

Abstract
Endothelial cells play a vital role in the maintenance of cardiovascular homeostasis. Epoxyeicosatrienoic acids (EETs), cytochrome P450 (CYP) epoxygenase metabolites of arachidonic acid in endothelial cells, possess potent and diverse biological effects within the vasculature. We evaluated the effects of overexpression of CYP epoxygenases on tumor necrosis factor (TNF)-α induced apoptosis in bovine aortic endothelial cells (BAECs). CYP epoxygenase overexpression significantly increased endothelial cell viability and inhibited TNF-α induction of endothelial cell apoptosis as evaluated by DNA laddering and FACS analysis. CYP epoxygenase overexpression also significantly inhibited caspase-3 activity and downregulation of Bcl-2 expression induced by TNF-α. The anti-apoptotic effects of CYP epoxygenase overexpression were significantly attenuated by inhibition of the PI3K/Akt and MAPK signaling pathways; however, inhibition of endothelial nitric oxide synthase activity had no effect. Furthermore, CYP epoxygenase overexpression significantly attenuated the extent of TNF-α induced ERK1/2 dephosphorylation in a time-dependent manner, and significantly increased PI3K expression and Akt phosphorylation in both the presence and absence of TNF-α. Collectively, these results suggest that CYP epoxygenase overexpression, which is known to increase EET biosynthesis, significantly protects endothelial cells from apoptosis induced by TNF-α. This effect is mediated, at least in part, through inhibition of ERK dephosphorylation and activation of PI3K/Akt signaling.
Keywords: apoptosis, endothelial cells, cytochrome P450, epoxygenase, epoxyeicosatrienoic acid, arachidonic acid
Introduction
Endothelial cells play a vital role in the maintenance of cardiovascular homeostasis by synthesizing and releasing endogenous vasodilators such as nitric oxide (NO) and prostacyclin (PGI2). In addition, these substances exhibit potent anti-inflammatory, anti-thrombotic and anti-apoptotic effects within the vasculature (1-3). Epoxyeicosatrienoic acids (EETs) are cytochrome P-450 (CYP) epoxygenase-derived metabolites of arachidonic acid that also possess potent vasodilatory properties. These eicosanoids are considered to be the leading candidates for endothelium-derived hyperpolarizing factor (EDHF) (4-6) because they hyperpolarize vascular smooth muscle cells by activating Ca2+-sensitive K+ channels (7, 8).
CYP epoxygenases of the CYP2C and CYP2J subfamilies actively metabolize arachidonic acid to various EET regio- and stereoisomers (9). In addition to EDHF-like properties, CYP epoxygenase-derived EETs have been shown in multiple in vitro and in vivo studies to possess other potent biological effects in the renal and cardiovascular systems. For example, EETs induce mitogenesis of renal epithelial cells (10,11) inhibit cytokine-induced vascular cell adhesion molecule expression and leukocyte adhesion to the vascular wall (12), inhibit vascular smooth muscle cell migration (13) and increase tissue plasminogen activator expression (14). More recently, our group has demonstrated that transfection of endothelial cells with various CYP epoxygenases increases EET biosynthesis and significantly up-regulates expression and activity of endothelial nitric oxide synthase (eNOS) (15).
The balance between endothelial cell survival and endothelial cell death is critical in various processes including vascular inflammation and remodeling. Indeed, endothelial cell apoptosis is hypothesized to be a key initiating event in the development of atherosclerosis and other vascular diseases (16-18). Recently, Dhanasekaran et al (19) reported that exogenous EETs increased human endothelial cell survival and attenuated apoptosis. However, the exact role CYP epoxygenase-derived EETs play in preventing endothelial cell apoptosis remains elusive. In the current study, we utilized cultured bovine aortic endothelial cells (BAECs) transfected with CYP epoxygenases CYP102 F87V, CYP2C11-CYPOR, and CYP2J2 to investigate the role of endogenously formed EETs on tumor necrosis factor (TNF)-α induced apoptosis. We demonstrate that these CYP epoxygenases markedly attenuate apoptosis and prevent both TNF-α activation of caspase-3 and TNF-α reduction of Bcl-2 expression. In addition, our data suggest that this anti-apoptotic effect is mediated, at least in part, through activation of the MAPK and PI3K/Akt signaling pathways.
MATERIALS AND METHODS
An expanded Materials and Methods section is available as supplement.
Cell Culture
BAECs were isolated from bovine aortas obtained from a local slaughterhouse by digestion with 0.25% trypsin and cultured in DMEM supplemented with 5 mM L-glutamine, 10% FBS and an antibiotic mixture containing penicillin (100 units/ml) and streptomycin (100 μg/ml). Cell viability was assessed using an MTT assay as described (20,21).
Recombinant Adeno-Associated Virus and Gene Transfection
Plasmids encoding CYP102 F87V, CYP2C11-CYPOR and CYP2J2 were kindly provided by Drs. Jorge Capdevila (Vanderbilt University) and Darryl Zeldin (NIEHS). The CYP102 F87V is a mutant P450 from Bacillus megaterium (P450BM3) in which phenylalanine 87 was replaced with valine, converting it to a highly regio- and stereoselective epoxygenase that biosynthesizes 14(S),15(R)-EET from arachidonic acid (22). CYP2C11-CYPOR is an active rat P450 epoxygenase fused with rat NADPH-cytochrome P450 oxidoreductase which synthesizes a regioisomeric mixture of 5−6-, 8,9-, 11,12- and 14,15-EETs (23). CYP2J2 is a human P450 that biosynthesizes primarily 8,9-, 11,12- and 14,15-EETs in human vascular endothelial cells (12). The recombinant adeno-associated virus (rAAV) vector pXXUF1, packaging plasmid pXX2, adenovirus helper plasmid pXX6, and a rAAV plasmid containing the GFP cDNA (GFP-pUF1) were generous gifts from Dr. Xiao Xiao (University of Pittsburgh). For rAAV packaging in vitro, epoxygenase cDNAs were subcloned into pXXUF1 and the rAAVs were produced as previously described (15, 24, 25). BAECs were infected with rAAV-CYP102 F87V, rAAV-CYP2C11-CYPOR, rAAV-CYP2J2 or rAAV-GFP (∼50 virions/cell) and cultured for one week to obtain maximal expression. The percent of cells infected by rAAV-GFP was over 50% according to routine microscopic observation.. Abundant P450 expression and increased EET biosynthesis after infection have been confirmed in our previous studies (9, 15). In some experiments, epoxygenase cDNAs were subcloned into the mammalian expression vector pCB6 to produce CYP102 F87V-pCB6, CYP2C11-CYPOR-pCB6 and CYP2J2-pCB6 (15). BAECs were then transfected using Superfect Transfection Reagent (Qiagen, Hilden, Germany) and cultured for 24h to obtain maximal expression. Cell viability was assessed 48h and one week following transfection.
