

Abstract
The bacterial CO-sensing heme protein CooA activates expression of genes whose products perform CO-metabolism by binding its target DNA in response to CO binding. The required conformational change has been proposed to result from CO-induced displacement of the heme and of the adjacent C-helix, which connects the sensory and DNA-binding domains. Support for this proposal comes from UV Resonance Raman (UVRR) spectroscopy, which reveals a more hydrophobic environment for the C-helix residue Trp110 when CO binds. In addition, we find a tyrosine UVRR response, which is attributable to weakening of a Tyr55-Glu83 H-bond that anchors the proximal side of the heme. Both Trp and Tyr responses are augmented in the heme domain when the DNA-binding domain has been removed, apparently reflecting loss of the inter-domain restraint. This augmentation is abolished by a Glu83Gln substitution, which weakens the anchoring H-bond. The CO recombination rate following photolysis of the CO adduct is similar for truncated and full-length protein, though truncation does increase the rate of CO association in the absence of photolysis; together these data indicate that truncation causes a faster dissociation of the endogenous Pro2 ligand. These findings are discussed in the light of structural evidence that the N-terminal tail, once released from the heme, selects the proper orientation of the DNA-binding domain, via docking interactions.
Introduction
Heme protein sensors have emerged as key biological transducers, whose enzymatic or DNA-binding activity is regulated by the binding of diatomic molecules, CO, NO or O2 [1]. There is great interest in elucidating their mechanisms of action. The transcription factor CooA is an exemplar of this class of proteins [2, 3]. When CO binds to its heme, CooA activates a suite of genes responsible for CO oxidation in CO-metabolizing bacteria.
Structural, spectroscopic and functional analysis has provided a detailed picture of CooA from Rhodospirillum rubrum (Rr), for which a mechanism of activation has been proposed [4–7]. The protein is a homodimer, showing cooperativity in CO binding [8]. Each chain contains an N-terminal sensory domain and a C-terminal DNA-binding domain, connected by a long C-helix [9]. The architecture is homologous to that of a cAMP-activated transcription factor, cAMP receptor protein (CRP) [10–12]. Figure 1 compares the structure of CO-free (inactive) CooA with that of cAMP-bound (active) CRP, which suggests the required reorientation of the DNA-binding domains. A remarkable feature of the CooA structure is that each heme is ligated by the N-terminal Pro2 residue of the opposite chain. This endogenous ligand is displaced by CO [13].
Fig. 1.

X-ray crystal structures of Rr-CooA (left, Protein Data Bank (PDB) code 1FT9) and CRP (PDB code 1G6N) showing the difference in orientation of the DNA binding domains, relative to the effector binding domains. In the CooA crystal, the C-helix connecting the effector and DNA binding domains is fully extended in the B chain but is bent over in the A chain (though not as much as in CRP), probably due to crystal forces [9].
Truncation of CooA at the C-helix interdomain connector yields a sensory domain tr-CooA, whose structure is essentially the same as in the full-length protein [14]. However, tr-CooA binds CO faster, and with higher affinity and cooperativity, reflecting a functional connection with the DNA-binding domain in the full-length protein.
Resonance Raman (RR) spectroscopy of key site-directed variants has identified side-chains that interact with the bound CO, and has revealed a characteristic weakening of the bond to the proximal histidine ligand (Fe-His77), an effect that is augmented by DNA binding [6, 7]. This weakening is attributed to displacement of the heme into an adjacent cavity, accompanied by motion of the C-helix. It is this motion that is proposed to permit the reorientation of the DNA-binding domains. A recent crystal structure of the CO-bound form of a variant locked into the active form (from Carboxydothermus hydrogenoformans [Ch]) fully supports this model [15]. In this structure, one of the subunits is without its heme, but the remaining subunit contains CO-bound heme, and displays the heme and C-helix displacement predicted on the basis of the RR analysis (Figure 2).
Fig. 2.

Overlay of CO-bound LL-ChCooA (orange) and CO-free RrCooA (blue), aligned via residues 29–99 in Ch- and the homologous residues 24–94 in Rr-CooA (A chain). (RMS = 1.30 Å for 71 Cα atoms). Shifts in the portions of the hemes, and of the opposite C-helix (carrying the UVRR indicator, Trp110), are indicated by pink arrows. In LL-ChCooA-CO, the Arg138-Glu59 (Arg143-Glu64) saltbridge is broken, the hinge is bent and the D-helix, which is connected to the DNA-binding domain, is reoriented. The heme displacement weakens the His77-Asn42 (His82-Asn47) H-bond. Also shown are the Tyr55-Glu83 (Tyr60-Gln88) contacts. The box is a close-up view of the heme pocket.
Kubo et al. [16] have provided additional support for the heme and C-helix displacement mechanism by showing that UV resonance enhancement of Raman bands associated with the sole tryptophan residue, Trp110, is augmented upon CO binding. This residue is on the C-helix, and its indole side chain is directed toward the heme. The UVRR intensification is attributed to a more hydrophobic environment produced by the heme and C-helix displacement. This interpretation is also supported by the Ch-CooA-CO structure, which shows the homologous C-helix residue, Leu115, displaced toward the heme (Figure 2).
In the present study we confirm the observation of Kubo et al., and show that the effect is further augmented in tr-CooA, suggesting relaxation of constraints imposed by the connection to the DNA-binding domain in the full-length protein. We have also discovered a tyrosine UVRR response to CO binding, which is also augmented in tr-CooA. This signal is attributed to weakening of a strong H-bond between Tyr55 and Glu83, which connects two loop segments in the β-sheet structure proximal to the heme (Figure 2); the homologous H-bond is weakened in the Ch-CooA-CO structure. This assignment was confirmed by a Glu83Gln replacement in tr-CooA, which diminishes the Tyr UVRR response. It also diminishes the Trp110 response, showing that the Tyr55-Glu83 H-bond is critical to the C-helix displacement.
