any time point examined. A two-way ANOVA examining locomotor activity failed to uncover any significant treatment effect or treatment x time interaction, nor was there any difference between PSI and control groups when activity data were collapsed across all sessions prior to analysis (see Figure 4).
Figure 4.

a). PSI administration did not result in a significant change in total movements at any time after PSI treatment relative to control subjects (N=7/gp); values on x-axis represent weeks following cessation of PSI treatment. b). Similarly, the average of locomotor activity collapsed over all time points was not significantly decreased in PSI-treated animals.
3. Discussion
We did not observe any evidence of PSI-induced disruption of the nigrostriatal dopamine neurons in response to systemic administration of the proteasome inhibitor. Treatment of ventral midbrain organotypic slice cultures with PSI was toxic to both dopaminergic and non-dopaminergic SN neurons, consistent with previous reports of the in vitro toxicity of PSI, and we observed a temporally-specific decrease in brain proteasomal activity after systemic PSI treatment. However, there was no decrease in either biochemical or anatomical markers of the integrity of nigrostriatal dopamine neurons, nor was there a change in behavior of PSI-treated rats.
There was no significant change in striatal dopamine concentrations in PSI-treated rats sacrificed 12 weeks after treatment with the proteasome inhibitor, nor were changes in dopamine metabolite concentrations observed. Similarly, no changes in striatal concentrations of norepinephrine or serotonin were uncovered. These data are consistent with a lack of effect of systemic PSI treatment on the nigrostriatal dopamine innervation.
We also examined the integrity of the dopamine innervtion of the midbrain and striatum using immunohistochemical methods. We saw no qualitative evidence of any changes in the anatomical integrity of the nigrostriatal dopamine system. We did not observe the presence of uibiquitin-positive aggregates in the SN (data not shown) following ubiquitin immunohistochemistry. The numbers of SN TH-ir dopamine neurons did not appear to differ between the control and PSI-treated animals, nor did the density and pattern of the striatal innervation appear altered. Although we did not use stereological methods for the anatomical studies, given the magnitude of the dopamine cell loss reported in the original paper of McNaught et al. (2004) as well as the two reports that found data consistent with PSI-induced DA cell loss (Schapira et al., 2006; Zeng et al., 2006) it is unlikely that a significant loss of SN dopaminergic neurons occured in our PSI-treated rats. Moreover, we failed to observe any significant decline in striatal dopamine concentrations in PSI-treated animals, suggesting that a stereological study of SN dopamine neurons was not warranted. Because striatal dopamine insufficiency is the proximate cause of parkinsonism, and we saw no decrease whatsoever in striatal dopamine concentration, our data do not support the contention that systemic administration of PSI produces an animal model of parkinsonism. Because degeneration of nigrostriatal dopamine neurons is the essential pathology of parkinsonism, we restricted our studies to the nigrostriatal system and did not examine other brain areas.
It is unclear why we failed to observe any evidence of nigrostriatal dopamine cell loss, yet McNaught and associates (2004) noted extensive and progressive loss of the striatal dopamine innervation. In a brief but lucid review of the conflicting studies on the effects of systemic PSI, Beal and Lang (2006) noted several possible explanations that might account for the differences observed by various authors. We examined many of these variables. We used PSI from the same vendor as reported in the initial study. Moreover, mass spectrometric testing confirmed the identity and stability of the PSI compound. We followed exactly the same treatment protocol in the same strain of rat as used by McNaught and investigators (2004). We examined only one time point (12 weeks) after PSI treatment for our biochemical and anatomical data, a time well after which McNaught and collaborators first found evidence of nigrostriatal dopamine degeneration, which they reported to be progressive. We studied the effects of only one PSI dose in vivo, but this dose was found by McNaught and colleagues to cause significant dopaminergic cell loss and behavioral changes.
It is unlikely that preparation of the proteasome inhibitor contributed to the discrepant results, because we observed potent effects of PSI in vitro, including comparable effects when using freshly-prepared PSI and solutions prepared two months earlier and then stored at −20° C. Because treatment of the organotypic slice cultures with the proteasome inhibitor caused neuronal toxicity but we did not see any significant effect on the nigrostriatal dopamine system in vivo, it is possible that brain penetration of PSI is limited. However, we observed a significant decrease in proteasome activity in whole brain samples of animals sacrificed two weeks after the end of the PSI treatment regimen, consistent with sufficient brain entry of the drug to cause functional changes in the proteasome. The change in whole brain activity was not matched by a corresponding decrease in SN activity, although a trend toward a decrease in proteasome activity was seen in the SN 2–3 hrs following cessation of PSI treatment. It is possible that inclusion of some extra-nigral tissue in the dissection of the SN may account for the non-significant trend toward a decrease. Our data thus indicates a transient decrease in brain proteasome activity following systemic PSI administration, consistent with brain entry of the proteasomal inhibitor; sustained decreases may be required in order for any toxicity of PSI to nigrostriatal dopamine neurons to be manifested.