14,15-DHET detection by ELISA
In order to reflect activity of metabolizing arachidonic acid into EETs, concentrations of the stable EET metabolite 14,15-dihydroxyeicosatrienoic acid (DHET) in cultured endothelial cells were determined by an ELISA kit (Detroit R&D). Briefly, the cells cultured in 100 mm Petri dishes were washed with chilled phosphate buffered saline (PBS) two times and then scraped using 1.0 ml of cold PBS containing triphenylphosphine. The samples were sonicated on ice and the protein quantity was determined. Eicosanoids were then extracted from the cell samples three times with ethyl acetate after acidification with acetic acid (to convert EETs into DHETs). After evaporation, saponification with 0.4N KOH in methanol and re-extraction, 14,15-DHET was dissolved in 30ul DMF and quantified by ELISA according to the manufacturer's instructions as previously described (26).
Treatments of BAECs
Apoptosis was induced by incubating transfected BAECs with TNF-α (5 ηg/ml) and actinomycin D (0.01 μg/μl, to inhibit RNA synthesis) for 24h. Inhibitor experiments involved 30 minutes of pre-treatment with the Akt inhibitor 1L-6-Hydroxymethyl-chiro-inositol-2-(R)-2-O-methyl-3-O-octadecylcarbonate (Akt I, 2 μM); the PI3K inhibitor LY294002 (20 μM), the MAPK inhibitor apigenin (20 μM) or the MEK inhibitor PD98059 (20 μM) (27). In some experiments, cells were also treated with the P450 inhibitor 17-ODYA (100 ηM) or with synthetic, HPLC-purified 14,15-EET (100 ηM) as described (28).
Assessment of Apoptosis
Apoptotic responses were assessed by three independent methods following treatment of BAECs with TNF-α plus actinomycin D. First, DNA fragmentation was assessed by gel electrophoresis as previously described (29). Second, cells were resuspended and stained with fluorescein isothiocyanate-conjugated annexin V and fluorescent dye propidium iodide (PI) and analyzed by flow cytometry (FACS, Vantage, BD, USA). The relative increase in apoptotic cells was calculated as a percentage. These percentages were calculated as the ratio of apoptotic cells in rAAV-CYP102 F87V versus rAAV-GFP infected cells for each signaling molecule inhibitor, and then normalized to the corresponding ratio in cells treated with no inhibitor. Third, caspase-3 activity was measured with a colorimetric assay kit using DEVD-p-nitroanilide as a substrate (30). The relative increase percentage of capase-3 activity was also calculated, as completed for the apoptotic cell calculation above.
Immunoblot Analyses
Lysates of cell and tissue were subjected to western blotting as described in the on-line data supplement. Expression was quantified by densitometry and normalized to β-actin expression, and then all groups were normalized to their respective vehicle or empty vector controls.
RESULTS
Cell Viability Assays
Viability of BAECs following transfection with CYP102 F87V, CYP2C11-CYPOR or CYP2J2 containing vectors was significantly greater than following transfection with empty pCB6 vector or a GFP-containing vector at the 48 hour (Fig. 1A) and rAAV-mediated transfections of CYP102 F87V, CYP2C11-CYPOR or CYP2J2 had very similar effect (data not shown). Treatment with synthetic 14,15-EET also significantly increased cell viability, and the CYP inhibitor 17-ODYA abolished the protective effects of CYP102 F87V transfection (Fig. 1B). The expression levels of transfected target genes in our cell system were previously reported (31). The 14,15-DHET levels in untransfected and GFP transfected cells were 1225±609 and 923±309 pg/mg cell protein, respectively. Significantly higher 14,15-DHET levels were observed in the CYP102 F87V (7164±1382 pg/mg cell protein), CYP2C11-CYPOR (6018±3154 pg/mg cell protein) and CYP2J2 (4886±1759 pg/mg cell protein) transfected cells (N=6, p<0.05). Importantly, these data indicate that epoxygenase gene overexpression markedly increased EET production in endothelial cells.
Figure 1.


Effect of CYP epoxygenase overexpression on BAEC viability by MTT assay. A, Cell viability quantified 48 hours after transfection with CYP2J2, CYP102 F87V or CYP2C11-CYPOR. Blank represents BAECs without any treatment. Control represents BAECs following transfection with the empty pCB6 vector. GFP represents BAECs following transfection with the pCB6 vector containing the GFP cDNA. Values represent mean±SEM of optical density obtained from the MTT assay. Each group was completed in triplicate. *p<0.05 and **p<0.01 compared to control. B, Cell viability quantified 48 hours after transfection with or without the CYP inhibitor 17-ODYA (100nM) or synthetic 14,15-EET (100nM). *p<0.05 and **p<0.01 compared to reference group as indicated.
CYP Epoxygenase Overexpression Attenuates TNF-α Induced Apoptosis
BAECs infected with rAAV-CYP2J2, rAAV-CYP102 F87V or rAAV-CYP2C11-CYPOR underwent significantly less apoptosis 24 hours after TNF-α treatment compared to uninfected BAECs. First, DNA fragmentation analysis revealed that CYP epoxygenase transfection reduced TNF-α induced DNA degradation in BAECs (Fig. 2). Second, flow cytometry revealed significant attenuation in the percentage of apoptotic cells (Annexin V positive, PI negative) after TNF-α treatment following transfection with CYP2J2, CYP101 F87V or CYP2C11 epoxygenases compared to control (17.0±1.1%, 8.3±1.6% and 12.0±1.9%, respectively, versus 31.4±2.0%, p<0.05) (Fig. 3A and 3B).
Figure 2.

