

Abstract
This review examines the role of spatial electrical heterogeneity within ventricular myocardium on the function of the heart in health and disease. The cellular basis for transmural dispersion of repolarization (TDR) is reviewed and the hypothesis that amplification of spatial dispersion of repolarization underlies the development of life-threatening ventricular arrhythmias associated with inherited ion channelopathies is evaluated. The role of TDR in the long QT, short QT and Brugada syndromes as well as catecholaminergic polymorphic ventricular tachycardia (CPVT) are critically examined. In the long QT Syndrome, amplification of TDR is often secondary to preferential prolongation of the action potential duration (APD) of M cells, whereas in the Brugada Syndrome, it is thought to be due to selective abbreviation of the APD of right ventricular (RV) epicardium. Preferential abbreviation of APD of either endocardium or epicardium appears to be responsible for amplification of TDR in the short QT syndrome. In catecholaminergic polymorphic VT, reversal of the direction of activation of the ventricular wall is responsible for the increase in TDR. In conclusion, the long QT, short QT, Brugada and catecholaminergic polymorphic VT syndromes are pathologies with very different phenotypes and etiologies, but which share a common final pathway in causing sudden cardiac death.
Keywords: Long QT Syndrome, Short QT Syndrome, Brugada Syndrome, Polymorphic Ventricular Tachycardia, Electrophysiology
It is a distinct honor and privilege to be invited to present the Wiggers lecture. My knowledge of Carl Wiggers derives in part from my discussions with Gordon K. Moe. Moe was my mentor and Wiggers was his. Moe completed one year of postdoctoral work in the laboratory of Carl J. Wiggers at the School of Medicine at the Western Reserve University in Cleveland, Ohio in 1940.
Among Wiggers' many seminal contributions to physiology and medicine was his elucidation of the mechanisms responsible for ventricular fibrillation. He described approaches to the prevention and treatment of these conditions (127). I am pleased to have the opportunity to present some of what we have done to further his important contributions to our field.
My principal focus will be on the heterogeneity that exists within the ventricular myocardium with respect to electrical activity, how this heterogeneity contributes to spatial dispersion of repolarization and the degree to which amplification of this spatial dispersion contributes to the development of life-threatening ventricular arrhythmias associated with inherited ion channelopathies (Table 1) such as the long QT, short QT and Brugada syndromes as well as catecholaminergic polymorphic ventricular tachycardia (CPVT). Preferential prolongation of the action potential duration (APD) of M cells is responsible for amplification of TDR is most cases of long QT Syndrome, whereas preferential abbreviation of the APD of right ventricular (RV) epicardium is believed to responsible for amplification of spatial dispersion of repolarization in the Brugada Syndrome. Recent reports suggest that preferential abbreviation of APD of either endocardium or epicardium is responsible for amplification of TDR in different forms of the short QT syndrome. Although triggered activity has long been invoked to explain arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia (CPVT), recent reports suggest that reversal of the direction of activation of the ventricular wall by triggered beats arising in epicardium can amplify TDR and thus contribute to precipitation of rapid polymorphic ventricular tachycardia (VT) that may underlie sudden death in CPVT.
Table 1.
Inherited Disorders Caused by Ion Channelopathies
| Rhythm | Inheritance | Locus | Ion Channel | Gene | ||
|---|---|---|---|---|---|---|
| Long QT syndrome | (RW) | TdP | AD | |||
| LQT1 | 11p15 | IKs | KCNQ1, KvLQT1 | |||
| LQT2 | 7q35 | IKr | KCNH2, HERG | |||
| LQT3 | 3p21 | INa | SCN5A, Nav1.5 | |||
| LQT4 | 4q25 | ANKB, ANK2 | ||||
| LQT5 | 21q22 | IKs | KCNE1, minK | |||
| LQT6 | 21q22 | IKr | KCNE2, MiRP1 | |||
| LQT7 | (Andersen-Tawil Syndrome) | 17q23 | IK1 | KCNJ2, Kir 2.1 | ||
| LQT8 | (Timothy Syndrome) | 6q8A | ICa | CACNA1C,Cav1.2 | ||
| LQT9 | 3p25 | INa | CAV3, Caveolin-3 | |||
| LQT10 | 11q23.3 | INa | SCN4B. Navb4 | |||
| LQT syndrome (JLN) | TdP | AR | 11p15 | IKs | KCNQ1, KvLQT1 | |
| 21q22 | IKs | KCNE1, minK | ||||
| Brugada syndrome | BrS1 | PVT | AD | 3p21 | INa | SCN5A, Nav1.5 |
| BrS2 | PVT | AD | 3p24 | INa | GPD1L | |
| BrS3 | PVT | AD | 12p13.3 | ICa | CACNA1C,CaV1.2 | |
| BrS4 | PVT | AD | 10p12.33 | ICa | CACNB2b, Cavβ2b | |
| Short QT syndrome | SQT1 | VT/VF | AD | 7q35 | IKr | KCNH2, HERG |
| SQT2 | 11p15 | IKs | KCNQ1, KvLQT1 | |||
| SQT3 | AD | 17q23.1-24.2 | IK1 | KCNJ2, Kir2.1 | ||
| SQT4 | 12p13.3 | ICa | CACNA1C,CaV1.2 | |||
| SQT5 | AD | 10p12.33 | ICa | CACNB2b, Cavβ2b | ||
| Catecholaminergic | VT CPVT1 | VT | AD | 1q42-43 | RyR2 | |
| CPVT2 | VT | AR | 1p13-21 | CASQ2 | ||
Abbreviations: AD: autosomal dominant, AR: autosomal recessive, JLN: Jervell and Lange–Nielsen, LQT: Long QT, RW: Romano-Ward, TdP: Torsade de Pointes, VF: ventricular fibrillation, VT: ventricular tachycardia, PVT: Polymorphic VT
Electrical Heterogeneity within the Ventricular Myocardium
Studies conducted over the past two decades have provided evidence in support of the thesis that ventricular myocardium is comprised of at least three electrophysiologically and functionally distinct cell types: epicardial, M and endocardial cells.(15; 17) These three principal ventricular myocardial cell types differ with respect to phase 1 and phase 3 repolarization characteristics (Fig. 1). Ventricular epicardial and M, but not endocardial, cells generally display a prominent phase 1, due to a large 4-aminopyridine (4-AP) sensitive transient outward current (Ito), giving the action potential a spike and dome or notched configuration. These regional differences in Ito, first suggested on the basis of action potential data (60), have now been directly demonstrated in canine (62), feline (50), rabbit (46), rat (35), ferret (32) and human (71; 126) ventricular myocytes.
Figure 1.

Ionic distinctions among epicardial, M and endocardial cells. A: Action potentials recorded from myocytes isolated from the epicardial, endocardial and M regions of the canine left ventricle. B: I-V relations for IK1 in epicardial, endocardial and M region myocytes. Values are mean ± S.D. C: Transient outward current (Ito) recorded from the three cell types. D: The average peak current-voltage relationship for Ito for each of the three cell types. Values are mean±S.D. E: Voltage-dependent activation of the slowly activating component of the delayed rectifier K+ current (IKs) (currents were elicited by the voltage pulse protocol shown in the inset; Na+-, K+- and Ca2+- free solution). F: Voltage dependence of IKs (current remaining after exposure to E-4031) and IKr (E-4031-sensitive current). Values are mean ± S.E. * p<0.05 compared with Epi or Endo. From references (61; 62; 139) with permission. G: Reverse-mode sodium-calcium exchange currents recorded in potassium- and chloride-free solutions at a voltage of −80 mV. INa-Ca was maximally activated by switching to sodium-free external solution at the time indicated by the arrow. H: Midmyocardial sodium-calcium exchanger density is 30% greater than endocardial density, calculated as the peak outward INa-Ca normalized by cell capacitance. Endocardial and epicardial densities were not significantly different. I: TTX-sensitive late sodium current. Cells were held at −80 mV and briefly pulsed to −45 mV to inactivate fast sodium current before stepping to −10 mV. J: Normalized late sodium current measured 300 msec into the test pulse was plotted as a function of test pulse potential. Modified from reference (139) with permission.
Differences in the magnitude of the action potential notch and corresponding differences in Ito have also been described between right and left ventricular epicardium (40). Similar interventricular differences in Ito have also been described for canine ventricular M cells (116). This distinction is thought to form the basis for why the Brugada syndrome, a channelopathy–mediated form of sudden death, is a right ventricular disease.