Our findings are arguably the most comprehensive set of data addressing the toxicity of PSI to nigrostriatal dopamine neurons since the original report by McNaught et al. (2004). Notably, we have provided data that addresses a key argument advanced by McNaught and Olanow (2006) in an attempt to explain the discrepant findings across different laboratories: differences in commercial production of PSI and the bioavailability of the compound. Previous studies, both positive and negative, failed to confirm the identity of PSI and show that it causes cell death in vitro. We verified the identity of the compound by mass spectrometry and showed that although the same batch and sample of PSI was toxic to nigral neurons in organotypic slice cultures, the proteasome inhibitor failed to lesion nigrostriatal dopamine neurons in vivo. Furthermore, because we demonstrated proteasomal inhibition in brain after systemic administration, it is clear that the PSI actually enters the brain, yet does not elicit any evidence of parkinsonism using anatomical, biochemical, or behavioral criteria.
Several lines of evidence suggest that UPS dysfunction may be involved in the pathogenesis of PD (Dawson and Dawson, 2003; Dauer and Przedborski, 2003; Fornai et al., 2003, 2005; Singleton et al., 2003; Sawada et al., 2004; Rideout et al., 2005). In particular, it appears that UPS dysfunction coupled with other stressors, such as oxidative stress and mitochondrial inhibition, is toxic to nigrostriatal dopamine neurons (Giasson et al., 2000; Hoglinger et al., 2003; Elkon et al., 2004; Betarbet et al., 2005). Although in vitro studies have found that UPS inhibitors can cause dopamine cell death, there has been a lack of data indicating that systemic treatment with UPS inhibitors alone causes specific dopamine neuron toxicity. In sharp contrast to in vitro studies, Inden et al. (2005) suggested that proteasomal inhibition may afford protection against 6-hydroxydopamine-induced SN cell loss, suggesting that the role of proteasomal inhibition in dopamine cell loss is likely to be more complex than initially envisioned (see Setsuie et al., 2005).
An animal model of parkinsonism that faithfully reproduces the pathological changes of PD in a time-dependent fashion would be a major advance over current models, and would be particularly useful for studies of treatments that may retard the progressive loss of SN dopamine neurons. As such, it will be important to identify any factors that might elicit dopamine cell loss in response to PSI, as reported by McNaught and colleagues (2004). Our data, however, are not consistent with the suggestion that UPS dysfunction alone is sufficient to cause degeneration of nigrostriatal dopamine neurons, and indicate that systemic administration of PSI does not reliably produce a rodent model of parkinsonism.
4. Experimental Procedures
PSI mass spectrometric analysis
In order to confirm the identity of the PSI we used, which was commercially obtained (Sigma-Aldrich Co., St. Louis, MO), PSI was subjected to MALDI TOF/TOF mass spectrometric analysis, using an Ultraflex II (Bruker Daltonics, Billerica MA). 1 μL of PSI in water (0.1% TFA):acetonitrile with 2,5-dihydroxy-benzoic acid was spotted on a MALDI plate. An MS spectrum was then acquired in reflector mode to accurately determine the molecular weight of the intact PSI molecule, and in LIFT mode for acquiring the MS/MS spectrum.
Organotypic Slice Cultures
Organotypic slice cultures of the ventral mesencephalon were prepared from neonatal (P0–P2) rats, as described by Stoppini et al. (1991). 300 μm thick coronal slices were placed in Millicell-CM culture inserts (0.4 μm, Millipore). The culture medium consisted of Basal Medium Eagle containing 25% Earle’s Balanced Salt Solution (25%), equine serum (Hyclone; Logan, UT; 25% first 3 days, 10% afterwards), 36 mM glucose, 25 mM Hepes-buffer, 250 M glutamax, and (for the first eight days in vitro) 10 ng/ml GDNF (Peprotech, Rocky Hill, NJ). Starting on day 8 in vitro, 10μM 5-fluoro-2′-deoxyuridine was added to the cultures to inhibit astrocytic overgrowth. Cultures were maintained in an incubator at 37°C and 5% CO2 and the media changed three times weekly. After two weeks in culture, PSI (40 nM – 5μM) or vehicle (70% ethanol) were added to the culture medium, and 24 hours later the media was changed. Three days later cell death was assessed by propidium iodide (PI) uptake (10 μM for 2 hours at 37°C).
Subjects and PSI treatment
Adult male Sprague Dawley rats (Harlan, Indianapolis, IL) were group housed on a 12 h light/dark cycle (lights on at 0:600), with food and water available ad libitum. Studies were performed in accordance with the NIH Guide for Care and Use of Laboratory Animals. Animals (N=7/gp) were injected subcutaneously with 3.0 mg/kg PSI or vehicle (70% ethanol) over a two-week period (M-W-F-M-W-F), as described by McNaught and associates (2004). PSI solutions were prepared immediately prior to injection.