Finally, we measured CO recombination to 5-coordinate CooA and tr-CooA following a photolysis pulse. The rate is hardly affected by truncation, showing that the increased rate of CO association to Pro2-bound protein is due to acceleration of Pro2 dissociation in tr-CooA.
These results are discussed in conjunction with the crystal structures to further develop the mechanism of CooA activation.
Methods
CooA expression and purification
The purification of full-length wild-type (WT) CooA and Leu120Ser variant was performed with our standard method as described previously [17]. The construction and purification of truncated CooA (tr-CooA) has been described previously [14]. The truncated Glu83Gln CooA variant (tr-Glu83Gln) was constructed via PCR amplification of tr-cooA with primers designed to incorporate the desired nucleotide change, as described elsewhere [18]. The tr-Glu83Gln CooA variant was purified using the same procedure for tr-CooA. In all cases, the final protein preparation was >95% pure based on SDS-PAGE. The heme content of CooA preparations was estimated using the extinction coefficient of WT Fe(II)-CO CooA, and protein concentration was measured using the BCA assay (Pierce).
Sample preparation
Purified CooA was diluted into an appropriate buffer (25 mM MOPS/0.5 M NaCl, pH 7.4 for tr-CooA and its E83Q variant; 25 mM MOPS/0.1 M NaCl, pH 7.4 for all others) to give heme concentrations of ~ 15–20 μM for visible RR, ~ 20–30 μM for visible time-resolved resonance Raman (TR3), and ~ 50–90 μM for UVRR experiments. For UVRR experiments, samples also contain ~ 0.1 M NaClO4 as an internal standard. Ferrous and ferrous-CO CooA samples were prepared as previously described [7].
Resonance Raman (RR) spectroscopy
Visible and UV-RR spectra were obtained, respectively, with excitation wavelengths of 406.7 nm from a Kr+ laser (Spectra Physics, 2080-RS), and 229 nm from an intracavity doubled-argon ion laser (Innova 300 FReD, Coherent Radiation, Palo Alto, CA) in a backscattering sample geometry. Photodissociation of the bound CO and sample degradation were minimized by using low laser power (~ 1 and ~ 0.5 mW at the sample for visible and UVRR experiments, respectively) and by spinning the sample. For the visible RR experiments, the scattered light was collected and focused onto a triple spectrograph (Spex 1877) equipped with a CCD detector (Roper Scientific, Model 7375-0001) operating at −110 °C. Spectra were calibrated with dimethyl formamide and dimethylsulfoxide-d6. For the UVRR experiments, a single spectrograph (Spex 1269) equipped with a UV enhanced CCD detector (Princeton Instruments, Model LN/CCD-1340/400) was used to collect the scattered light. UVRR spectra were calibrated using acetone.
For the visible TR3 measurements, the second harmonic of a Q-switched Nd:YLF laser (Photonics Industries International, GM-30–527) was used to pump a Ti:sapphire laser (Photonics International TU-UV), which gave a narrowed laser frequency output (<0.1 cm−1) tunable between 810 and 920 nm. The Ti:S laser output (~ 25 ns at 1 kHz) was frequency doubled using a non-linear lithium triborate crystal to produce a 438-nm probe. The second harmonic output of a Q-switched Nd:YLF (527 nm) was used to produce pump pulses (~ 250 ns at 1 kHz). The optimum pump laser power to achieve maximum photolysis was 600 milliwatts. Photolysis due to the probe laser itself was minimized by keeping the power at 1 milliwatt. A DG535 delay generator (Stanford Research Systems, Inc.) controlled the time delay between the two laser pulses (600 ns to 250 μs). The beams were overlapped and then focused with a pair of cylindrical lenses onto the sample. The sample solution was contained in a sealed spinning NMR tube with a small magnetic stirring bar and was effectively mixed using a second magnet placed outside the tube. The NMR tube was saturated with CO at a concentration of 1 mM. The scattered light was collected and focused onto double spectrograph (Spex 14018) equipped with a liquid nitrogen cooled CCD detector (Roper Scientific, Model 7375-0001). Twelve 10-s acquisition scans were averaged. Spectra were calibrated with dimethyl formamide. The spectra were deconvoluted using the GRAMS/AI version 7.00 software (Thermo Galactic) and analyzed as described previously [8].
Results
UVRR Trp and Tyr signals
Excitation at 229 nm selectively enhances Raman bands arising from sidechain vibrations of tryptophan and tyrosine residues in proteins [19]. The band intensities are responsive to the sidechain environments, especially solvation and H-bonding, which shift the resonant electronic transitions, thereby altering the resonance enhancement factors. The 229 nm-excited UVRR spectra of CooA and its CO adduct are similar to those of other proteins (Figure 3). Kubo et al. [16] reported that Trp band intensities are diminished when CO binds, suggesting increased hydrophobicity of the Trp environment. This result supported the previously proposed heme/C-helix displacement model [4–6], inasmuch as the only Trp residue, Trp110, is on the C-helix, not far from the heme, and the displacements were expected to further protect the Trp110 indole sidechain from solvent. The Ch-CooA-CO structure [15] supports this expectation, since the sidechain of the residue at the homologous position, Leu115, is closer to the hydrophobic CO-binding pocket than is Trp110 in Rr-CooA (Cα···Fe = 9.9 and 11.5 Å, respectively – Figure 2).
Fig. 3.

RrCooA UVRR spectra with and without CO, and the difference spectra (CooA-CO minus CooA) for the indicated variants (5 × amplification). Band frequencies and assignments are labeled.
We confirmed Kubo et al.’s finding (Figure 3). Subtracting the CooA-CO UVRR spectrum from that of CooA leaves residual intensity for all the Trp bands (labeled Wn – see ref. [20] for assignments). Table 1 lists intensity differences of the most prominent bands, W16 and W3, measured via band deconvolution of the parent spectra, and expressed as a fraction of the CO adduct intensity. The estimated uncertainty is 1 %. The augmentation on binding CO differs for different Trp bands, reflecting different responses of the vibrational mode displacements to environmental change.
Table 1.