Effect of CYP epoxygenase overexpression on TNF-α induced DNA fragmentation. Cells were infected with rAAV-GFP, rAAV-CYP2C11-CYPOR, rAAV-CYP102 F87V, or rAAV-CYP2J2, and one week later were incubated with TNF-α for 24 hours in the presence of Actinomycin D. DNA (20 μg/lane) extracted from the BAECs was electrophoresed on a 2% agarose gel to detect DNA ladder formation. This is representative photo of 6 repeats and control refers to BAECs without infection or with rAAV-GFP infection.
Figure 3.


Effect of CYP epoxygenase overexpression on apoptosis induced by TNF-α as assessed by flow cytometry. A, Representative results of three independent experiments evaluating TNF-α induced apoptosis by annexin-V-FITC/propidium iodide (PI) staining and flow cytometry. Cells were infected with rAAV-CYP2J2, rAAV-CYP102 F87V, or rAAV-CYP2C11-CYPOR, and one week later were incubated with TNF-α for 24 hours in the presence of Actinomycin D. Control refers to BAECs infected with rAAV-GFP. Cells with negative staining of both PI and annexin V are living. Cells with PI-negative and annexin V-positive staining are early apoptotic cells. Cells with PI-positive and annexin V-positive staining are primarily in a late stage of apoptosis. B, showing percentage of apoptotic cells from flow cytometry analysis above and the values are mean±SEM from five independent experiments; **p<0.01 compared to control. C, Representative results of three independent experiments evaluating the effects of signaling molecule inhibitors on TNF-α induced apoptosis in CYP epoxygenase infected BAECs. Cells infected with either rAAV-GFP or rAAV-CYP102 F87V one week prior were incubated with inhibitors of Akt (Akt I), PI3K (LY294002), MAPK (apigenin), MEK (PD98059), or no inhibitor for 30 minutes prior to TNF-α treatment. D, Percentage of apoptotic cells in panel C. Values shown are mean±SEM from five independent experiments. *p<0.05, **p<0.01 compared to rAAV-GFP cells without inhibitor treatment; #p<0.05, ##p<0.01 compared to rAAV-CYP102 F87V cells without inhibitor treatment.
In order to identify the signaling pathways responsible for the observed anti-apoptotic effects of CYP epoxygenase overexpression, we also characterized TNF-α induced apoptosis in BAECs pretreated with various cell signaling pathway inhibitors. Pretreatment of rAAV-GFP infected BAECs with Akt I, LY294002, apigenin, and PD98059 significantly enhanced TNF-α-induced apoptosis compared to rAAV-GFP cells without inhibitor treatment (Fig. 3C and 3D). Likewise, TNF-α-induced apoptosis was significantly increased in cells infected with rAAV-CYP102 F87V when treated with these signaling pathway inhibitors compared to rAAV-CYP102 F87V infected cells without inhibitor treatment (Fig. 3C and 3D). Importantly, inhibition of the MAPK and PI3K/Akt signaling pathways increased TNF-α-induced apoptosis to a significantly greater extent in rAAV-CYP102 F87V versus rAAV-GFP infected BAECs, demonstrating that the anti-apoptotic effect of CYP epoxygenase overexpression is mediated, at least in part, through these two pathways.
CYP Epoxygenase Overexpression Inhibits Caspase-3 Activity
Treatment of BAECs with TNF-α significantly increased caspase-3 activity; however, infection with rAAV-CYP2J2, rAAV-CYP102 F87V, and rAAV-CYP2C11-CYPOR significantly attenuated the TNF-α-induced increase in caspase-3 activity compared to uninfected and rAAV-GFP infected cells (Fig. 4A). TNF-α also induced a significant increase in caspase-3 activity in both rAAV-GFP and rAAV-CYP102 F87V infected BAECs when incubated with Akt I, LY294002, apigenin and PD98059 compared to the respective cells without inhibitor treatment (Fig. 4B). However, inhibition of the MAPK and PI3K/Akt signaling pathways enhanced the TNF-α-induced increase in caspase-3 activity to a significantly greater extent in rAAV-CYP102 F87V versus rAAV-GFP infected BAECs, further demonstrating the role of these signaling pathways in the anti-apoptotic effect of CYP epoxygenase overexpression.
Figure 4.


Effect of CYP epoxygenase overexpression on caspase-3 activity. A, Caspase-3 activity measurements in cells infected with rAAV-GFP, rAAV-CYP2J2, rAAV-CYP102 F87V, or rAAV-CYP2C11-CYPOR, and one week later incubated with TNF-α for 24 hours. Control refers to BAECs without infection as 100% and The values of other samples were normalized to the untreated controls and expressed as mean±SEM of percentage according values from four independent experiments; *p<0.05, **p<0.01 compared to untreated control, ##p<0.01 compared to control treated with TNF-α; B, Caspase-3 activity measurements evaluating the effects of cell signaling molecule inhibitors on TNFα-induced apoptosis. Cells infected with rAAV-CYP102 F87V or rAAV-GFP one week prior were incubated with an inhibitor of Akt (Akt I), PI3K (LY294002), MAPK (apigenin), MEK (PD98059), or no inhibitor 30 minutes prior to TNF-α treatment. Control without inhibitor treatment was assigned 100% and the values of other samples are expressed as percentages of the control (all values are from six independent experiments); **p<0.01 compared to control. ##p<0.01 compared to rAAV-CYP102 F87V cells without inhibitor treatment.
CYP Epoxygenase Overexpression Inhibits Bcl-2 Downregulation
Overexpression of CYP2C11-CYPOR, CYP102 F87V or CYP2J2 in BAECs caused significant increase in Bcl-2 protein expression at baseline, but also significantly inhibited the TNF-α-induced downregulation of Bcl-2 protein expression even though TNF-α incubation downregulated Bcl-2 expression (data not shown). Further experiments demonstrated that TNF-α treatment resulted in a time-dependent downregulation of Bcl-2 protein expression over 48 hours; however, transfection with the three CYP epoxygenases significantly inhibited this Bcl-2 downregulation at each timepoint (Fig. 5A and 5B).
Figure 5.