Brain proteasome activity
Animals were treated with vehicle or PSI as described above and were sacrificed 2–3 hours after the last PSI injection (N=4/gp), 2 weeks after the last injection (N=4/gp), or 12 weeks after cessation of PSI treatment (N=7/gp). The SN was dissected from 1.0 mm thick coronal slices as previously described (Deutch et al., 1985), and the remainder of the brain designated whole brain. Samples were stored at −80° C until assayed for determination of proteasome peptidase activities. Samples were homogenized (1:10 w/v) in an ice-chilled buffer containing: 20 mM Tris-HCl (pH 7.2), 1 mM EDTA, 1mM sodium azide, 1 mM DTT, 0.1% NP40, and a 1:200 protease inhibitor cocktail (Sigma) (Braun et al., 2005). Homogenates were centrifuged at 14000g for 5 min at 4°C and the supernatants were used for determination of proteasome peptidase activity. Suc-Leu-Leu-Val-Try-aminomethylcoumarin [AMC] (Sigma), Boc-Leu-Arg-Arg-AMC (Sigma), and Z-Leu-Leu-Glu-AMC (Calbiochem) were used to estimate chymotryptic-, tryptic-, and caspase-like proteasomal activities, respectively, as per the report of McNaught et al (2004). Substrates were incubated with cell lysate at 37°C in assay buffer (as above, except omitting the protease inhibitor cocktail and detergent) for 30min; the lysate was preincubated with 1 μM MG132 (Peptides International, Louisville, KY) for one hour as a control. The released AMC was detected by fluorescence emission at 460 nm (excitation at 390 nm). The values reported for proteasome peptidase activities are expressed as fluorescence units (FU) per ug of total protein per min incubation time, after subtraction for non-specific peptide hydrolysis (control reactions). Measurements were performed in 96 well plates, which required 5 μl (corresponding to ~10 μg total protein) per well. The samples were assayed in triplicate for all three activities, therefore requiring 45 μl of cell lysate from each animal (roughly 90μg total protein). Samples derived from small brain areas, such as the SN, yielded 700–1000 μg of total protein. This approach allowed for sufficient material to measure proteasome activities from small brain areas from a single animal.
Immunohistochemistry and biochemistry
Twelve weeks after the last PSI injection, rats were sacrificed and the brains removed and blocked along the mid-saggital line. Monoamine levels in punch-dissected samples of the dorsolateral striatum from one hemisphere were measured by HPLC with electrochemical detection (Deutch and Cameron, 1992). The other hemisphere was post-fixed in 4% paraformaldehyde, cryoprotected, and 40 μm-thick frozen coronal sections cut through the forebrain and midbrain. Immunohistochemical methods were used to reveal tyrosine hydroxylase-immunoreactive neurons (-ir) neurons and axons, using a monoclonal anti-tyrosine hydroxylase antibody (1:3000; Immunostar, Hudson, WI), and ubiquitin-immunoreactive aggregates using a monoclonal anti-ubiquitin antibody (1:500; Chemicon, Temecula CA) as previously described (Deutch and Cameron, 1992).
Behavior
Locomotor activity was assessed in photocell cages (San Diego Instruments, San Diego, CA) equipped with four beams spaced 9 cm apart along the length of the activity monitor. Animals were tested during the animals’ dark phase, between 20:00 (two hours after light offset) and 04:00 (two hours prior to dark offset). The animals were first tested three weeks after the last PSI injection (e.g., five weeks after the start of treatment with the proteasome inhibitor), and thereafter tested every three weeks until sacrificed. Activity was monitored in 10 min bins for a total of 80 min. The number of sequential beam breaks was recorded as locomotor activity, while the sum of repeated disruptions of a single beam (small movements) and locomotor activity was designated as total movement.