Percentage UVRR intensity differences induced by CO binding, for selected bands of the indicated Rr-CooA variants.
| Modes | ΔI/ICooA-CO (%)a |
|||
|---|---|---|---|---|
| WT | L120S | E83Q-TR | TR | |
| Tryptophan | ||||
| W16 | 10 | 5 | 8 | 17 |
| W3 | 5 | 4 | 5 | 8 |
| Tyrosine | ||||
| Y9a | 3 | 0 | 6 | 9 |
Measured by subtraction of the CooA from the CooA-CO bands, after deconvolution from the parent spectra. The uncertainty is estimated to be 1 %.
These intensity increments are diminished in the Leu120Ser variant of CooA (Figure 3 and Table 1). This behavior is consistent with independent evidence from heme-resonant RR spectra for lesser heme displacement in Leu120Ser [7] (see below). The Leu120 sidechain forms part of the hydrophobic cavity toward which the heme slides upon CO binding (Figure 2), and substitution by the polar Ser has been suggested to lower the driving force for the heme displacement [7].
However, the increments are augmented in tr-CooA (Figure 3, Table 1), indicating an increase in the extent of C-helix displacement upon truncation. As discussed below, this structural effect correlates with increased CO affinity and binding rate.
We also detect small but reproducible difference bands for WT CooA associated with Tyr vibrations (Figure 3), especially Y9a (see ref [21] for assignments). These did not appear in Kubo et al.’s data, which had somewhat lower signal/noise. The Tyr difference signals are more prominent when the UVRR spectrum of tr-CooA is subtracted from that of CO-free tr-CooA (Figure 3). Rr-CooA has four Tyr residues per monomer, while the tr-CooA has two. However, only Tyr55 is buried from solvent and the environments of the surface Tyr residues are unlikely to be affected by the conformational change associated with CO binding. Tyr55 forms an H-bond to Glu83 (O···O distance = 3.4Å) in the CO-free Rr-CooA [9], and in tr-CooA [14], which should affect the UVRR intensity [22].
We hypothesized that the CO-induced conformational change might weaken this H-bond, inducing an increase in resonance enhancement, as documented for other proteins with H-bonded Tyr residues [22]. The smaller number of surface Tyr residues on tr-CooA accounts in part for the greater relative prominence of the putative Tyr55 difference signature. However, the percentage change in the Y9a intensity is triple that of the full-length protein (Table 1), whereas a doubling would have been expected on the basis of the number of Tyr’s.
The Tyr55 hypothesis is strengthened by the Ch-CooA-CO structure, in which the O···O distance of the homologous H-bond pair, Tyr60···Gln88 (Figure 2), is lengthened to 4.3Å. However, the weaker H-bond might result from the altered conformation, or from Gln being a weaker H-bond acceptor than Glu. To explore the issue further, we replaced Glu83 in tr-CooA with Gln. The difference spectrum for the tr-Glu83Gln variant (Figure 3) shows twice the Y9a percentage intensity as WT Rr-CooA (Table 1), the ratio expected from truncation if the Tyr55···Gln83 H-bond is weakened to the same extent as the Tyr55···Glu83 H-bond in full-length protein.
The Trp110 percentage difference intensity is the same for tr-Glu83Gln as for WT Rr-CooA (Figure 3 and Table 1), indicating the same extent of C-helix displacement. However, as noted above, the increments for tr-CooA itself are higher, and the Tyr 9a difference is higher than in tr-Glu83Gln. This means that the Tyr55···Glu83 H-bond is connected to the C-helix displacement, and that the Glu83Gln substitution abolishes the truncation-induced increase in the C-helix displacement.
νFeC and νCO
Fe-C and C-O stretching RR bands were recorded at 407 nm (Figure 4), in resonance with the heme Soret band. These data have been reported previously for WT CooA-CO, with and without bound DNA, and for the L120S variant [7]. For tr-CooA-CO, the band positions are similar to those of WT protein, while they are both 2 cm−1 lower for the Glu83Gln variant.
Fig. 4.

Heme-resonant FeC and CO stretching RR bands for the indicated RrCooA-CO variants.
The νFeC/νCO data for CooA variants are consistent with a pattern of variable strength of the proximal Fe-His bond [6, 7]. The points on the νFeC/νCO plot (Figure 5) deviate from the standard backbonding correlation for myoglobin variants along a horizontal line, at a position indicating a hydrophobic CO binding pocket. This is the behavior expected if the Fe-His bond is weakened when the His H-bond to a protein acceptor is weakened. The resulting diminution in the His anion character diminishes Fe-CO backbonding, thereby strengthening the C-O bond and weakening the Fe-C bond, but this weakening is compensated by concomitant Fe-C strengthening due to lessened σ competition from the His ligand. Consequently νCO increases with little change in νFeC [23, 24]. The crystal structure of Ch-CooA-CO [15] shows that proximal His H-bonding does indeed weaken upon CO binding, the N···O distance to the Asn acceptor increasing to 4.7 Å from the 2.7 Å seen in the crystal structure of CO-free Rr-CooA [9].
Fig. 5.

νFe-C/νCO back-bonding plot showing data for CO adducts of myoglobin variants (□) [6] and of the CooA variants in this (●) and previous (○, [7]) studies. The CooA variants deviate horizontally from the Mb line, as expected for weakening of the Fe-His bond. The lower and upper inserts show the proximal histidine H-bond arrangements in Mb and CooA, respectively.
However, the extent of Fe-His bond weakening is variable, increasing (higher νCO) when DNA binds to WT CooA, but decreasing for the variants Leu120Ser and Leu120Phe [7]. This behavior was interpreted as reflecting variations in the extent of heme/C-helix displacement. This displacement is augmented by DNA binding, which stabilizes the fully active conformation of CooA, and it is inhibited by the Leu120Ser or Leu120Phe substitutions, near the heme. The νFeC and νCO positions are the same in tr-CooA-CO as in WT CooA-CO, indicating the same extent of heme displacement. In the tr-Glu83Gln variant, however, νCO is shifted down 2 cm−1, consistent with inhibition of the heme displacement. This observation indicates that the Glu83Gln substitution in tr-CooA reduces both the heme and the C-helix displacement.