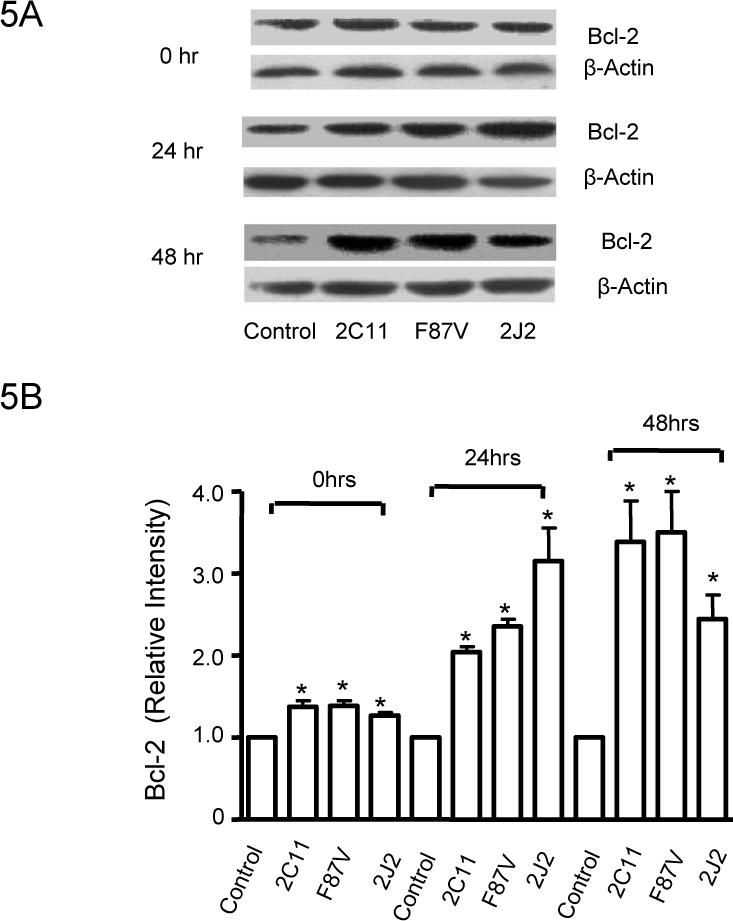
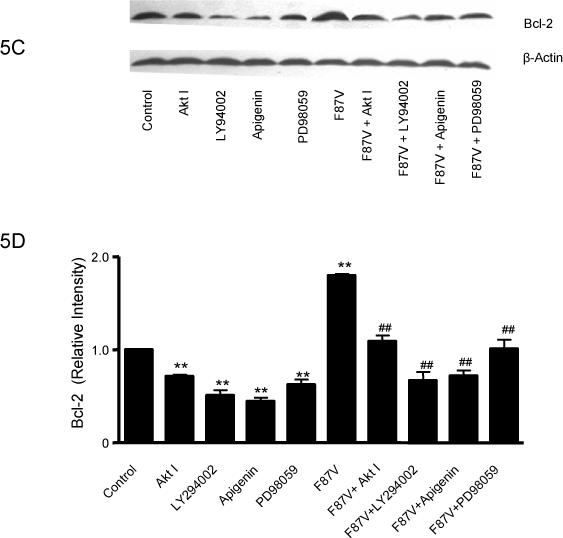
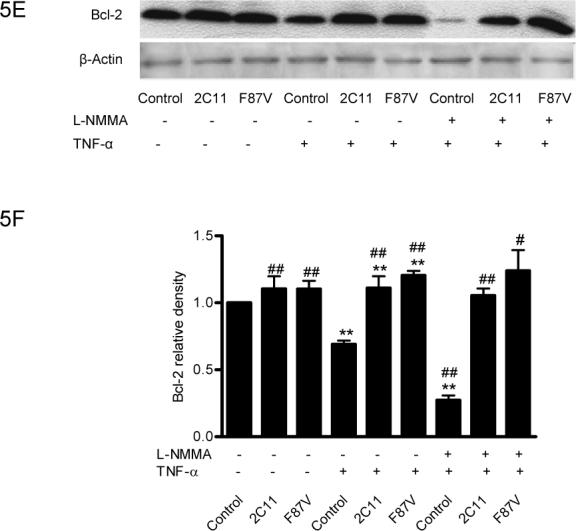
Effect of CYP epoxygenase overexpression on Bcl-2 expression. A, Bcl-2 and β-actin expression by immunoblot after stimulation with TNF-α for 0, 24, or 48 hours. Control represents transfection with the empty pCB6 vector. B, Densitometry of Bcl-2 protein expression relative to β-actin expression, normalized to control of the individual time points. Values shown represent mean±SEM from three independent experiments; **p<0.01 compared to corresponding control. C, Bcl-2 and β-actin expression by immunoblot in pCB6 and CYP102 F87V transfected BAECs after incubation with an inhibitor of Akt (Akt I), PI3K (LY294002), MAPK (apigenin), or MEK (PD98059) 30 minutes prior to TNF-α treatment. D, Densitometry of Bcl-2 protein expression relative to β-actin expression, normalized to control. Values shown are mean±SEM from 4 or 5 independent experiments; **p<0.01 compared to control cells without inhibitor treatment; ##p<0.01 compared to CYP102 F87V transfected cells without inhibitor treatment. E, Bcl-2 and β-actin expression by immunoblot. After transfection with CYP2C11-CYPOR or CYP102 F87V for 24 hours, L-NMMA (2mM) was incubated for 2 hours prior to TNF-α treatment. Control represents transfection with the empty pCB6 vector. F, Densitometry of Bcl-2 protein expression relative to β-actin expression, normalized to control, Values shown are mean±SEM from three independent experiments; Molecular weights of Bcl-2 and beta-actin are 28K and 43K, respectively. #p<0.05, ##p<0.01 compared to corresponding treatment control group. **p<0.01 compared to corresponding L-NMMA (−) and TNF-α (−) control group.