Myocytes isolated from the epicardial region of the left ventricular wall of the rabbit show a higher density of cAMP-activated chloride current when compared to endocardial myocytes (105). Ito2, initially ascribed to a K+ current, is now thought to be primarily due to the calcium-activated chloride current (ICl(Ca)) is also thought to contribute to the action potential notch, but it is not known whether this current, differs among the three ventricular myocardial cell types.(137) Wang and co-workers reported a larger L-type calcium channel current (ICa) in canine endocardial vs. epicardial ventricular myocytes,(119) although other studies have failed to detect any difference in ICa among cells isolated from epicardium, M, and endocardial regions of the canine left ventricular wall.(21; 36)
Between the surface epicardial and endocardial layers are M cells and transitional cells. The M Cell, Masonic Midmyocardial Moe Cell, discovered in the early 1990's, was named in memory of Gordon K. Moe.(15; 18; 94) The hallmark of the M cell is the ability of its action potential to prolong more than that of epicardium or endocardium in response to a slowing of rate or in response to agents that prolong APD (Fig. 2). (17; 19; 94)
Figure 2.
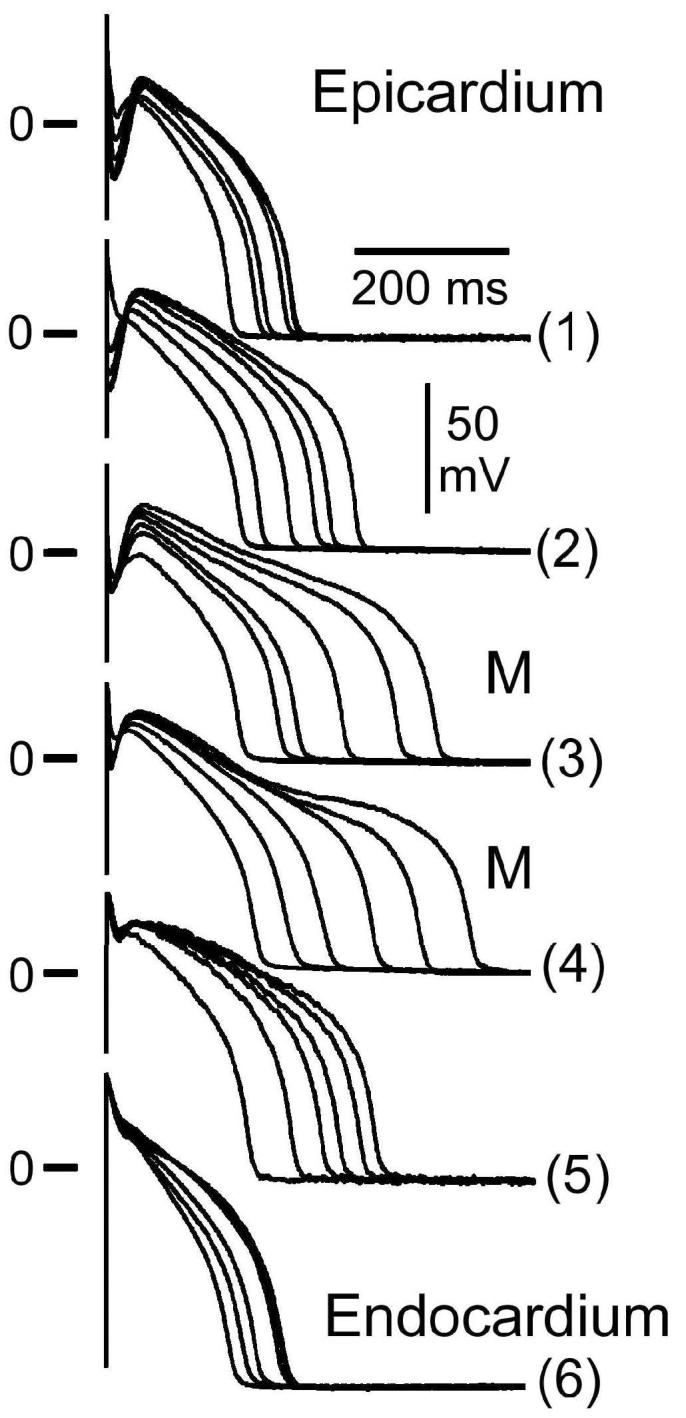
Transmembrane activity recorded from cells isolated from the epicardial (Epi), M and endocardial (Endo) regions of the canine left ventricle at basic cycle lengths (BCL) of 300 to 5000 msec (steady-state conditions). The M and transitional cells were enzymatically dissociated from the midmyocardial region. Deceleration- induced prolongation of APD in M cells is much greater than in epicardial and endocardial cells. The spike and dome morphology is also more accentuated in the epicardial cell. From (62), with permission.
Histologically, M cells are similar to epicardial and endocardial cells. Electrophysiologically and pharmacologically, they appear to be a hybrid between Purkinje and ventricular cells.(11) Like Purkinje fibers, M cells show a prominent APD prolongation and develop early afterdepolarizations (EAD) in response to IKr blockers, whereas epicardium and endocardium do not. Like Purkinje fibers, M cells develop delayed afterdepolarizations (DAD) more readily in response to agents that calcium load or overload the cardiac cell. α1 adrenoceptor stimulation produces APD prolongation in Purkinje fibers, but abbreviation in M cells, and little or no change in endocardium and epicardium (31).
Distribution of M cells within the ventricular wall has been investigated in greatest detail in the left ventricle of the canine heart. Although transitional cells are found throughout the wall in the canine left ventricle, M cells displaying the longest action potentials (at BCLs ≥ 2000 msec) are often localized in the deep subendocardium to midmyocardium in the anterior wall, (134) deep subepicardium to midmyocardium in the lateral wall (94) and throughout the wall in the region of the right ventricular (RV) outflow tracts.(15) M cells are also present in the deep cell layers of endocardial structures, including papillary muscles, trabeculae and the interventricular septum (96). Unlike Purkinje fibers, M cells are not found in discrete bundles or islets (96; 97), although there is evidence that they may be localized in discrete muscle layers. Cells with the characteristics of M cells have been described in the canine, guinea pig, rabbit, pig and human ventricles (16; 19; 20; 30; 42; 43; 59; 61; 62; 66; 81; 85; 88; 89; 93-97; 99; 103; 123; 125; 131; 134).
Myocytes enzymatically dissociated from the different layers of the left ventricular wall typically display APD values that differ by more than 200 msec at slow rates of stimulation (basic cycle lengths ≥ 2000 msec). In the intact ventricular wall, this dispersion of APD is less pronounced (25-55 msec) because of electrotonic interaction among the different cell types. The transmural increase in APD from epi- to endocardium is relatively gradual, except between the epicardium and subepicardium where there is often a sharp increase in APD (Fig. 3). This has been shown to be due to an increase in tissue resistivity in this region (134), which may be related to the sharp transition in cell orientation in this region as well as to reduced expression of connexin 43,(75; 129) which is principally responsible for intracellular communication in ventricular myocardium. Moreover, LeGrice et al. have shown that the density of collagen is heterogeneously distributed across the ventricular wall. (58) A greater density of collagen in the deep subepicardium may also contribute to the resistive barrier in this region of the wall, limiting the degree of electrotonic interaction between myocardial layers. The degree of electrotonic coupling, together with the intrinsic differences APD, contribute to transmural dispersion of repolarization in the ventricular myocardium. (115)
Figure 3.

Transmural distribution of action potential duration and tissue resistivity across the ventricular wall. A: Schematic diagram of the coronary-perfused canine LV wedge preparation. Transmembrane action potentials are recorded simultaneously from epicardial (Epi), M region (M) and endocardial (Endo) sites using three floating microelectrodes. A transmural ECG is recorded along the same transmural axis across the bath, registering the entire field of the wedge. B: Histology of a transmural slice of the left ventricular wall near the epicardial border. The region of sharp transition of cell orientation coincides with the region of high tissue resistivity depicted in panel D and the region of sharp transition of action potential duration illustrated in panel C. C: Distribution of conduction time (CT), APD90 and repolarization time (RT = APD90 + CT) in a canine left ventricular wall wedge preparation paced at BCL of 2000 msec. A sharp transition of APD90 is present between epicardium and subepicardium. Epi: epicardium; M: M Cell; Endo: endocardium. RT: repolarization time; CT: conduction time. D: Distribution of total tissue resistivity (Rt) across the canine left ventricular wall. Transmural distances at 0% and 100% represent epicardium and endocardium, respectively. * p<0.01 compared with Rt at mid-wall. Tissue resistivity increases most dramatically between deep subepicardium and epicardium. Error bars represent SEM (n=5). From (11; 134) with permission.
In the dog, the ionic basis for these features of the M cell include the presence of a smaller slowly activating delayed rectifier current (IKs) (61), a larger late sodium current (late INa) (138) and a larger Na-Ca exchange current (INa-Ca) (139). In the canine heart, the rapidly activating delayed rectifier (IKr) and inward rectifier (IK1) currents are similar in the three transmural cell types. Transmural and apico-basal differences in the density of IKr channels have been described in the ferret heart (26). IKr message and channel protein are much larger in the ferret epicardium. IKs is larger in M cells isolated from the right vs. left ventricles of the dog (116).
These ionic distinctions sensitize the M cells to a variety of pharmacological agents. Agents that block the rapidly activating delayed rectifier current (IKr), IKs or that increase calcium channel current (ICa) or late INa generally produce a much greater prolongation of the APD of the M cell than of epicardial or endocardial cells leading to amplification of transmural dispersion of repolarization.