CO Photolysis and rebinding, monitored via heme RR bands
The rate of rebinding after CO photolysis was measured for WT CooA and tr-CooA. As in our earlier study of WT CooA [8], we used time-resolved heme-resonant RR spectroscopy to separately monitor CO-bound, Pro2-bound and 5-coordinate (5-c) heme (the immediate photolysis product). Part of the motivation of this experiment was to see if rebinding of the endogenous Pro2 ligand to the 5-c heme could be monitored by lengthening the photolysis pulse to 250 ns (from the earlier 25 ns); this was accomplished by utilizing a Q-switched YLF laser, with sufficient power at 527 nm to completely photolyze CooA-CO via excitation in the heme Q absorption bands. Because of efficient geminate recombination, the maximum fraction of 5-c photoproduct at the start of time-resolved measurements was 30 % with a 25 ns photolysis pulse [8]. By lengthening the pulse we were able to increase this yield to 45 %. We also shifted the probe wavelength to 438 nm (from 429 nm) to better enhance the 5-c heme signal (Soret band maximum at ~ 440 nm, vs 425 and 422 nm for CooA and CooA-CO).
These changes improved the signal strength substantially, allowing more precise monitoring of the time course. The time-resolved RR spectra are shown in Figure 6 and the time course of the species population is shown in Figure 7. As before [8], the strong porphyrin ν4 RR band envelope was deconvoluted into contributions from heme-CO, heme-Pro2 and 5-c heme, and the band intensities were converted to populations after correction for differential resonance enhancement (equal for CooA and CooA-CO and 1.5-fold higher for 5-c heme). The time-course is well-described by single exponential curves for loss of 5-c heme and formation of heme-CO, both with 70 μs time constants, and proceeding to 90 % completion by the last time point, 250 μs. Since the solution was saturated with CO (1000 μM), the corresponding recombination rate constant is k’CO = 14 μM−1s−1.
Fig. 6.
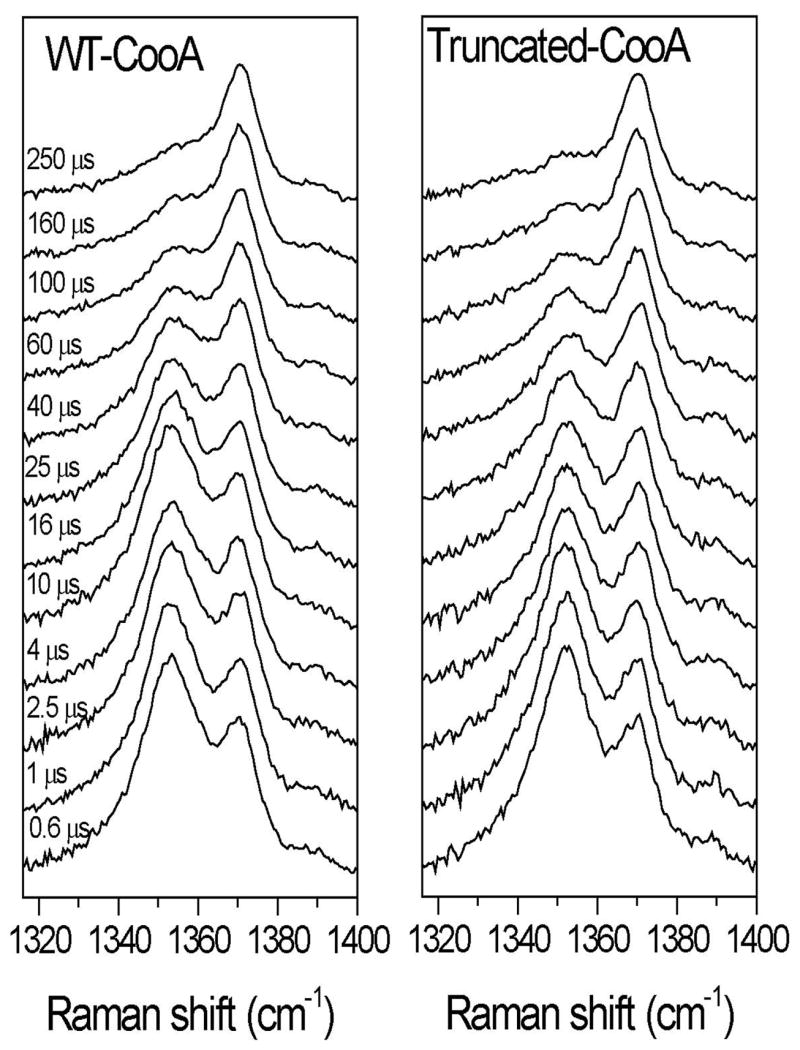
Time-resolved resonance Raman spectra (438-nm probe and 527-nm pump) showing the ν4 bands obtained after photolysis of CooA-CO at the indicated delay times.
Fig. 7.

Changes in the percentages of CooA-CO, CooA, and CooA (5c) as a function of time, based on deconvolution of the Raman ν4 bands. The inset shows the band deconvolution into components (see “Methods”).
The heme-Pro2 population was undetectable over the 250 μs time course. This negative observation is consistent with our previous analysis [8] of pulse-probe absorption data, at longer times and lower CO concentration, which indicated that Pro2 recombination occurs on the millisecond time scale (kPro ~ 1000 s−1).
The previous short-pulse RR (and absorption) data yielded biexponential decays [8], interpreted as two rebinding processes with k’CO = 67-32 and 2 μM−1s−1 (Aono and coworkers [25] reported three exponential phases, with k’CO = 35, 6.8 and 1.2 μM−1s−1.) With the current, higher signal/noise data, we see no evidence for a second, slower process, and find a single rebinding phase with a rate constant intermediate between the previously reported fast and slow phases. However, it is possible that our time-resolution was insufficient to distinguish two phases, since, because of the longer photolysis pulse, our first time point was 600 ns.