Pretreatment of BAECs with Akt I, LY294002, apigenin, or PD98059 prior to TNF-α treatment significantly reduced Bcl-2 protein expression in both control and CYP102 F87V transfected cells (Fig. 5C and 5D). Moreover, this signaling inhibitor-mediated reduction in Bcl-2 expression was significantly more pronounced in the CYP102 F87V transfected cells compared to control cells, suggesting that the MAPK and PI3K/Akt signaling pathways mediate, at least in part, the effect of CYP epoxygenase overexpression on Bcl-2 expression. In addition, incubation of control cells with the eNOS inhibitor, NG-monomethyl-L-arginine (L-NMMA), prior to TNF-α treatment significantly decreased Bcl-2 protein expression compared to treatment with TNF-α alone; however, L-NMMA pretreatment did not significantly attenuate the increase in Bcl-2 expression observed in TNF-α treated cells transfected with CYP2C11-CYPOR or CYP102 F87V (Fig. 5E and 5F). Together, these findings suggest that the anti-apoptotic effect of CYP epoxygenase overexpression in BAECs is likely not mediated via CYP epoxygenase- induced upregulation of eNOS expression.
Effects of CYP Epoxygenase Overexpression on ERK1/2 and PI3K/Akt Signaling
Treatment of BAECs with TNF-α induced a significant time-dependent dephosphorylation of extracellular signal-regulated kinases (ERK)1/2 over 4 hours in control cells; however, transfection with CYP2C11-CYPOR or CYP102 F87V significantly attenuated the extent of ERK1/2 dephosphorylation at these time points (Fig. 6A and 6B). Moreover, phosphorylation of ERK1/2 was significantly increased in cells transfected with the CYP epoxygenases compared to control cells prior to TNF-α treatment. Overexpression of CYP2J2, CYP102 F87V, and CYP2C11-CYPOR also significantly increased PI3K protein expression in both the presence (Fig. 7A and 7B) and absence (Fig. 7C and 7D) of TNF-α treatment compared to uninfected and rAAV-GFP infected control cells. Similarly, phosphorylation of Akt was significantly increased in BAECs transfected with the three CYP epoxygenases in both the presence (Fig. 7E and 7F) and absence (Figure 7G and 7H) of TNF-α treatment. Collectively, these findings demonstrate that CYP epoxygenase overexpression stimulates phosphorylation of ERK1/2 and PI3K/Akt signaling molecules in BAECs, and this may contribute to the observed anti-apoptotic effects of CYP epoxygenase overexpression in endothelial cells.
Figure 6.

Effect of CYP epoxygenase overexpression on ERK1/2 phosphorylation. A, Phospho-ERK1/2 (p-ERK) and total ERK1/2 expression by immunoblot. Cells were infected with rAAV-GFP, rAAV-CYP2C11-CYPOR or rAAV-CYP102 F87V. One week later, they were incubated with TNF-α for 0, 1, 2, or 4 hours and the cells were lysed. Control refers to BAECs infected with rAAV-GFP. B, Densitometry of p-ERK protein expression relative to total ERK expression, normalized to the control/0 hours ratio. Values shown are mean±SEM of three independent experiments; *p<0.05, **p<0.01 compared to corresponding groups at 0 hours; & and &&, # and ##, $ and $$ indicate p<0.05 and 0.01 for comparisons to control for the 0, 1, and 2 hour time points, respectively.
Figure 7.
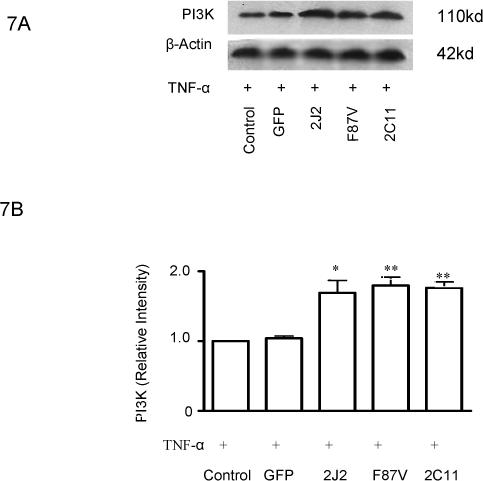
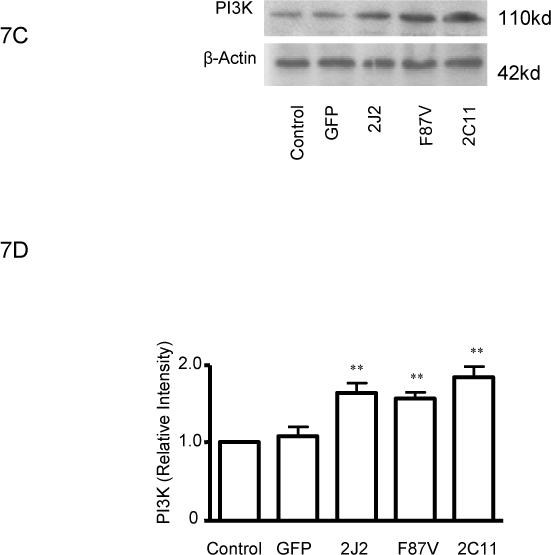
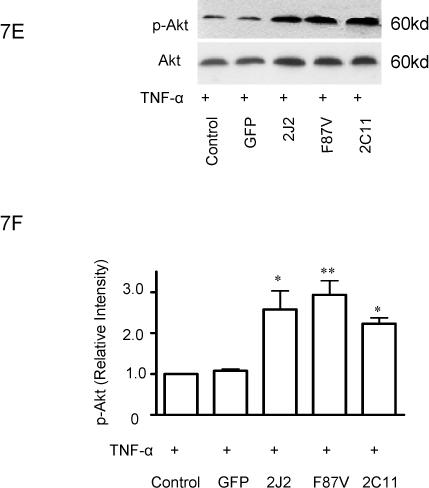
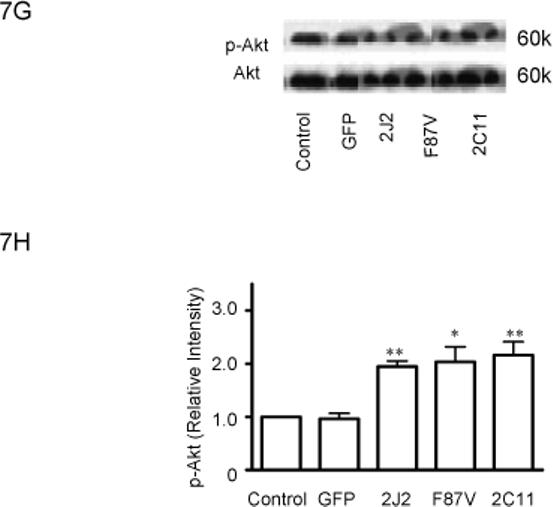
Effect of CYP epoxygenase overexpression on PI3K expression and Akt phosphorylation. A and C, PI3K and β-actin expression by immunoblot. Cells were infected with rAAV-GFP, rAAV-CYP2J2, rAAV-CYP102 F87V, or rAAV-CYP2C11-CYPOR, respectively, and one week later incubated with (A) or without (C) TNF-α for 24 hours. Control refers to BAECs without transfection. B and D, Densitometry of PI3K protein expression relative to β-actin expression normalized to control after incubation with (B) or without (D) TNF-α for 24 hours. Values are mean±SEM from three independent experiments; *p<0.05, **p<0.01 compared to control. E and G, Phospho-Akt (p-Akt) and total Akt expression by immunoblot after incubation with (E) or without (G) TNF-α for 24 hours. F and H, Densitometry of p-Akt protein expression relative to total Akt expression normalized to control after incubation with (F) or without (H) TNF-α for 24 hours. Values are mean±SEM from three independent experiments; *p<0.05, **p<0.01 compared to control.