Amplification of transmural heterogeneities normally present in the early and late phases of the action potential can lead to the development of a variety of arrhythmias, including long QT, Brugada and short QT syndromes as well as Catecholaminergic VT.
The Long QT Syndrome
As its name implies, the long QT syndrome (LQTS) is characterized by prolongation of the interval between the start of the QRS and the end of the T wave in the electrocardiogram (ECG). The long QT syndromes are phenotypically and genotypically diverse, but have in common the appearance of a long QT interval in the ECG, an atypical polymorphic ventricular tachycardia known as Torsade de Pointes (TdP), and, in many but not all cases, a relatively high risk for sudden cardiac death.(70; 82; 136) Ten genotypes of the congenital long QT syndrome have been identified. They are distinguished by mutations in at least eight different ion channel genes, a structural anchoring protein and a caveolin protein located on chromosomes 3, 4, 6, 7, 11, 17 and 21 (Table 1). (37; 67; 74; 102; 120; 121)
Two recently genotyped forms of LQTS are associated with multi-organ disease. Andersen–Tawil syndrome,(74) also referred to as LQT7, is characterized by skeletal muscle periodic paralysis, frequent ectopy, but relatively rare episodes of TdP, secondary to loss of function mutations in KCNJ2, which encodes Kir2.1, the channel conducting the inward rectifier current, IK1. Timothy syndrome, also referred to as LQT8, is a rare congenital disorder characterized by multi-organ dysfunction including prolongation of the QT interval, lethal arrhythmias, webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and autism. Timothy syndrome has been linked to loss of voltage-dependent inactivation due to mutations in CACNA1C, the gene that encodes Cav1.2, the α subunit of the calcium channel.(101) The most recent genes associated with LQTS are CAV3 which encodes caveolin-3 and SCN4B which encodes NaVB4, an auxiliary subunit of the cardiac sodium channel. Caveolin-3 spans the plasma membrane twice, forming a hairpin structure on the surface, and is the main constituent of caveolae, small invaginations in the plasma membrane. Mutations in CAV3 and SCNB4 both produce a gain of function in late INa , causing an LQT3-like phenotype.(41; 114)
LQTS shows both autosomal recessive and autosomal dominant patterns of inheritance: 1) a rare autosomal recessive disease associated with deafness (Jervell and Lange-Nielsen), caused by 2 genes that encode for the slowly activating delayed rectifier potassium channel (KCNQ1 and KCNE1); and 2) a much more common autosomal dominant form known as the Romano Ward syndrome, caused by mutations in 10 different genes (Table 1). Eight of the 10 genes encode cardiac ion channels.
Acquired LQTS refers to a QT prolongation caused by exposure to drugs that prolong the duration of the ventricular action potential(22) or QT prolongation secondary to cardiomyopathies including dilated or hypertrophic cardiomyopathy, as well as to abnormal QT prolongation associated with bradycardia or electrolyte imbalance.(65; 100; 108; 111; 117) The acquired form of the disease is far more prevalent than the congenital form, and in some cases may have a genetic predisposition.(80)
Accentuation of spatial dispersion of repolarization within the ventricular myocardium has been identified as the principal arrhythmogenic substrate in both acquired and congenital LQTS. The amplification of spatial dispersion of refractoriness can take the form of an increase of transmural, trans-septal or apico-basal dispersion of repolarization. This exaggerated intrinsic heterogeneity together with early and delayed afterdepolarization (EAD and DAD)-induced triggered activity, both caused by reduction in net repolarizing current, underlie the substrate and trigger for the development of Torsade de Pointes arrhythmias observed under LQTS conditions.(14; 23) Models of the LQT1, LQT2, and LQT3 forms of the long QT syndrome have been developed using the canine arterially perfused left ventricular wedge preparation (Fig. 4).(91) These models suggest that in these three forms of LQTS, preferential prolongation of the M cell APD leads to an increase in the QT interval as well as an increase in TDR, which contributes to the development of spontaneous as well as stimulation-induced Torsade de Pointes (TdP) (Fig. 5). (86; 87; 90; 109; 110) The spatial dispersion of repolarization is further exaggerated by sympathetic influences in LQT1 and LQT2, accounting for the great sensitivity of patients with these genotypes to adrenergic stimuli (Figs. 4 and 5).
Figure 4.

LQT1, LQT2, and LQT3 models of LQTS. Panels A–C shows action potentials simultaneously recorded from endocardial (Endo), M and epicardial (Epi) sites of arterially-perfused canine left ventricular wedge preparations together with a transmural ECG. BCL = 2000 msec. Transmural dispersion of repolarization across the ventricular wall, defined as the difference in the repolarization time between M and epicardial cells, is denoted below the ECG traces. LQT1 model was mimicked using Isoproterenol + chromanol 293B − an IKs blocker. LQT2 was created using the IKr blocker d-sotalol + low [K+]o. LQT3 was mimicked using the seas anemone toxin ATX-II to augments late INa. Panels D–F: Effect of isoproterenol in the LQT1, LQT2 and LQT3 models. In LQT1, isoproterenol (Iso) produces a persistent prolongation of the APD90 of the M cell and of the QT interval (at both 2 and 10 minute), whereas the APD90 of the epicardial cell is always abbreviated, resulting in a persistent increase in TDR (D). In LQT2, isoproterenol initially prolongs (2 minute) and then abbreviates the QT interval and the APD90 of the M cell to the control level (10 minute), whereas the APD90 of epicardial cell is always abbreviated, resulting in a transient increase in TDR (E). In LQT3, isoproterenol produced a persistent abbreviation of the QT interval and the APD90 of both M and epicardial cells (at both 2 and 10 minute), resulting in a persistent decrease in TDR (F). *P < .0005 vs. Control; † P < .0005, †† P < .005, ††† P < .05, vs. 293B, d-Sotalol (d-Sot) or ATX-II. (Modified from references (86; 87; 90)with permission).
Figure 5.

Proposed cellular mechanism for the development of Torsade de Pointes in the long QT syndromes.
Differences in the time course of repolarization of the three predominant myocardial cell types contributes prominently to the inscription of the T wave of the ECG. Voltage gradients developing as a result of the different time course of repolarization of phases 2 and 3 in the three cell types give rise to opposing voltage gradients on either side of the M region, which are in part responsible for the inscription of the T wave.(131) In the case of an upright T wave, the epicardial response is the earliest to repolarize and the M cell action potential is the latest. Full repolarization of the epicardial action potential coincides with the peak of the T wave and repolarization of the M cells is coincident with the end of the T wave. The duration of the M cell action potential therefore determines the QT interval, whereas the duration of the epicardial action potential determines the QTpeak interval. The interval between the peak and end of the T wave (Tpeak–Tend interval) in precordial ECG leads is suggested to provide an index of transmural dispersion of repolarization.(15) Recent studies provide guidelines for the estimation of transmural dispersion of repolarization in the case of more complex T waves, including negative, biphasic and triphasic T waves.(44) In these cases, the interval from the nadir of the first component of the T wave to the end of the T wave provides an approximation of transmural dispersion of repolarization.