The data for tr-CooA-CO (Figures 6 and 7) are very similar, except that the fitted time constant is slightly shorter, 58 μs. However, the difference is within the uncertainty of the measurements (+/− 10 μs for each run). Thus there is no significant difference between rebinding rates for full-length and truncated proteins.
Discussion
Heme and C-helix displacement
On the basis of mutational studies combined with RR spectroscopy we have proposed a heme/C-helix displacement model for the mechanism of CooA activation. In this model, displacement of the endogenous Pro2 ligand by CO induces an upward movement (in the orientation of Figures 1 and 2) of the heme into an adjacent hydrophobic cavity. This movement induces a concerted displacement of the C-helix toward the opposite heme, forming the CO-binding pocket. The C-helix displacement rearranges contacts at the ‘hinge’ between the C and D helices, allowing the DNA-binding domains to swing into the proper orientation for binding. The concerted motions required for both subunits (the initially bound Pro2 ligand also belongs to the opposite chain) accounts for the cooperativity in CO binding that is observed experimentally [8].
The recent crystal structure of a variant of Ch-CooA-CO that is poised in the active form [15] strongly supports this model. Rr- and Ch-CooA are highly similar, and superposition (Figure 2) of the Rr-CooA (blue) and Ch-CooA-CO (orange) structures, reveals exactly the displacements anticipated by the activation model. On the basis of mutational evidence regarding the proximity of Leu116 (Rr-CooA), the upward motion of the heme was predicted [6] to be ~2 Å. The heme displacement seen in Figure 2 (arrow) is 2.05 Å, and the homologous Ch-CooA residue, Leu121, is indeed part of the CO-binding pocket. Displacement of the opposite C-helix toward the heme was posited, in order to bring Rr-CooA residues Gly117 and Ile113 into contact with the bound CO. Such a displacement is evident in Figure 2, and the homologous Ch-CooA residues, Gly122 and Val118 are again part of the binding pocket. As a result of the C-helix displacement Leu120 moves toward the heme, and its replacement by the bulky Phe or the polar Ser would be expected to inhibit the heme displacement, as inferred previously from the νFeC/νCO data [7], and now from the Trp110 UVRR data for Leu120Ser.
The Ch-CooA-CO structure also reveals a weakened H-bond between the proximal His residue and a nearby Asn sidechain, again providing support for our analysis of the spectroscopic data, with respect to νFeC/νCO variation. The horizontal νFeC/νCO deviations from the standard backbonding line for CooA variants (Figure 4) is evidence for weakening of the Fe-His bond, as discussed above. Our hypothesis was that heme displacement weakens this bond, either by mechanical tension, or by weakening the proximal His H-bonding. However, recent Density Functional Theory (DFT) calculations [24] indicate that mechanical tension should produce vertical displacement (selective νFeC increase) from the backbonding line, while H-bond weakening does indeed produce horizontal displacement (selective νCO increase). The structural comparison in Figure 2 confirms this interpretation. The N···O distance is 2.7 Å for the His77-Asn42 contact in CO-free Rr-CooA, but 4.7 Å for the homologous His82-Asn47 contact in Ch-CooA-CO.
(There had been some doubt about the H-bond interpretation, because replacing Asn42 with the non-H-bonding residue Ala, failed to weaken the Fe-His bond, as measured by the νFe-His stretching frequency in a variant with significant 5-coordinate heme population [7]. However, it was recognized that this negative result might be explained by a water molecule entering the proximal region in the Asn42Ala variant, and forming a H-bond with His77, in place of Asn42.)
Truncation of the heme domain
The CO-induced UVRR intensity gain of Trp modes, discovered by Kubo et al. [16], is an independent monitor of C-helix displacement, which brings the Trp110 sidechain into the hydrophobic CO binding pocket (Figure 2). It is therefore of considerable interest that the extent of the Trp intensity change is higher for tr-CooA than it is for the full-length protein (Table 1), even though νFeC and νCO are the same. We infer that the CO-induced heme displacement is unaltered when CooA is truncated, but that the C-helix displacement is increased. Evidently, C-helix displacement is restrained in the full-length protein, and this restraint is released upon truncation. Truncation occurs at residue 131, just before the ‘hinge’ for bending between the C and D helices [14]. Thus restraint in the full-length protein is likely associated with this bending motion, which is required for proper alignment of the DNA-binding domain. As pointed out previously [14], the energy required by this conformational change is evident in the diminished CO affinity (2-fold) and cooperativity (n ~ 1.5 vs ~2) of the full-length, relative to the truncated protein. The augmented C-helix displacement after truncation can be seen as the mechanical manifestation of the energy release.
Truncation also increases the rate of CO association with CooA (10-fold), as measured in stopped-flow experiments [14]. In the current work, we find essentially no change in the rate of CO recombination after photolysis of CooA-CO. Thus the CO binding pocket appears to be unaffected by the truncation. This result supports the inference [14] that acceleration of CO association results from faster dissociation of the endogenous Pro2 ligand in the truncated heme domain. The Rr-CooA crystal structure [9] reveals that the ligating N-terminus, Pro2, is in non-bonded contacts with C-helix residues Ile113, Leu116 and Gly117 (which are also part of the CO binding pocket, as noted above.) The loss of C-helix constraints upon truncation may loosen these contacts, facilitating Pro2 dissociation.
C-helix displacement and domain reorientation
A number of contacts between different regions of CooA have been identified that may mediate domain reorientation [9]. One of these is a salt-bridge between the hinge-region residue Arg138 and Glu59, located at the tip of the β4/5 hairpin loop, which extends from the heme domain (Figure 2). One strand of this loop extends to the heme ligand, His77, providing a mechanical connection between Glu59 and the heme. The displacement of the heme in the Ch-CooA-CO structure is accompanied by movement of the β4/5 hairpin away from the C-helix (Figure 2), and the homologous Arg143-Glu64 salt-bridge is broken, allowing the hinge to bend. This salt-bridges would contribute to the energy penalty for the CO-induced conformation change, and is eliminated in tr-CooA. Highlighting the importance of the β4/5 hairpin connection is mutational evidence linking the C-helix residue 128 and β4/5 hairpin residue 61 in constitutively active variants of the homologous CRP protein [26].