DISCUSSION
Endothelial cells play an integral role in the regulation of vascular function via the synthesis and release of several vasoactive factors including NO and prostacyclin. EETs are epoxygenase metabolites of arachidonic acid synthesized in the endothelium which possess potent vasodilatory properties (4-6). These CYP epoxygenases, which are known to increase the synthesis of EETs, also play critical roles in the regulation of vascular and bronchial smooth muscle tone, cellular proliferation, peptide hormone secretion and ionic transport (9). Importantly, EETs possess potent effects within the vasculature via regulation of intravascular inflammation and homeostasis (12, 14); however, their role in regulating endothelial cell apoptosis has not been characterized. Dhanasekaran et al recently demonstrated the capacity of EETs to enhance human endothelial cell survival by inhibiting pathways of apoptosis in cell lines of human lung microvascular endothelial cells and coronary artery endothelial cells (19). Moreover, they reported that application of the PI3K inhibitor wortmannin abolished this protective effect, suggesting that activation of PI3K/Akt signaling is integrally involved in the EET-mediated cytoprotection (19). However, it is not clear whether endogenous EETs have anti-apoptotic effects in endothelial cells. Dhanasekaran's study demonstrated the protective effect of EETs in a serum-deprivation-induced apoptosis model, but the anti-apoptotic effects of EETs in models involving the induction of apoptosis by inflammatory cytokines such as TNF-α, remains unknown.
We sought to evaluate the potential influence of CYP epoxygenase overexpression and increased EET biosynthesis on endothelial cell apoptosis induced by the inflammatory cytokine TNF-α. Our findings demonstrate that CYP epoxygenase overexpression significantly increases BAEC viability, as observed previously by our group (31). Moreover, CYP epoxygenase overexpression significantly protects BAECs from TNF-α induced apoptosis, as evaluated by DNA laddering and FACS analysis. Moreover, CYP epoxygenase overexpression significantly inhibits caspase-3 activity and down-regulation of Bcl-2 expression induced by TNF-α in this model of endothelial injury. Collectively, our data implicate the CYP epoxygenase pathway as an important regulator of endothelial cell apoptosis. Although the observed anti-apoptotic effects related to epoxygenase overexpression could be due to increased synthesis of a non-EET product, we demonstrated that increased epoxygenase expression was also associated with increased 14,15-DHET production (a stable EET metabolite), consistent with our previous studies using these transfected epoxygenases. Moreover, our findings parallel the findings from other studies where EETs were administered exogenously. Importantly, our findings also demonstrate the important role of ERK dephosphorylation and activation of PI3K/Akt signaling in the anti-apoptotic effects of CYP epoxygenase overexpression.
Apoptosis is an enzymatically controlled and energy dependent form of programmed cell death, and caspases are a group of enzymes vital to the regulation of this process (32). A diverse array of intrinsic or extrinsic stimuli regulates endothelial cell apoptosis by modulating the balance between the pro-apoptotic caspases and various anti-apoptotic proteins such as Bcl-2 (16-18). We observed that CYP epoxygenase overexpression significantly inhibited caspase-3 activity and the time-dependent down-regulation of Bcl-2 expression induced by TNF-α, suggesting that EETs not only inhibit activity of pro-apoptotic proteins but also maintain levels of anti-apoptotic proteins. Moreover, these anti-apoptotic effects were significantly attenuated by inhibition of the PI3K/Akt and MAPK signaling pathways, suggesting they were mediated, at least in part, by activation of these pathways. Previous studies have indicated that increased eNOS expression also has anti-apoptotic effects (33, 34). Our prior work has demonstrated that exogenous EET treatment or CYP expoxygenase overexpression significantly increase eNOS expression and activity in BAECs (15). In order to determine if up-regulation of eNOS expression mediated the anti-apoptotic effects of CYP epoxygenase overexpression, we pre-incubated BAECs with the eNOS inhibitor L-NMMA. We found that eNOS inhibition did not attenuate the inhibitory effect of CYP epoxygenase overexpression on TNF-α induced Bcl-2 down-regulation, suggesting that the observed anti-apoptotic effects of CYP epoxygenase overexpression are independent of eNOS.
It has been reported that EETs participate in the regulation of several intracellular signaling pathways (9, 14, 35, 36). For example, 14,15-EET activates ERK during EET-induced mitogenesis (37). Moreover, transfection of porcine coronary arteries with CYP2C8 resulted in increased ERK1/2 phosphorylation compared to untreated cells (38). In endothelial cells, MAP kinase (ERK1/2)-dependent phosphorylation stabilized Bcl-2 and prevented its proteasome-dependent degradation, consequently increasing cell survival (17, 39). In the current study, phosphorylation of ERK1/2 was increased following CYP epoxygenase overexpression compared to control cells, similar to our previous observations (31), suggesting a role for MAPK signaling in the observed anti-apoptotic effect. While CYP expoxygenase transfection activated ERK1/2, it did not prevent the time-dependent dephosphorylation of ERK1/2 induced by TNF-α, implying the existence of other important signaling pathways. One such anti-apoptotic pathway involves signaling through PI3K/Akt (40-42) resulting in increased Bcl-2 expression (43) and prevention of mitochondrial cytochrome C release (44-46). Interestingly, CYP epoxygenase overexpression in BAECs also increased PI3K expression and Akt phosphorylation, as has been previously observed in cancer cells (26). Moreover, inhibition of PI3K and Akt signaling significantly attenuated the CYP epoxygenase-mediated increase in Bcl-2 expression, indicating a role for the PI3K/Akt pathway in EET-mediated anti-apoptotic signaling in endothelial cells. Collectively, our results demonstrate that CYP epoxygenase overexpression significantly attenuates TNF-α induced apoptosis in BAECs via both ERK1/2 and PI3K/Akt signaling.