Because the precordial leads are the only ECG leads designed to view the electrical field across the ventricular wall, Tpeak-Tend is thought to be most representative of TDR in these leads. Tpeak-Tend intervals measured in the limb leads are unlikely to provide an index of TDR, but may provide a measure of global dispersion within the heart.(73; 135) Because TDR can be highly variable among different regions within the heart, it is also inadvisable to average Tpeak-Tend among several leads or to measure Tpeak and Tend in different leads.(135) Because LQTS is principally a left ventricular disorder, TDR is likely to be greatest in the left ventricular wall or septum and thus be best reflected in left precordial leads or V3, respectively.(130) In contrast, because Brugada syndrome is a right ventricular disorder, TDR is greatest in the right ventricular free wall and thus is best reflected in the right precordial leads.(33)
Tpeak-Tend interval does not provide an absolute measure of transmural dispersion in vivo, but changes in this parameter are thought to reflect changes in spatial dispersion of repolarization and thus may be prognostic of arrhythmic risk under a variety of conditions.(49; 84; 106; 107; 122; 128) Takenaka et al. recently demonstrated exercise-induced accentuation of the Tpeak-Tend interval in LQT1 patients, but not LQT2.(106) These observations coupled with those of Schwartz et al.(83), demonstrating an association between exercise and risk for TdP in LQT1, but not LQT2, patients, once again point to the potential value of Tpeak–Tend in forecasting risk for the development of TdP. Direct evidence in support of Tpeak-Tend as an index to predict TdP in patients with long QT syndrome was provided by Yamaguchi and co-workers.(130) These authors concluded that Tpeak-Tend is more valuable than QTc and QT dispersion as a predictor of Torsade de Pointes (TdP) in patients with acquired LQTS. Shimizu et al. demonstrated that Tpeak-Tend, but not QTc, predicted sudden cardiac death in patients with hypertrophic cardiomyopathy.(84) Most recently, Watanabe et al. demonstrated that prolonged Tpeak-Tend is associated with inducibility as well as spontaneous development of ventricular tachycardia (VT) in high risk patients with organic heart disease.(122)
The available data support the hypothesis that TDR rather QT prolongation underlies the principal substrate for the development of TdP.(6; 8; 23; 39; 47) Our working hypothesis for the development of LQTS-related TdP presumes the presence of electrical heterogeneity in the form of transmural dispersion of repolarization under baseline conditions and the amplification of TDR by agents that reduce net repolarizing current via a reduction in IKr or IKs or augmentation of ICa or late INa (Fig. 5). Conditions leading to a reduction in IKr or augmentation of late INa produce a preferential prolongation of the M cell action potential. As a consequence, the QT interval prolongs and is accompanied by a dramatic increase in transmural dispersion of repolarization, thus creating a vulnerable window for the development of reentry. The reduction in net repolarizing current also predisposes to the development of EAD-induced triggered activity in M and Purkinje cells, which provide the extrasystole that triggers TdP when it falls within the vulnerable period. β adrenergic agonists further amplify transmural heterogeneity (transiently) in the case of IKr block, but reduce it in the case of INa agonists. (59; 90)
Although agents that block IKr and which increase late INa clearly augment TDR, not all agents that prolong the QT interval increase TDR. Amiodarone, a potent antiarrhythmic agent used in the management of both atrial and ventricular arrhythmias, is rarely associated with TdP. Chronic administration of amiodarone produces a greater prolongation of APD in epicardium and endocardium, but less of an increase, or even a decrease at slow rates, in the M region, thereby reducing TDR.(98) In a dog model of chronic complete atrioventricular block and acquired LQTS, 6 weeks of amiodarone was shown to produce a major QT prolongation without producing TdP. In contrast, after 6 weeks of dronedarone, TdP occurred in 4 of 8 dogs with the highest spatial dispersion of repolarization (105±20 ms).(112)
Sodium pentobarbital is another agent that prolongs the QT interval but reduces TDR. Pentobarbital has been shown to produce a dose-dependent prolongation of the QT interval, accompanied by a reduction in TDR. (93) TdP is not observed under these conditions, nor can it be induced with programmed electrical stimulation. Amiodarone and pentobarbital have in common the ability to block IKs, IKr, and late INa. This combination produces a preferential prolongation of the APD of epicardium and endocardium so that the QT interval is prolonged, but TDR is actually reduced and TdP does not develop. Cisapride is another agent that blocks both inward and outward currents. In the canine left ventricular wedge preparation, cisapride produces a biphasic dose-dependent prolongation of the QT interval and TDR. TDR peaks at 0.2 μM, and it is only at this concentration that TdP is observed. Higher concentrations of cisapride lead to an abbreviation of TDR and elimination of TdP, even though QT is further prolonged.(39) This finding suggests that the spatial dispersion of repolarization is more important than the prolongation of the QT interval in determining the substrate for TdP.
Block of IKs with chromanol 293B also increases QT without augmenting TDR. Chromanol 293B prolongs APD of the 3 cell types homogeneously, neither increasing TDR nor widening the T wave. TdP is never observed under these conditions. The addition of β adrenergic agonist, however, abbreviates the APD of epicardial and endocardial cells but not that of the M cell, resulting in a marked accentuation of TDR and the development of TdP.(90)
Brugada Syndrome
The Brugada syndrome is another inherited channelopathy in which amplification of TDR leads to the development of polymorphic VT and sudden cardiac death.(7) The syndrome is characterized by an elevated ST segment or J wave appearing in the right precordial leads (V1-V3), often followed by a negative T wave. First described in 1992, the syndrome is associated with a high incidence of sudden cardiac death secondary to a rapid polymorphic VT or VF.(27) The ECG characteristics of the Brugada syndrome are dynamic and often concealed, but can be unmasked by potent sodium channel blockers such as ajmaline, flecainide, procainamide, disopyramide, propafenone and pilsicainide.(28; 77; 92)
In at least 15% of Brugada Syndrome (BrS) probands, the syndrome is associated with mutations in SCN5A, the gene that encodes the α subunit of the cardiac sodium channel (34). Over one hundred mutations in SCN5A have been linked to the syndrome in recent years (see (9) for references; also see http://www.fsm.it/cardmoc ). Only a fraction of these mutations have been studied in expression systems and shown to result in loss of function due either to: 1) failure of the sodium channel to express; 2) a shift in the voltage- and time-dependence of sodium channel current (INa) activation, inactivation or reactivation; 3) entry of the sodium channel into an intermediate state of inactivation from which it recovers more slowly or 4) accelerated inactivation of the sodium channel. Mutations in the SCN5A gene account for approximately 15% of Brugada syndrome probands. Of note, negative SCN5A results generally do not rule out causal gene mutations, since the promoter region, cryptic splicing mutations or presence of gross rearrangements are generally not part of routine investigation. A recent report by Hong et al. (54) provided the first report of a dysfunctional sodium channel created by an intronic mutation giving rise to cryptic splice site activation in SCN5A in a family with the Brugada syndrome. The deletion of fragments of segments 2 and 3 of Domain IV of SCN5A caused complete loss of function. Bezzina and co-workers recently provided interesting evidence in support of the hypothesis that an SCN5A promoter polymorphism common in Asians modulates variability in cardiac conduction, and may contribute to the high prevalence of Brugada syndrome in the Asian population.(25) Sequencing of the SCN5A promoter identified a haplotype variant consisting of 6 polymorphisms in near-complete linkage disequilibrium that occurred at an allele frequency of 22% in Asian subjects and was absent in whites and blacks.
Weiss et al. described a second locus on chromosome 3, close to but distinct from SCN5A, linked to the syndrome (124) in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis. The gene was recently identified in a preliminary report as the Glycerol-3-Phosphate Dehydrogenase 1-Like Gene (GPD1L) and the mutation in GPD1L was shown to result in a reduction of INa.(63)
The third and fourth genes associated with the Brugada syndrome were recently identified and shown to encode the α1 (CACNA1C) and β (CACNB2b) subunits of the L-type cardiac calcium channel. Mutations in the α and β subunits of the calcium channel also lead to a shorter than normal QT interval, in some cases creating a new clinical entity consisting of a combined Brugada/Short QT syndrome. (13)
The development of extrasystolic activity and polymorphic VT in the Brugada syndrome has been shown to be due to amplification of heterogeneities intrinsic to the early phases (phase 1-mediated notch) of the action potential of cells residing in different layers of the right ventricular wall of the heart (Fig. 6). Rebalancing of the currents active at the end of phase 1 can lead to accentuation of the action potential notch in right ventricular epicardium, which is responsible for the augmented J wave and ST segment elevation associated with the Brugada syndrome (see (5; 7) for references). Under physiologic conditions, the ST segment isoelectric due to the absence of major transmural voltage gradients at the level of the action potential plateau. Accentuation of the right ventricular action potential notch under pathophysiological conditions leads to exaggeration of transmural voltage gradients and thus to accentuation of the J wave or to an elevation of the J point (Fig. 6). If the epicardial action potential continues to repolarize before that of endocardium, the T wave remains positive, giving rise to a saddleback configuration of the ST segment elevation. Further accentuation of the notch is accompanied by a prolongation of the epicardial action potential causing it to repolarize after endocardium, thus leading to inversion of the T wave. (48; 132)
Figure 6.

Cellular basis for electrocardiographic and arrhythmic manifestation of Brugada Syndrome. Each panel shows transmembrane action potentials from one endocardial (top) and two epicardial sites together with a transmural ECG recorded from a canine coronary-perfused right ventricular wedge preparation. A: Control (BCL 400 msec). B: Combined sodium and calcium channel block with terfenadine (5 μM) accentuates the epicardial action potential notch creating a transmural voltage gradient that manifests as a ST segment elevation or exaggerated J wave in the ECG. C: Continued exposure to terfenadine results in all-or-none repolarization at the end of phase 1 at some epicardial sites but not others, creating a local epicardial dispersion of repolarization (EDR) as well as a transmural dispersion of repolarization (TDR). D: Phase 2 reentry occurs when the epicardial action potential dome propagates from a site where it is maintained to regions where it has been lost giving rise to a closely coupled extrasystole. E: Extrastimulus (S1-S2 = 250 msec) applied to epicardium triggers a polymorphic VT. F: Phase 2 reentrant extrasystole triggers a brief episode of polymorphic VT. (Modified from reference (48), with permission)
Despite the appearance of a typical Brugada ECG, accentuation of the RV epicardial action potential (AP) notch alone does not give rise to an arrhythmogenic substrate. The arrhythmogenic substrate may develop with a further shift in the balance of current leading to loss of the action potential dome at some epicardial sites but not others. A marked transmural dispersion of repolarization develops as a consequence, creating a vulnerable window, which when captured by a premature extrasystole can trigger a reentrant arrhythmia. Because loss of the action potential dome in epicardium is generally heterogeneous, epicardial dispersion of repolarization develops as well. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local re-excitation via phase 2 reentry, leading to the development of a closely-coupled extrasystole capable of capturing the vulnerable window across the ventricular wall, thus triggering a circus movement reentry in the form of VT/VF (Figs. 6 and 7).(1; 64; 132) Support for these hypotheses derives from experiments involving the arterially perfused right ventricular wedge preparation(1; 48; 68; 69; 132) and from studies in which monophasic action potential (MAP) electrodes where positioned on the epicardial and endocardial surfaces of the right ventricular outflow tract (RVOT) in patients with the Brugada syndrome.(10; 55)
Figure 7.