The present results suggest that another contributing contact may be the Tyr55-Glu83 H-bond (Figure 2), whose weakening accounts for the CO-induced intensity loss in Tyr UVRR bands, in both full-length and truncated CooA. Replacement of the H-bond acceptor, Glu83 with Gln has a dramatic effect on the spectral characteristics of tr-CooA. The augmented C-helix displacement is abolished, as seen by the return of the Trp110 UVRR difference intensity to that of WT-CooA. But in addition, the extent of heme displacement is reduced (lower νCO) to about the same extent as in the Leu120Ser variant of the full-length protein. We interpret this to mean that the CO-induced displacement of the heme and of the C-helix is diminished when the Tyr55···Glu83 H-bond is weakened by the Gln substitution. Tyr55 is on the second strand of the β4/5 hairpin, and the H-bond to Glu83 connects it to another β loop, downstream from the His77 ligand (Figure 2). Thus weakening this H-bond may attenuate the connection between the heme and β4/5 hairpin, and diminish the displacement when CO binds. Although weakened, the substitute Tyr55···Gln83 H-bond is nevertheless further weakened as a result of this partial displacement, as evident from the Y8a UVRR enhancement, which is the same as in full-length WT CooA.
We speculate that the relaxed ligand specificity of Ch-CooA compared to that of Rr-CooA, e.g. the ability of the former but not the latter to bind imidazole [27], may result from substitution of Gln for Glu at the position 88 (homologous to position 83 in Rr-CooA) weakening the proximal constraint to heme motion. It is interesting that in the crystal structure of imidazole-bound Ch-CooA, recently reported by Komori et al. [27], the heme is found to be rotated, rather than displaced, unlike that in Ch-CooA-CO.
Role of the N-terminus in activation
Bending at the hinge region between helices C and D is a necessary but not sufficient condition for the proper alignment of the DNA-binding domains in CooA. Thus the A subunit in the CO-free Rr-CooA structure has a bent hinge region (Figure 1), in contrast to the B subunit. But the DNA-binding domain of neither subunit is oriented properly for binding to DNA (Figure 1). The hinge region bending in the A subunit was attributed to the influence of crystal contacts [9]. Likewise the hinge region is bent, but the DNA-binding domains are improperly aligned in the crystal structure of Ch-CooA-Im [27]. Thus it is apparent that, once the hinge bends, a variety of conformations are available to the DNA-binding domain.
Borjigin et al. [15] have proposed that the N-terminus might play a role in activation. In the adduct-free protein, the N-terminus binds the heme of the opposite chain, but displacement by CO was shown by RR spectroscopy to expel the N-terminus from the region of the binding pocket [6]. In Ch-CooA-CO, the N-terminal tail is sandwiched between the DNA-binding domain and the heme domain, held in place by several tertiary contacts [15]. These contacts are proposed to lock the DNA-binding domain in the active conformation. Such a role is supported by the suggestion of Komori et al. [27] that in the imidazole adduct of Ch-CooA, an H-bond from the backbone carbonyl of Met5, near the N-terminus, to the Nε atom of the bound imidazole, restrains the conformation change required for activity. This H-bond would in fact prevent docking of the N-terminal tail between the heme and DNA-binding domains.
The picture that emerges from these considerations is that CO binding to CooA releases the bound N-terminus, and induces concerted heme and C-helix displacements. These are impelled by desolvation forces that drive the heme further into the protein interior, providing a hydrophobic binding pocket for CO. The displacements release constraints at the β4/5 hairpin allowing the hinge to bend. However, the protein is then in a ‘flexible’ state, with a range of orientations available to the N-terminal tail and to the DNA-binding domain. It has been reported that electron density maps on crystals of CooA variants, often give weak density for the DNA-binding domain, reflecting disorder [15], and a small angle X-ray scattering study has indicated surprisingly little shape variation between CooA and CooA-CO [28]. However, docking of the N-terminal tail between the heme- and DNA-binding domains can lock the latter in the proper orientation for DNA-binding.
It is consistent with this ‘flexible’ view of CooA that DNA binding to CooA-CO augments the heme/C-helix displacement, as revealed by the νFeC/νCO data [6], and by an increase in the Trp110 UVRR intensity change [16]. In the absence of DNA, CooA-CO appears to be in a conformational equilibrium between active and inactive forms. The new crystallographic data suggests that CO binding frees the N-terminal tail, which induces activity by docking between the domains in the active form. There appears to be a dynamic equilibrium between docked and undocked molecules, which is drawn toward the docked conformation by DNA binding.
Supporting this view is the finding that substitutions which leave Rr-CooA constitutively active (without CO binding), also induce maximal heme/C-helix displacement in the CO adduct, with or without bound DNA [7]. These substitutions tilt the energetics toward the fully active form. They involve replacement of a C-helix segment just before the hinge, residues 121–126 (TSCMRT), with A(or R)YLLRL. As discussed previously [4, 7], these substitutions improve the “leucine zipper” contact between the two C-helices; in particular the two Cys123 residues are at the critical ‘d’ position of the heptad repeat, and are replaced by Leu. In addition both Cys123 and Met124 form part of the hydrophobic cavity into which the heme is proposed to slide [5], and their replacement by Leu increases the computed cavity size and makes it more hydrophobic [7]. The new Ch-CooA-CO structure, which is also of a variant with substitutions on the C-helix, suggests that these substitutions also improve the docking of the N-terminus in the active form. Borjigin et al. [15] noted a close contact between Leu7, near the N-terminus, and an introduced Leu127 residue (Asn127 in the wild-type protein – the introduction of Leu at this position and at the adjacent position render Ch-CooA constitutively active [15]). The residue at the homologous position to Leu7 in Ch-CooA is the N-terminal Pro2 of Rr-CooA, while the residue at the homologous position to Leu127 of Rr-CooA, Thr122, is replaced by Tyr in A/RYLLRL of Rr-CooA. When modeled into the Ch-CooA-CO structure (Figure 8), the Pro2 carbonyl and Tyr122 side-chain can be brought into proximity (2.15 Å O···O distance). The resulting H-bond could favor docking of the N-terminus, at the expense of heme binding; the A/RYLLRL variants are known to have significant populations of 5-coordinate heme. The wild-type residue at position 122 is Ser, which is a weaker H-bond donor than Tyr. It may nevertheless be strong enough to favor the docked conformation, once the N-terminus is released by CO binding.