Acknowledgments
We are indebted to Shannon Cornett in the Vanderbilt Mass Spectrometry Core Facility for verifying the identity of the PSI compound. This work was supported in part by the National Parkinson Foundation Center of Excellence at Vanderbilt University and PO1 NS 44282, and by the Canadian Institutes of Health Research and the Parkinsonism Society of Canada.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beal F, Lang A. The proteasomal inhibition model of Parkinson’s disease: “Boon or bust”? Ann Neurol. 2006;60:158–161. doi: 10.1002/ana.20939. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Ubiquitin-proteasome system and Parkinson’s diseases. Exp Neurol. 2005;191:S17–27. doi: 10.1016/j.expneurol.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Bove J, Zhou C, Jackson-Lewis V, Taylor J, Chu Y, Rideout HJ, Wu DC, Kordower JH, Petrucelli L, Przedborski S. Proteasome inhibition and Parkinson’s disease modeling. Ann Neurol. 2006;60:260–264. doi: 10.1002/ana.20937. [DOI] [PubMed] [Google Scholar]
- Braun HA, Umbreen S, Groll M, Kuckelkorn U, Mlynarczuk I, Wigand ME, Drung I, Kloetzel PM, Schmidt B. Tripeptide mimetics inhibit the 20 S proteasome by covalent bonding to the active threonines. J Biol Chem. 2005;280:28394–28401. doi: 10.1074/jbc.M502453200. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:89–90. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Cameron DS. Pharmacological characterization of dopamine systems in the nucleus accumbens core and shell. Neuroscience. 1992;46:49–56. doi: 10.1016/0306-4522(92)90007-o. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Elkon H, Melamed E, Offen D. Oxidative stress, induced by 6-hydroxydopamine, reduces proteasome activities in PC12 cells: implications for the pathogenesis of Parkinson’s disease. J Mol Neurosci. 2004;24:387–400. doi: 10.1385/JMN:24:3:387. [DOI] [PubMed] [Google Scholar]
- Fornai F, Lenzi P, Gesi M, Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti CL, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG, Paparelli A. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci. 2003;23:8955–8966. doi: 10.1523/JNEUROSCI.23-26-08955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and α-synuclein. Proc Natl Acad Sci USA. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giason BL, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Carrard G, Michel PP, Medja F, Lombes A, Ruberg M, Friguet B, Hirsch EC. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J Neurochem. 2003;86:1297–307. doi: 10.1046/j.1471-4159.2003.01952.x. [DOI] [PubMed] [Google Scholar]
- Inden M, Kondo J, Kitamura Y, Takata K, Nishimura K, Taniguchi T, Sawada H, Shimohama S. Proteasome inhibitors protect against degeneration of nigral dopaminergic neurons in hemiparkinsonian rats. J Pharmacol Sci. 2005;97:203–211. doi: 10.1254/jphs.fp0040525. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Kanaan NM, Chu Y, Suresh Babu R, Stansell J, 3rd, Terpstra BT, Sortwell CE, Steece-Collier K, Collier TJ. Failure of proteasome inhibitor adminstration to provide a model of Parkinson’s disease in rats and monkeys. Ann Neurol. 2006;60:264–268. doi: 10.1002/ana.20935. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, Reaney SH, Chou VP, Johnston LC, McCormack AL, Johston J, Langston JW, Di Monte DA. Lack of nigrostriatal pathology in a rat model of proteasome inhibition. Ann Neurol. 2006;60:256–260. doi: 10.1002/ana.20938. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Mytilineou C, Jonbaptiste R, Yabut J, Shashidharan P, Jenner P, Olanow CW. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002;81:301–306. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Olanow CW. Proteasome inhibitor-induced model of Parkinson’s disease. Ann Neurol. 2006;60:243–247. doi: 10.1002/ana.20936. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- Miwa H, Kubo T, Suzuki A, Nishi K, Kondo T. Retrograde dopaminergic neuron degeneration following intrastriatal proteasome inhibition. Neurosci Lett. 2005;380:93–98. doi: 10.1016/j.neulet.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant α-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin IC, Savalle M, Stefanis L. Dopaminergic neuron in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not upregulate iHSP70 following proteasomal inhibition. J Neurochem. 2005;93:1304–1313. doi: 10.1111/j.1471-4159.2005.03124.x. [DOI] [PubMed] [Google Scholar]
- Sawada H, Kohno R, Kihara T, Izumi Y, Sakka N, Ibi M, Nakanishi M, Nakamizo T, Yamakawa K, Shibasaki H, Yamamoto N, Akaike A, Inden M, Kitamura Y, Taniguchi T, Shimohama S. Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes α-synuclein inclusions. J Biol Chem. 2004;279:10710–10719. doi: 10.1074/jbc.M308434200. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cleeter MW, Muddle JR, Workman JM, Cooper JM, King RH. Proteasome inhibition causes loss of nigral tyrosine hydroxylase neurons. Ann Neurol. 2006;60:253–255. doi: 10.1002/ana.20934. [DOI] [PubMed] [Google Scholar]
- Setsuie R, Kabuta T, Wada K. Does proteosome inhibition decrease or accelerate toxin-induced dopaminergic neurodegeneration? J Pharmacol Sci. 2005;97:457–460. doi: 10.1254/jphs.ltj05003x. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α–Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–73. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Zeng BY, Bukhatwa S, Hakima A, Rose S, Jenner P. Reproducible nigral cell loss after systemic proteasomal inhibitor administration in rats. Ann Neurol. 2006;60:248–252. doi: 10.1002/ana.20932. [DOI] [PubMed] [Google Scholar]