Proposed mechanism for the Brugada syndrome. A shift in the balance of currents serves to amplify existing heterogeneities by causing loss of the action potential dome at some epicardial, but not endocardial sites. A vulnerable window develops as a result of the dispersion of repolarization and refractoriness within epicardium as well as across the wall. Epicardial dispersion leads to the development of phase 2 reentry, which provides the extrasystole that captures the vulnerable window and initiates VT/VF via a circus movement reentry mechanism. Modified from (4), with permission.
Short QT Syndrome
The Short QT syndrome (SQTS) is a recently identified inherited channelopathy (53). SQTS is characterized by a QTc ≤ 360 msec and high incidence of VT/VF in infants, children and young adults.(52) The familial nature of this sudden death syndrome was highlighted by Gaita et al. in 2003.(51) The first genetic defect responsible for the short QT syndrome (SQTS1) involved two different missense mutations (substitution of one amino acid for another) resulting in the same amino acid substitution in HERG (N588K), which caused a gain of function in the rapidly activating delayed rectifier channel, IKr .(29). A second gene was reported by Bellocq et al. (SQTS2)(24); a missense mutation in KCNQ1 (KvLQT1) caused a gain of function in IKs. A third gene (SQT3) involves KCNJ2, the gene that encodes the inward rectifier channel. Mutations in KCNJ2 caused a gain of function in IK1, leading to an abbreviation of QT interval. SQT3 is associated with QTc intervals <330 msec, not quite as short as SQT1, and SQT2. Two additional genes recently linked to SQTS encode the α1 (CACNA1C) and β (CACNB2b) subunits of the L-type cardiac calcium channel. SQT4 caused by mutations in the α subunit of calcium channel have been shown to lead to QT interval <360 ms, whereas SQT5 caused by mutations in the β subunit of the calcium channel are characterized by QT intervals of 330-360 msec.(13) Mutations in the α and β subunits of the calcium channel may also lead to ST segment elevation, creating a combined Brugada/Short QT syndrome. (13)
The ECG commonly displays tall peaked symmetrical T waves in SQT1, 2 and 3, due to acceleration of phase 3 repolarization. An augmented Tpeak-Tend interval associated with this electrocardiographic feature of the syndrome suggests that TDR is significantly increased (Fig. 8). Evidence in support of the hypothesis derives from studies of a left ventricular wedge model of the short QT syndrome demonstrating that an increase in outward repolarizing current can preferentially abbreviate endocardial/M cell action potential, thus increasing TDR and creating the substrate for reentry.(45) The potassium channel opener pinacidil used in this study caused a heterogeneous abbreviation of APD among the different cell types spanning the ventricular wall, thus creating the substrate for the genesis of VT under conditions associated with short QT intervals. Polymorphic VT could be readily induced with programmed electrical stimulation. The increase in TDR was further accentuated by isoproterenol, leading to easier induction and more persistent VT/VF. Of note, an increase of TDR to values greater than 55 msec was associated with inducibility of VT/VF. In LQTS models, a TDR of >90 msec is required to induce TdP. The easier inducibility in SQTS is due to the reduction in the wavelength (refractory period × conduction velocity) of the reentrant circuit, which reduces the pathlength required for maintenance of reentry.(45)
Figure 8.

Proposed mechanism for arrhythmogenesis in the short QT syndrome. An increase in net outward current due to a reduction in late inward current or augmentation of outward repolarizing current serves to abbreviate action potential duration heterogeneously leading to an amplification of transmural dispersion of repolarization and the creation of a vulnerable window for the development of reentry. Reentry is facilitated both by the increase in TDR and abbreviation of refractoriness.
TDR as the Common Denominator in Channelopathy-induced Sudden Cardiac Death
The three inherited sudden cardiac death syndromes discussed differ with respect to the characteristics of the QT interval (Fig. 9). In the long QT syndrome, QT increases as a function of disease or drug concentration. In the Brugada syndrome it remains largely unchanged and in the short QT syndrome QT interval decreases as a function of disease of drug. What these three syndromes have in common is an amplification of TDR, which results in the development of polymorphic ventricular tachycardia and fibrillation when dispersion of repolarization and refractoriness reaches the threshold for reentry. Whe polymorphic VT occurs in the setting of long QT, we refer to it as TdP. The threshold for reentry decreases as APD and refractoriness are reduced and the pathlength required for establishing a reentrant wave is progressively reduced.
Figure 9.

The role of transmural dispersion of repolarization (TDR) in channelopathy-induced sudden cardiac death. In the long QT syndrome, QT increases as a function of disease or drug concentration. In the Brugada syndrome it remains largely unchanged and in the short QT syndrome QT interval decreases as a function of disease or drug. The three syndromes have in common the ability to amplify TDR, which results in the development of TdP when dispersion reaches the threshold for reentry. The threshold for reentry decreases as APD and refractoriness are reduced. Modified from (12), with permission.
Catecholaminergic Polymorphic VT
Can arrhythmogenesis generally attributed to triggered activity be aggravated by amplification of TDR? Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare, autosomal dominant or recessive inherited disorder, predominantly affecting children or adolescents with structurally normal hearts. It is characterized by bidirectional ventricular tachycardia (BVT), monomorphic and polymorphic VT (PVT), and a high risk of sudden cardiac death (30-50% by the age of 20 to 30 years).(57; 104) Mutations in genes encoding the cardiac ryanodine receptor 2 (RyR2) or calsequestrin 2 (CASQ2) in patients have been associated with this phenotype.(56; 76; 78; 79) Mutations in RyR2 cause autosomal dominant CPVT, whereas mutations in CASQ2 are responsible for either an autosomal recessive or dominant form of CPVT.
Numerous studies have provided evidence pointing to delayed afterdepolarization (DAD)-induced triggered activity (TA) as the mechanism underlying monomorphic or bidirectional VT in patients with this syndrome. The cellular mechanisms underlying the various ECG phenotypes, and the transition of monomorphic VT to polymorphic VT or VF, were recently elucidated with the use of low dose caffeine to mimic the defective calcium homeostasis encountered under conditions that predispose to CPVT.
The combination of isoproterenol and caffeine was found to lead to the development of DAD-induced triggered activity arising from the epicardium, endocardium or M region. Alternation of epicardial and endocardial source of ectopic activity gave rise to a bidirectional VT. The triggered activity-induced monomorphic, bidirectional and slow polymorphic VT would be expected to be hemodynamically well tolerated because of the relatively slow rate of these rhythms and are unlikely to be the cause of sudden death in these syndromes.
Ectopic activity or VT in the model arose from epicardium and was associated with an increased Tpeak-Tend interval and augmented transmural dispersion of repolarization due to reversal of the normal transmural activation sequence. The increase in TDR created a vulnerable window across the ventricular wall that when invaded by a premature extrasystole permitted the precipitation of a very rapid polymorphic VT, that would be expected to lead hemodynamic compromise.(72) Thus, even in a syndrome in which arrhythmogenesis is traditionally ascribed to triggered activity, sudden cardiac death may be due to amplification of TDR, giving rise to reentrant VT/VF.
Conclusion
Amplification of spatial dispersion of refractoriness in ventricular myocardium, particularly when due to augmentation of transmural dispersion of repolarization, can predispose to the development of potentially lethal reentrant arrhythmias in a variety of ion channelopathies including long QT, short QT and Brugada syndromes as well as catecholaminergic polymorphic ventricular tachycardia. These same principles apply to arrhythmogenesis associated with hypertrophic and dilated cardiomyopathies (2; 3; 113; 118) as well as some arrhythmias associated with ischemia and reperfusion.(38; 133)
Acknowledgments
Grants
Supported by grant HL47678 from NHLBI and grants from the American Heart Association and NYS and Florida Grand Lodges F. & A.M.