Fig. 8.
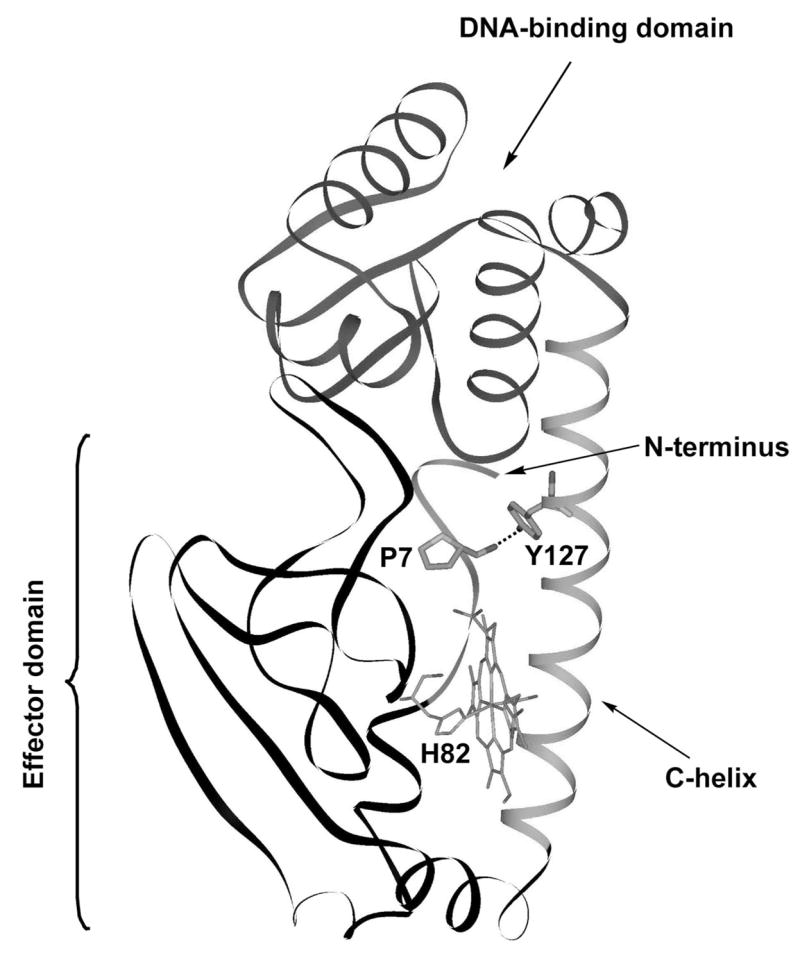
View of LL-ChCooA-CO (chain A) [15], with Pro and Tyr modeled as substitutions for Leu7 and Leu127, showing the proposed H-bond docking of the N-terminal tail in the active conformation of (A/R)YLLRL variants of RrCooA, in which Pro2 and Tyr122 are the position 7 and 127 homologs.
However, other contacts are certainly involved [15] and the docking interactions are flexible enough to allow for the deletion of two residues near the Rr-CooA N-terminus, or the addition of an extra one, with retention of some, albeit reduced, activity [29].
In summary, the CooA activation mechanism appears to involve release of the heme-bound N-terminus and its subsequent docking between the heme and the DNA-binding domains (see Figure 5 scheme in ref [15]), orienting the latter for DNA-binding. In between these steps, a wide range of domain orientations are available to the protein. This flexibility is facilitated by breaking of contacts between the β4/5 loop on the heme domain, and the ‘hinge’ region between the C and D helices. These contacts break when the heme and the C-helices are displaced as a result of hydrophobic forces, after the N-terminus dissociates from the heme. Dissociation of the N-terminus accompanies CO binding, but can also be induced by residue substitutions that promote heme/C-helix displacement and/or docking of the N-terminus.
Conclusions
CO promotes CooA activation by displacing the N-terminal ligand and inducing a concerted heme and C-helix displacement. The heme displacement is monitored by the νFeC/νCO frequencies, which reflect weakening of the bond to the proximal His ligand, as a result of tension on a His-Asn H-bond when the heme is displaced. It also results in weakening of a Tyr55-Glu83 H-bond, on the proximal side of the heme, which can be detected by changes in the tyrosine UVRR intensity. The C-helix displacement is monitored by the UVRR intensity of the Trp110 residue, which is drawn toward the hydrophobic CO-binding pocket.
The C-helix displacement breaks restraining contacts between the C-helix and the heme domain β4/5 loop, allowing the hinge to bend. Loss of these restraints upon truncation of the heme domain results in a 10-fold increase in the rate of Pro2 dissociation from the heme, reflecting loosened non-bonded contacts with surrounding C-helix residues. In addition, the CO affinity is doubled and the C-helix displacement increases.
Bending the hinge region between the C- and D-helices in the full-length protein allows a range of conformations for the DNA-binding domain. The conformation that allows DNA to bind can be stabilized by docking of the N-terminal tail between the DNA-binding and heme domains. By displacing the heme and C-helix, CO binding leaves CooA in an equilibrium between ‘flexible’ and ‘docked’ conformations. The equilibrium is shifted toward ‘docked’ by DNA binding itself, and also by substitutions that stabilize the docking and displacement contacts.