Chen et al. reported that 14,15-EET inhibits apoptosis induced by serum withdrawal or H2O2 in LLCPKcl4 cells, a renal proximal tubule-like epithelial cell line (47). In the current study, we provide evidence that endogenously synthesized EETs prevent endothelial cell apoptosis induced by TNF-α. Endothelial cell apoptosis represents a form of endothelial injury that may significantly influence endothelial function, as well as vascular inflammation and homeostasis. Indeed, endothelial cell apoptosis may be an important initiating event in a variety of cardiovascular diseases such as atherosclerosis and hypertension (16-18). TNF-α, a multifunctional cytokine, promotes endothelial cell apoptosis and elicits inflammatory responses by increasing expression and secretion of adhesion molecules such as E-selectin, ICAM-1 and VCAM-1 (16, 18, 48, 49), which have been implicated in the pathogenesis of atherosclerosis. Our results suggest that CYP epoxygenase overexpression and increased EET biosynthesis may also inhibit the atherogenic effects of TNF-α by preventing endothelial cell apoptosis, demonstrating yet another potent vascular effect of EETs independent of their EDHF properties. Thus, it appears that CYP epoxygenase-derived EETs may have atheroprotective effects in the endothelium via multiple synergistic mechanisms. Importantly, future experiments will be necessary to investigate the potential contribution of other (i.e., non-EET) metabolites of the CYP epoxygenase pathway to these biological effects.
In conclusion, the current study reveals a novel role for CYP epoxygenase-derived EETs in endothelial cell survival. The anti-apoptotic effect of EETs markedly attenuated TNF-α induced apoptosis of endothelial cells and increased cell viability. These protective effects appear to involve both attenuation of the TNF-α-mediated decrease in the anti-apoptotic protein Bcl-2 and activation of the pro-apoptotic caspase-3, as well activation of the pro-survival PI3K/Akt and ERK signaling pathways. Thus, it appears that increased EET biosynthesis contributes to the survival of endothelial cells and maintenance of cardiovascular homeostasis. The apoptosis-suppressive effect of EETs may have important clinical implications and modulation CYP epoxygenase-mediated EET biosynthesis may represent a novel therapeutic strategy for the treatment and/or prevention of cardiovascular diseases.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation Committee (No. 30270561, and 30430320), National “973” grant (No. 2006CB503801), and the Intramural Research Program of the NIH, NIEHS. We are grateful to Dr. Jorge Capdevila for providing the CYP102 F87V and CYP2C11-CYPOR cDNAs and corresponding polyclonal antibodies. Shilin Yang, Li Lin and Ji-Xiong Chen contributed equally to this work.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
No authors have conflicts of interest to disclose.
REFERENCES
- 1.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 2.Shimokawa H. Primary endothelial dysfunction: atherosclerosis. J Mol Cell Cardiol. 1999;31:23–37. doi: 10.1006/jmcc.1998.0841. [DOI] [PubMed] [Google Scholar]
- 3.Vanhoutte PM, Mombouli JV. Vascular endothelium: vasoactive mediators. Prog Cardiovasc Dis. 1996;39:229–238. doi: 10.1016/s0033-0620(96)80003-x. [DOI] [PubMed] [Google Scholar]
- 4.Miura H, Gutterman DD. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P-450 monooxygenase and Ca2+-activated K+ channels. Circ Res. 1998;83:501–507. doi: 10.1161/01.res.83.5.501. [DOI] [PubMed] [Google Scholar]
- 5.Fleming I. Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol Res. 2004;49:525–533. doi: 10.1016/j.phrs.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 7.Feletou M, Vanhoutte PM. The alternative: EDHF. J Mol Cell Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- 8.McGuire JJ, Ding H, Triggle CR. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s). Can J Physiol Pharmacol. 2001;79:443–470. [PubMed] [Google Scholar]
- 9.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 10.Chen JK, Falck JR, Reddy KM, Capdevila J, Harris RC. Epoxyeicosatrienoic acids and their sulfonimide derivatives stimulate tyrosine phosphorylation and induce mitogenesis in renal epithelial cells. J Biol Chem. 1998;273:29254–29261. doi: 10.1074/jbc.273.44.29254. [DOI] [PubMed] [Google Scholar]
- 11.Chen JK, Capdevila J, Harris RC. Overexpression of C-terminal Src kinase blocks 14, 15-epoxyeicosatrienoic acid-induced tyrosine phosphorylation and mitogenesis. J Biol Chem. 2000;275:13789–13792. doi: 10.1074/jbc.275.18.13789. [DOI] [PubMed] [Google Scholar]
- 12.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J, Sui X, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ Res. 2002;90:1020–1027. doi: 10.1161/01.res.0000017727.35930.33. [DOI] [PubMed] [Google Scholar]
- 14.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Lin L, Jiang J, Wang Y, Lu ZY, Bradbury JA, Lih FB, Wang DW, Zeldin DC. Up-regulation of endothelial nitric-oxide synthase by endothelium-derived hyperpolarizing factor involves mitogen-activated protein kinase and protein kinase C signaling pathways. J Pharmacol Exp Ther. 2003;307:753–764. doi: 10.1124/jpet.103.052787. [DOI] [PubMed] [Google Scholar]
- 16.Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann N Y Acad Sci. 2001;947:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 17.Choy JC, Granville DJ, Hunt DW, McManus BM. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol. 2001;33:1673–1690. doi: 10.1006/jmcc.2001.1419. [DOI] [PubMed] [Google Scholar]
- 18.Stefanec T. Endothelial apoptosis: could it have a role in the pathogenesis and treatment of disease? Chest. 2000;117:841–854. doi: 10.1378/chest.117.3.841. [DOI] [PubMed] [Google Scholar]