Abbreviations
- 4-AP
4-aminopyridine
- AP
action potential
- AD
autosomal dominant
- APD
action potential duration
- APD90
APD values at 90 percent repolarization
- AR
autosomal recessive
- BCL
basic cycle lengths
- BrS
Brugada Syndrome
- BVT
bidirectional ventricular tachycardia
- CASQ2
calsequestrin 2
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CT
conduction time
- DAD
delayed afterdepolarization
- d-Sot
d-Sotalol
- EAD
early afterdepolarization
- EDR
epicardial dispersion of repolarization
- Endo
endocardial
- Epi
epicardial
- GPD1L
Glycerol-3-Phosphate Dehydrogenase 1-Like Gene
- ICl(Ca))
calcium-activated chloride current
- IK1
inward rectifier current
- INa
sodium channel current
- INa-Ca
sodium-calcium exchange current
- ICa
calcium channel current
- IKr
rapidly activating delayed rectifier current
- IKs
slowly activating delayed rectifier current
- Iso
isoproterenol
- Ito
transient outward current
- I-V
current-voltage
- JLN
Jervell and Lange–Nielsen
- Late INa
late sodium current
- LQTS
long QT syndromes
- LQT7
Andersen–Tawil syndrome
- LQT8
Timothy syndrome
- LV
left ventricular
- MAP
monophasic action potential
- PVT
polymorphic VT
- RT
repolarization time
- Rt
tissue resistivity
- RV
right ventricular
- RVOT
right ventricular outflow tract
- RW
Romano-Ward
- RyR2
ryanodine receptor 2
- SQTS
Short QT syndrome
- TA
triggered activity
- TdP
Torsade de Pointes
- TDR
transmural dispersion of repolarization
- VF
ventricular fibrillation
- VT
ventricular tachycardia
Footnotes
Disclosures
There are no conflicts of interest.
Reference List
- 1.Aiba T, Shimizu W, Hidaka I, Uemura K, Noda T, Zheng C, Kamiya A, Inagaki M, Sugimachi M, Sunagawa K. Cellular basis for trigger and maintenance of ventricular fibrillation in the Brugada syndrome model: high-resolution optical mapping study. J Am Coll Cardiol. 2006;47:2074–2085. doi: 10.1016/j.jacc.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 2.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 3.Akar FG, Tomaselli GF. Conduction abnormalities in nonischemic dilated cardiomyopathy: basic mechanisms and arrhythmic consequences. Trends Cardiovasc Med. 2005;15:259–264. doi: 10.1016/j.tcm.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Antzelevitch C. The Brugada Syndrome: Diagnostic Criteria and Cellular Mechanisms. Eur Heart J. 2001;22:356–363. doi: 10.1053/euhj.2000.2461. [DOI] [PubMed] [Google Scholar]
- 5.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–272. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 6.Antzelevitch C. Drug-induced channelopathies. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology. From Cell to Bedside. W.B. Saunders; New York: 2004. pp. 151–157. [Google Scholar]
- 7.Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas GP. Electrophysiologic effects of ranolazine: a novel anti-anginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada Syndrome: From Bench to Bedside. Blackwell Futura; Oxford: 2005. [Google Scholar]
- 10.Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada Syndrome. A Decade of Progress. Circ Res. 2002;91:1114–1119. doi: 10.1161/01.res.0000046046.53721.90. [DOI] [PubMed] [Google Scholar]
- 11.Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: Physiological, pharmacological and clinical implications. In: Page E, Fozzard HA, Solaro RJ, editors. Handbook of Physiology. Section 2 The Cardiovascular System. Oxford University Press; New York: 2001. pp. 654–692. [Google Scholar]
- 12.Antzelevitch C, Oliva A. Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Intern Med. 2006;259:48–58. doi: 10.1111/j.1365-2796.2005.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Jr., Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the Long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego JM, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 16.Antzelevitch C, Sicouri S. Clinical relevance of cardiac arrhythmias generated by afterdepolarizations. Role of M cells in the generation of U waves, triggered activity and torsade de pointes. J Am Coll Cardiol. 1994;23:259–277. doi: 10.1016/0735-1097(94)90529-0. [DOI] [PubMed] [Google Scholar]
- 17.Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 18.Anyukhovsky EP, Sosunov EA, Gainullin RZ, Rosen MR. The controversial M cell. J Cardiovasc Electrophysiol. 1999;10:244–260. doi: 10.1111/j.1540-8167.1999.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 19.Anyukhovsky EP, Sosunov EA, Rosen MR. Regional differences in electrophysiologic properties of epicardium, midmyocardium and endocardium: In vitro and in vivo correlations. Circulation. 1996;94:1981–1988. doi: 10.1161/01.cir.94.8.1981. [DOI] [PubMed] [Google Scholar]
- 20.Balati B, Varro A, Papp JG. Comparison of the cellular electrophysiological characteristics of canine left ventricular epicardium, M cells, endocardium and Purkinje fibres. Acta Physiol Scand. 1998;164:181–190. doi: 10.1046/j.1365-201X.1998.00416.x. [DOI] [PubMed] [Google Scholar]
- 21.Banyasz T, Fulop L, Magyar J, Szentandrassy N, Varro A, Nanasi PP. Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc Res. 2003;58:66–75. doi: 10.1016/s0008-6363(02)00853-2. [DOI] [PubMed] [Google Scholar]
- 22.Bednar MM, Harrigan EP, Anziano RJ, Camm AJ, Ruskin JN. The QT interval. Prog Cardiovasc Dis. 2001;43:1–45. doi: 10.1053/pcad.2001.21469. [DOI] [PubMed] [Google Scholar]
- 23.Belardinelli L, Antzelevitch C, Vos MA. Assessing Predictors of drug-induced Torsade de Pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Bellocq C, Van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, Baro I, Wilde AA. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394–2397. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- 25.Bezzina CR, Shimizu W, Yang P, Koopmann TT, Tanck MW, Miyamoto Y, Kamakura S, Roden DM, Wilde AA. Common Sodium Channel Promoter Haplotype in Asian Subjects Underlies Variability in Cardiac Conduction. Circulation. 2006;113:338–344. doi: 10.1161/CIRCULATIONAHA.105.580811. [DOI] [PubMed] [Google Scholar]
- 26.Brahmajothi MV, Morales MJ, Reimer KA, Strauss HC. Regional localization of ERG, the channel protein responsible for the rapid component of the delayed rectifier, K+ current in the ferret heart. Circ Res. 1997;81:128–135. doi: 10.1161/01.res.81.1.128. [DOI] [PubMed] [Google Scholar]
- 27.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 28.Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–515. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 29.Brugada R, Hong K, Dumaine R, Cordeiro JM, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden Death associated with Short QT-Syndrome linked to Mutations in HERG. Circulation. 2003;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 30.Burashnikov A, Antzelevitch C. Acceleration-induced action potential prolongation and early afterdepolarizations. J Cardiovasc Electrophysiol. 1998;9:934–948. doi: 10.1111/j.1540-8167.1998.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 31.Burashnikov A, Antzelevitch C. Differences in the electrophysiologic response of four canine ventricular cell types to α1-adrenergic agonists. Cardiovasc Res. 1999;43:901–908. doi: 10.1016/s0008-6363(99)00124-8. [DOI] [PubMed] [Google Scholar]
- 32.Campbell DL, Rasmusson RL, Qu YH, Strauss HC. The calcium-independent transient outward potassium current in isolated ferret right ventricular myocytes. I. Basic characterization and kinetic analysis. J Gen Physiol. 1993;101:571–601. doi: 10.1085/jgp.101.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castro HJ, Antzelevitch C, Tornes BF, Dorantes SM, Dorticos BF, Zayas MR, Quinones Perez MA, Fayad RY. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol. 2006;47:1828–1834. doi: 10.1016/j.jacc.2005.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schultze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 35.Clark RB, Bouchard RA, Salinas-Stefanon E, Sanchez-Chapula J, Giles WR. Heterogeneity of action potential waveforms and potassium currents in rat ventricle. Cardiovasc Res. 1993;27:1795–1799. doi: 10.1093/cvr/27.10.1795. [DOI] [PubMed] [Google Scholar]
- 36.Cordeiro JM, Greene L, Heilmann C, Antzelevitch D, Antzelevitch C. Transmural heterogeneity of calcium activity and mechanical function in the canine left ventricle. Am J Physiol Heart Circ Physiol. 2004;286:H1471–H1479. doi: 10.1152/ajpheart.00748.2003. [DOI] [PubMed] [Google Scholar]
- 37.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 38.Di Diego JM, Antzelevitch C. Cellular basis for ST-segment changes observed during ischemia. J Electrocardiol. 2003;36(Suppl):1–5. doi: 10.1016/j.jelectrocard.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Di Diego JM, Belardinelli L, Antzelevitch C. Cisapride-induced transmural dispersion of repolarization and torsade de pointes in the canine left ventricular wedge preparation during epicardial stimulation. Circulation. 2003;108:1027–1033. doi: 10.1161/01.CIR.0000085066.05180.40. [DOI] [PubMed] [Google Scholar]
- 40.Di Diego JM, Sun ZQ, Antzelevitch C. Ito and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol. 1996;271:H548–H561. doi: 10.1152/ajpheart.1996.271.2.H548. [DOI] [PubMed] [Google Scholar]
- 41.Domingo AM, Kaku T, Tester DJ, Torres PI, Itty A, Ye B, Valdivia CR, Makielski JC, Quintero SC, Luna TT, Ackerman MJ. AB16-6: Sodium channel ß4 subunit mutation causes congenital long QT syndrome. Heart Rhythm. 2006;3:S34. Ref Type: Abstract. [Google Scholar]