Acknowledgments
This work was supported by NIH grant GM 33576 (to TGS), NIH grant GM53228 (to GPR) and GM42614 (to TLP). We thank Mary Conrad for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gilles-Gonzalez MA, Gonzalez G. J Inorg Biochem. 2005;99:1–22. doi: 10.1016/j.jinorgbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Roberts GP, Kerby RL, Youn H, Conrad M. J Inorg Biochem. 2005;99:280–292. doi: 10.1016/j.jinorgbio.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 3.Aono S. Acc Chem Res. 2003;36:825–831. doi: 10.1021/ar020097p. [DOI] [PubMed] [Google Scholar]
- 4.Kerby RL, Youn H, Thorsteinsson MV, Roberts GP. J Mol Biol. 2003;325:809–823. doi: 10.1016/s0022-2836(02)01203-2. [DOI] [PubMed] [Google Scholar]
- 5.Youn H, Kerby RL, Roberts GP. J Biol Chem. 2003;278:2333–2340. doi: 10.1074/jbc.M210825200. [DOI] [PubMed] [Google Scholar]
- 6.Coyle CM, Puranik M, Youn H, Nielsen SB, Williams RD, Kerby RL, Roberts GP, Spiro TG. J Biol Chem. 2003;278:35384–35393. doi: 10.1074/jbc.M301000200. [DOI] [PubMed] [Google Scholar]
- 7.Ibrahim M, Kerby RL, Puranik M, Wasbotten IH, Youn H, Roberts GP, Spiro TG. J Biol Chem. 2006;281:29165–29173. doi: 10.1074/jbc.M605568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puranik M, Nielsen SB, Youn H, Hvitved AN, Bourassa JL, Case MA, Tengroth C, Balakrishnan G, Thorsteinsson MV, Groves JT, McLendon GL, Roberts GP, Olson JS, Spiro TG. J Biol Chem. 2004;279:21096–21108. doi: 10.1074/jbc.M400613200. [DOI] [PubMed] [Google Scholar]
- 9.Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Nat Struct Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- 10.Weber IT, Steitz TA. J Mol Biol. 1987;198:311–326. doi: 10.1016/0022-2836(87)90315-9. [DOI] [PubMed] [Google Scholar]
- 11.Passner JM, Schultz SC, Steitz TA. J Mol Biol. 2000;304:847–859. doi: 10.1006/jmbi.2000.4231. [DOI] [PubMed] [Google Scholar]
- 12.Parkinson G, Wilson C, Gunasekera A, Ebright YW, Ebright RE, Berman HM. J Mol Biol. 1996;260:395–408. doi: 10.1006/jmbi.1996.0409. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto K, Ishikawa H, Takahashi S, Ishimori K, Morishima I, Nakajima H, Aono S. J Biol Chem. 2001;276:11473–11476. doi: 10.1074/jbc.C100047200. [DOI] [PubMed] [Google Scholar]
- 14.Kuchinskas M, Li HY, Conrad M, Roberts G, Poulos TL. Biochemistry. 2006;45:7148–7153. doi: 10.1021/bi052609o. [DOI] [PubMed] [Google Scholar]
- 15.Borjigin M, Li HY, Lanz ND, Kerby RL, Roberts GP, Poulos TL. Acta Crystallogr, Sect D: Biol Crystallogr. 2007;63:282–287. doi: 10.1107/S0907444906051638. [DOI] [PubMed] [Google Scholar]
- 16.Kubo M, Inagaki S, Yoshioka S, Uchida T, Mizutani Y, Aono S, Kitagawa T. J Biol Chem. 2006;281:11271–11278. doi: 10.1074/jbc.M513261200. [DOI] [PubMed] [Google Scholar]
- 17.Shelver D, Thorsteinsson MV, Kerby RL, Chung SY, Roberts GP, Reynolds MF, Parks RB, Burstyn JN. Biochemistry. 1999;38:2669–2678. doi: 10.1021/bi982658j. [DOI] [PubMed] [Google Scholar]
- 18.Chiang LW, Kovari I, Howe MM. PCR methods and applications. 1993;2:210–217. doi: 10.1101/gr.2.3.210. [DOI] [PubMed] [Google Scholar]
- 19.Austin JC, Rodgers KR, Spiro TG. Metallobiochemistry, Part C. In: Riordan JF, Vallee BL, editors. Methods in Enzymology. Vol. 226. Academic Press Inc; San Diego: 1993. pp. 374–396. [Google Scholar]
- 20.Takeuchi H, Harada I. Spectrochim Acta, Part A. 1986;42:1069–1078. [Google Scholar]
- 21.Takeuchi H, Watanabe N, Harada I. Spectrochim Acta, Part A. 1988;44:749–761. [Google Scholar]
- 22.Hildebrandt PG, Copeland RA, Spiro TG, Otlewski J, Laskowski M, Prendergast FG. Biochemistry. 1988;27:5426–5433. doi: 10.1021/bi00415a007. [DOI] [PubMed] [Google Scholar]
- 23.Franzen S. J Am Chem Soc. 2002;124:13271–13281. doi: 10.1021/ja017708d. [DOI] [PubMed] [Google Scholar]
- 24.Xu C, Spiro TG. J Am Chem Soc. 2007 Submitted. [Google Scholar]
- 25.Uchida T, Ishikawa H, Ishimori K, Morishima I, Nakajima H, Aono S, Mizutani Y, Kitagawa T. Biochemistry. 2000;39:12747–12752. doi: 10.1021/bi0011476. [DOI] [PubMed] [Google Scholar]
- 26.Youn H, Kerby RL, Conrad M, Roberts GP. J Biol Chem. 2006;281:1119–1127. doi: 10.1074/jbc.M509421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komori H, Inagaki S, Yoshioka S, Aono S, Higuchi Y. J Mol Biol. 2007;367:864–871. doi: 10.1016/j.jmb.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama S, Fujisawa T, Ishimori K, Morishima I, Aon S. J Mol Biol. 2004;341:651–668. doi: 10.1016/j.jmb.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 29.Clark RW, Youn H, Parks RB, Cherney MM, Roberts GP, Burstyn JN. Biochemistry. 2004;43:14149–14160. doi: 10.1021/bi0487948. [DOI] [PubMed] [Google Scholar]