- 19.Dhanasekaran A, Al-Saghir R, Lopez B, Zhu D, Gutterman DD, Jacobs ER, Medhora M. Protective effects of epoxyeicosatrienoic acids on human endothelial cells from the pulmonary and coronary vasculature. Am J Physiol Heart Circ Physiol. 2006;291:H517–31. doi: 10.1152/ajpheart.00953.2005. [DOI] [PubMed] [Google Scholar]
- 20.Duriez PJ, Wong F, Dorovini-Zis K, Shahidi R, Karsan A. A1 functions at the mitochondria to delay endothelial apoptosis in response to tumor necrosis factor. J Biol Chem. 2000;275:18099–18107. doi: 10.1074/jbc.M908925199. [DOI] [PubMed] [Google Scholar]
- 21.Karsan A, Yee E, Harlan JM. Endothelial cell death induced by tumor necrosis factor-alpha is inhibited by the Bcl-2 family member, A1. J Biol Chem. 1996;271:27201–27204. doi: 10.1074/jbc.271.44.27201. [DOI] [PubMed] [Google Scholar]
- 22.Graham-Lorence S, Truan G, Peterson JA, Falck JR, Wei S, Helvig C, Capdevila JH. An active site substitution, F87V, converts cytochrome P450 BM-3 into a regio- and stereoselective (14S,15R)-arachidonic acid epoxygenase. J Biol Chem. 1997;272:1127–1135. doi: 10.1074/jbc.272.2.1127. [DOI] [PubMed] [Google Scholar]
- 23.Qu W, Rippe RA, Ma J, Scarborough P, Biagini C, Fiedorek FT, Travlos GS, Parker C, Zeldin DC. Nutritional status modulates rat liver cytochrome P450 arachidonic acid metabolism. Mol Pharmacol. 1998;54:504–513. doi: 10.1124/mol.54.3.504. [DOI] [PubMed] [Google Scholar]
- 24.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005;65:4707–4715. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- 27.Hu Y, Qiao L, Wang S, Rong SB, Meuillet EJ, Berggren M, Gallegos A, Powis G, Kozikowski AP. 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J Med Chem. 2000;43:3045–3051. doi: 10.1021/jm000117y. [DOI] [PubMed] [Google Scholar]
- 28.Grider JS, Falcone JC, Kilpatrick EL, Ott CE, Jackson BA. P450 arachidonate metabolites mediate bradykinin-dependent inhibition of NaCl transport in the rat thick ascending limb. Can J Physiol Pharmacol. 1997;75:91–96. [PubMed] [Google Scholar]
- 29.Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med. 2000;6:564–567. doi: 10.1038/75045. [DOI] [PubMed] [Google Scholar]
- 30.Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic Acid Epoxygenase Metabolites Stimulate Endothelial Cell Growth and Angiogenesis via Mitogen-Activated Protein Kinase and Phosphatidylinositol 3-Kinase/Akt Signaling Pathways. J Pharmacol Exp Ther. 2005;314:522–532. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- 32.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 33.Raveendran M WJ, Senthil D, Wang J, Utama B, Shen Y, Dudley D, Zhang Y, Wang XL. Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS Lett. 2005;579:733–740. doi: 10.1016/j.febslet.2004.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimmeler SZA. Nitric oxide-an endothelial cell survival factor. Cell Death Differ. 1999;6:964–968. doi: 10.1038/sj.cdd.4400581. [DOI] [PubMed] [Google Scholar]
- 35.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 36.Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–15676. doi: 10.1074/jbc.M110806200. [DOI] [PubMed] [Google Scholar]
- 37.Chen JK, Wang DW, Falck JR, Capdevila J, Harris RC. Transfection of an active cytochrome P450 arachidonic acid epoxygenase indicates that 14,15-epoxyeicosatrienoic acid functions as an intracellular second messenger in response to epidermal growth factor. J Biol Chem. 1999;274:4764–4769. doi: 10.1074/jbc.274.8.4764. [DOI] [PubMed] [Google Scholar]
- 38.Fleming I, Fisslthaler B, Michaelis UR, Kiss L, Popp R, Busse R. The coronary endothelium-derived hyperpolarizing factor (EDHF) stimulates multiple signalling pathways and proliferation in vascular cells. Pflugers Arch. 2001;442:511–518. doi: 10.1007/s004240100565. [DOI] [PubMed] [Google Scholar]
- 39.Rossig L, Haendeler J, Hermann C, Malchow P, Urbich C, Zeiher AM, Dimmeler S. Nitric oxide down-regulates MKP-3 mRNA levels: involvement in endothelial cell protection from apoptosis. J Biol Chem. 2000;275:25502–25507. doi: 10.1074/jbc.M002283200. [DOI] [PubMed] [Google Scholar]
- 40.Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:402–409. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- 41.Haga M, Chen A, Gortler D, Dardik A, Sumpio BE. Shear stress and cyclic strain may suppress apoptosis in endothelial cells by different pathways. Endothelium. 2003;10:149–157. doi: 10.1080/10623320390233463. [DOI] [PubMed] [Google Scholar]
- 42.Papapetropoulos A, Fulton D, Mahboubi K, Kalb RG, O'Connor DS, Li F, Altieri DC, Sessa WC. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- 43.Kumar P, Miller AI, Polverini PJ. p38 MAPK mediates gamma-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol Chem. 2004;279:43352–43360. doi: 10.1074/jbc.M405777200. [DOI] [PubMed] [Google Scholar]
- 44.Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–3049. doi: 10.1161/01.CIR.0000130647.29030.90. [DOI] [PubMed] [Google Scholar]
- 45.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 46.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen JK, Capdevila J, Harris RC. Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol. 2001;21:6322–6331. doi: 10.1128/MCB.21.18.6322-6331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–962. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Madge LA, Pober JS. A phosphatidylinositol 3-kinase/Akt pathway, activated by tumor necrosis factor or interleukin-1, inhibits apoptosis but does not activate NFkappaB in human endothelial cells. J Biol Chem. 2000;275:15458–15465. doi: 10.1074/jbc.M001237200. [DOI] [PubMed] [Google Scholar]