- 42.Drouin E, Charpentier F, Gauthier C, Laurent K, Le Marec H. Electrophysiological characteristics of cells spanning the left ventricular wall of human heart: Evidence for the presence of M cells. J Am Coll Cardiol. 1995;26:185–192. doi: 10.1016/0735-1097(95)00167-x. [DOI] [PubMed] [Google Scholar]
- 43.El-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome: Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- 44.Emori T, Antzelevitch C. Cellular basis for complex T waves and arrhythmic activity following combined I(Kr) and I(Ks) block. J Cardiovasc Electrophysiol. 2001;12:1369–1378. doi: 10.1046/j.1540-8167.2001.01369.x. [DOI] [PubMed] [Google Scholar]
- 45.Extramiana F, Antzelevitch C. Amplified Transmural Dispersion of Repolarization as the Basis for Arrhythmogenesis in a Canine Ventricular-Wedge Model of Short-QT Syndrome. Circulation. 2004;110:3661–3666. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- 46.Fedida D, Giles WR. Regional variations in action potentials and transient outward current in myocytes isolated from rabbit left ventricle. J Physiol (Lond ) 1991;442:191–209. doi: 10.1113/jphysiol.1991.sp018789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fenichel RR, Malik M, Antzelevitch C, Sanguinetti MC, Roden DM, Priori SG, Ruskin JN, Lipicky RJ, Cantilena LR. Drug-induced torsade de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475–495. doi: 10.1046/j.1540-8167.2004.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fish JM, Antzelevitch C. Role of Sodium and Calcium Channel Block in Unmasking the Brugada Syndrome. Heart Rhythm. 2004;1:210–217. doi: 10.1016/j.hrthm.2004.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frederiks J, Swenne CA, Kors JA, van Herpen G, Maan AC, Levert JV, Schalij MJ, Bruschke AV. Within-subject electrocardiographic differences at equal heart rates: role of the autonomic nervous system. Pflugers Arch. 2001;441:717–724. doi: 10.1007/s004240000487. [DOI] [PubMed] [Google Scholar]
- 50.Furukawa T, Myerburg RJ, Furukawa N, Bassett AL, Kimura S. Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ Res. 1990;67:1287–1291. doi: 10.1161/01.res.67.5.1287. [DOI] [PubMed] [Google Scholar]
- 51.Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- 52.Gussak I, Brugada P, Brugada J, Antzelevitch C, Osbakken M, Bjerregaard P. ECG phenomenon of idiopathic and paradoxical short QT intervals. Cardiac Electrophysiol Rev. 2002;6:49–53. doi: 10.1023/a:1017931020747. [DOI] [PubMed] [Google Scholar]
- 53.Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102. doi: 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- 54.Hong K, Guerchicoff A, Pollevick GD, Oliva A, Dumaine R, de Zutter M, Burashnikov E, Wu YS, Brugada J, Brugada P, Brugada R. Cryptic 5′ splice site activation in SCN5A associated with Brugada syndrome. J Mol Cell Cardiol. 2005;38:555–560. doi: 10.1016/j.yjmcc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 55.Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, Aihara N, Kamakura S, Kobayashi J, Kosakai Y. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–334. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 56.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 57.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children: A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 58.LeGrice IJ, Smaill BH, Chai LZ, Edgar SG, Gavin JB, Hunter PJ. Laminar structure of the heart 1: cellular organization and connective tissue architecture in ventricular myocardium. Am J Physiol. 1996;269:H571–H582. doi: 10.1152/ajpheart.1995.269.2.H571. [DOI] [PubMed] [Google Scholar]
- 59.Li GR, Feng J, Yue L, Carrier M. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol. 1998;275:H369–H377. doi: 10.1152/ajpheart.1998.275.2.H369. [DOI] [PubMed] [Google Scholar]
- 60.Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res. 1988;62:116–126. doi: 10.1161/01.res.62.1.116. [DOI] [PubMed] [Google Scholar]
- 61.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. Circ Res. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 62.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 63.London B, Sanyal S, Michalec M, Pfahnl AE, Shang LL, Kerchner BS, Lagana S, Aleong RG, Mehdi H, Gutmann R, Weiss R, Dudley SC. AB16-1: A mutation in the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) causes Brugada syndrome. Heart Rhythm. 2006;3:S32. Ref Type: Abstract. [Google Scholar]
- 64.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 65.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 66.McIntosh MA, Cobbe SM, Smith GL. Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure. Cardiovasc Res. 2000;45:397–409. doi: 10.1016/s0008-6363(99)00360-0. [DOI] [PubMed] [Google Scholar]
- 67.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 68.Morita H, Zipes DP, Lopshire J, Morita ST, Wu J. T wave alternans in an in vitro canine tissue model of Brugada syndrome. Am J Physiol Heart Circ Physiol. 2006;291:H421–H428. doi: 10.1152/ajpheart.01259.2005. [DOI] [PubMed] [Google Scholar]
- 69.Morita H, Zipes DP, Morita ST, Wu J. Temperature modulation of ventricular arrhythmogenicity in a canine tissue model of Brugada syndrome. Heart Rhythm. 2007;4:188–197. doi: 10.1016/j.hrthm.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 70.Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer JW, Hall WJ, Weitkamp LR, Vincent GM, Garson A, Robinson JL, Benhorin J, Choi S. The Long QT Syndrome: Prospective Longitudinal Study of 328 Families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 71.Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–177. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- 72.Nam G-B, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Opthof T, Coronel R, Wilms-Schopman FJ, Plotnikov AN, Shlapakova IN, Danilo P, Jr., Rosen MR, Janse MJ. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: T(p-e) interval does not reflect transmural dispersion. Heart Rhythm. 2007;4:341–348. doi: 10.1016/j.hrthm.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 74.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 75.Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol. 2004;286:H2001–H2009. doi: 10.1152/ajpheart.00987.2003. [DOI] [PubMed] [Google Scholar]
- 76.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–e26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 77.Priori SG, Napolitano C, Gasparini M, Pappone C, Della BP, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 78.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 79.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 80.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 81.Rodriguez-Sinovas A, Cinca J, Tapias A, Armadans L, Tresanchez M, Soler-Soler J. Lack of evidence of M-cells in porcine left ventricular myocardium. Cardiovasc Res. 1997;33:307–313. doi: 10.1016/s0008-6363(96)00205-2. [DOI] [PubMed] [Google Scholar]
- 82.Schwartz PJ. The idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109:399–411. doi: 10.1016/0002-8703(85)90626-x. [DOI] [PubMed] [Google Scholar]
- 83.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 84.Shimizu M, Ino H, Okeie K, Yamaguchi M, Nagata M, Hayashi K, Itoh H, Iwaki T, Oe K, Konno T, Mabuchi H. T-peak to T-end interval may be a better predictor of high-risk patients with hypertrophic cardiomyopathy associated with a cardiac troponin I mutation than QT dispersion. Clin Cardiol. 2002;25:335–339. doi: 10.1002/clc.4950250706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 86.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing Torsade de Pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 87.Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: Effects of β-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and Torsade de Pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 88.Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: effects of β-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 89.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under Long QT conditions. Circulation. 1999;99:1499–1507. doi: 10.1161/01.cir.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 90.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 91.Shimizu W, Antzelevitch C. Effects of a K(+) Channel Opener to Reduce Transmural Dispersion of Repolarization and Prevent Torsade de Pointes in LQT1, LQT2, and LQT3 Models of the Long-QT Syndrome. Circulation. 2000;102:706–712. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi A, Aihara N, Takaki H, Sunagawa K, Kamakura S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–1329. doi: 10.1046/j.1540-8167.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- 93.Shimizu W, McMahon B, Antzelevitch C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsade de pointes in models of acquired and congenital long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:156–164. doi: 10.1111/j.1540-8167.1999.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 94.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res. 1991;68:1729–1741. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 95.Sicouri S, Antzelevitch C. Drug-induced afterdepolarizations and triggered activity occur in a discrete subpopulation of ventricular muscle cell (M cells) in the canine heart: Quinidine and Digitalis. J Cardiovasc Electrophysiol. 1993;4:48–58. doi: 10.1111/j.1540-8167.1993.tb01211.x. [DOI] [PubMed] [Google Scholar]
- 96.Sicouri S, Antzelevitch C. Electrophysiologic characteristics of M cells in the canine left ventricular free wall. J Cardiovasc Electrophysiol. 1995;6:591–603. doi: 10.1111/j.1540-8167.1995.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 97.Sicouri S, Fish J, Antzelevitch C. Distribution of M cells in the canine ventricle. J Cardiovasc Electrophysiol. 1994;5:824–837. doi: 10.1111/j.1540-8167.1994.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 98.Sicouri S, Moro S, Litovsky SH, Elizari MV, Antzelevitch C. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. J Cardiovasc Electrophysiol. 1997;8:1269–1279. doi: 10.1111/j.1540-8167.1997.tb01018.x. [DOI] [PubMed] [Google Scholar]
- 99.Sicouri S, Quist M, Antzelevitch C. Evidence for the presence of M cells in the guinea pig ventricle. J Cardiovasc Electrophysiol. 1996;7:503–511. doi: 10.1111/j.1540-8167.1996.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 100.Sipido KR, Volders PG, De Groot SH, Verdonck F, Van de WF, Wellens HJ, Vos MA. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–2144. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 101.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 102.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 103.Stankovicova T, Szilard M, De Scheerder I, Sipido KR. M cells and transmural heterogeneity of action potential configuration in myocytes from the left ventricular wall of the pig heart. Cardiovasc Res. 2000;45:952–960. doi: 10.1016/s0008-6363(99)00418-6. [DOI] [PubMed] [Google Scholar]
- 104.Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–2042. doi: 10.1016/s0735-1097(99)00461-1. [DOI] [PubMed] [Google Scholar]
- 105.Takano M, Noma A. Distribution of the isoprenaline-induced chloride current in rabbit heart. Pflugers Arch. 1992;420:223–226. doi: 10.1007/BF00374995. [DOI] [PubMed] [Google Scholar]
- 106.Takenaka K, Ai T, Shimizu W, Kobori A, Ninomiya T, Otani H, Kubota T, Takaki H, Kamakura S, Horie M. Exercise stress test amplifies genotype-phenotype correlation in the LQT1 and LQT2 forms of the long-QT syndrome. Circulation. 2003;107:838–844. doi: 10.1161/01.cir.0000048142.85076.a2. [DOI] [PubMed] [Google Scholar]
- 107.Tanabe Y, Inagaki M, Kurita T, Nagaya N, Taguchi A, Suyama K, Aihara N, Kamakura S, Sunagawa K, Nakamura K, Ohe T, Towbin JA, Priori SG, Shimizu W. Sympathetic stimulation produces a greater increase in both transmural and spatial dispersion of repolarization in LQT1 than LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol. 2001;37:911–919. doi: 10.1016/s0735-1097(00)01200-6. [DOI] [PubMed] [Google Scholar]
- 108.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 109.Ueda N, Zipes DP, Wu J. Functional and transmural modulation of M cell behavior in canine ventricular wall. Am J Physiol Heart Circ Physiol. 2004;287:H2569–H2575. doi: 10.1152/ajpheart.00526.2004. [DOI] [PubMed] [Google Scholar]
- 110.Ueda N, Zipes DP, Wu J. Prior ischemia enhances arrhythmogenicity in isolated canine ventricular wedge model of long QT 3. Cardiovasc Res. 2004;63:69–76. doi: 10.1016/j.cardiores.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 111.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Opstal JM, Schoenmakers M, Verduyn SC, De Groot SH, Leunissen JD, Der Hulst FF, Molenschot MM, Wellens HJ, Vos MA. Chronic Amiodarone evokes no Torsade de Pointes arrhythmias despite QT lengthening in an animal model of acquired Long-QT Syndrome. Circulation. 2001;104:2722–2727. doi: 10.1161/hc4701.099579. [DOI] [PubMed] [Google Scholar]
- 113.van Opstal JM, Verduyn SC, Leunissen HD, De Groot SH, Wellens HJ, Vos MA. Electrophysiological parameters indicative of sudden cardiac death in the dog with chronic complete AV-block. Cardiovasc Res. 2001;50:354–361. doi: 10.1016/s0008-6363(01)00226-7. [DOI] [PubMed] [Google Scholar]
- 114.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant Caveolin-3 Induces Persistent Late Sodium Current and Is Associated With Long-QT Syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 115.Viswanathan PC, Rudy Y. Cellular arrhythmogenic effects of the congenital and acquired long QT syndrome in the heterogeneous myocardium. Circulation. 2000;101:1192–1198. doi: 10.1161/01.cir.101.10.1192. [DOI] [PubMed] [Google Scholar]
- 116.Volders PG, Sipido KR, Carmeliet E, Spatjens RL, Wellens HJ, Vos MA. Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation. 1999;99:206–210. doi: 10.1161/01.cir.99.2.206. [DOI] [PubMed] [Google Scholar]
- 117.Volders PG, Sipido KR, Vos MA, Spatjens RL, Leunissen JD, Carmeliet E, Wellens HJ. Downregulation of delayed rectifier K(+) currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999;100:2455–2461. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- 118.Vos MA, Jungschleger JG. Transmural repolarization gradients in vivo: the flukes and falls of the endocardium. Cardiovasc Res. 2001;50:423–425. doi: 10.1016/s0008-6363(01)00271-1. [DOI] [PubMed] [Google Scholar]
- 119.Wang HS, Cohen IS. Calcium channel heterogeneity in canine left ventricular myocytes. J Physiol. 2003;547:825–833. doi: 10.1113/jphysiol.2002.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Van Raay TJ, Shen J, Timothy KW, Vincent GM, De Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 121.Wang Q, Shen J, Splawski I, Atkinson DL, Li ZZ, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 122.Watanabe N, Kobayashi Y, Tanno K, Miyoshi F, Asano T, Kawamura M, Mikami Y, Adachi T, Ryu S, Miyata A, Katagiri T. Transmural dispersion of repolarization and ventricular tachyarrhythmias. J Electrocardiol. 2004;37:191–200. doi: 10.1016/j.jelectrocard.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 123.Weirich J, Bernhardt R, Loewen N, Wenzel W, Antoni H. Regional- and species-dependent effects of K+-channel blocking agents on subendocardium and mid-wall slices of human, rabbit, and guinea pig myocardium. Pflugers Archive - European Journal of Physiology. 1996;431:R 130. Ref Type: Abstract. [Google Scholar]
- 124.Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome. A novel gene locus on chromosome 3. Circulation. 2002;105:707–713. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 125.Weissenburger J, Nesterenko VV, Antzelevitch C. Transmural heterogeneity of ventricular repolarization under baseline and long QT conditions in the canine heart in vivo: Torsades de Pointes develops with halothane but not pentobarbital anesthesia. J Cardiovasc Electrophysiol. 2000;11:290–304. doi: 10.1111/j.1540-8167.2000.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 126.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–482. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 127.Wiggers CJ. The mechanism and nature of ventricular fibrillation. Am Heart J. 1940;20:399–412. [Google Scholar]
- 128.Wolk R, Stec S, Kulakowski P. Extrasystolic beats affect transmural electrical dispersion during programmed electrical stimulation. Eur J Clinical Invest. 2001;31:293–301. doi: 10.1046/j.1365-2362.2001.00817.x. [DOI] [PubMed] [Google Scholar]
- 129.Yamada KA, Kanter EM, Green KG, Saffitz JE. Transmural distribution of connexins in rodent hearts. J Cardiovasc Electrophysiol. 2004;15:710–715. doi: 10.1046/j.1540-8167.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 130.Yamaguchi M, Shimizu M, Ino H, Terai H, Uchiyama K, Oe K, Mabuchi T, Konno T, Kaneda T, Mabuchi H. T wave peak-to-end interval and QT dispersion in acquired long QT syndrome: a new index for arrhythmogenicity. Clin Sci (Lond) 2003;105:671–676. doi: 10.1042/CS20030010. [DOI] [PubMed] [Google Scholar]
- 131.Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long QT syndrome. Circulation. 1998;98:1928–1936. doi: 10.1161/01.cir.98.18.1928. [DOI] [PubMed] [Google Scholar]
- 132.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 133.Yan GX, Joshi A, Guo D, Hlaing T, Martin J, Xu X, Kowey PR. Phase 2 Reentry as a Trigger to Initiate Ventricular Fibrillation During Early Acute Myocardial Ischemia. Circulation. 2004;110:1036–1041. doi: 10.1161/01.CIR.0000140258.09964.19. [DOI] [PubMed] [Google Scholar]
- 134.Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially-perfused canine left ventricular wedge preparations. Circulation. 1998;98:1921–1927. doi: 10.1161/01.cir.98.18.1921. [DOI] [PubMed] [Google Scholar]
- 135.Yuan S, Kongstad O, Hertervig E, Holm M, Grins E, Olsson B. Global repolarization sequence of the ventricular endocardium: monophasic action potential mapping in swine and humans. PACE. 2001;24:1479–1488. doi: 10.1046/j.1460-9592.2001.01479.x. [DOI] [PubMed] [Google Scholar]
- 136.Zipes DP. The long QT interval syndrome: A Rosetta stone for sympathetic related ventricular tachyarrhythmias. Circulation. 1991;84:1414–1419. doi: 10.1161/01.cir.84.3.1414. [DOI] [PubMed] [Google Scholar]
- 137.Zygmunt AC. Intracellular calcium activates chloride current in canine ventricular myocytes. Am J Physiol. 1994;267:H1984–H1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 138.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 139.Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]