

Abstract
Dendritic cells (DCs) are bone marrow–derived leukocytes that function as potent antigen presenting cells capable of initiating T cell–dependent responses from quiescent lymphocytes. DC pulsed with tumor-associated antigen (TAA) peptide or protein have recently been demonstrated to elicit antigen-specific protective antitumor immunity in a number of murine models. Transduction of DCs with TAA genes may allow stable, prolonged antigen expression as well as the potential for presentation of multiple, or unidentified, epitopes in association with major histocompatibility complex class I and/or class II molecules. To evaluate the potential efficacy of retrovirally transduced DCs, bone marrow cells harvested from BALB/c mice were transduced with either a model antigen gene encoding β-galactosidase (β-gal) or a control gene encoding rat HER-2/neu (Neu) by coculture with irradiated ecotropic retroviral producer lines. Bone marrow cells were differentiated into DC in vitro using granulocyte/macrophage colony-stimulating factor and interleukin-4. After 7 d in culture, cells were 45–78% double positive for DC phenotypic cell surface markers by FACS® analysis, and DC transduced with β-gal were 41–72% positive for β-gal expression by X-gal staining. In addition, coculture of β-gal transduced DC with a β-gal–specific T cell line (CTLx) resulted in the production of large amounts of interferon-γ, demonstrating that transduced DCs could process and present endogenously expressed β-gal. DC transduced with β-gal and control rat HER-2/neu were then used to treat 3-d lung metastases in mice bearing an experimental murine tumor CT26.CL25, expressing the model antigen, β-gal. Treatment with β-gal–transduced DC significantly reduced the number of pulmonary metastatic nodules compared with treatment with Hank's balanced salt solution or DCs transduced with rat HER-2/neu. In addition, immunization with β-gal–transduced DCs resulted in the generation of antigen-specific cytotoxic T lymphocytes (CTLs), which were significantly more reactive against relevant tumor targets than CTLs generated from mice immunized with DCs pulsed with the Ld-restricted β-gal peptide. The results observed in this rapidly lethal tumor model suggest that DCs transduced with TAA may be a useful treatment modality in tumor immunotherapy.
Dendritic cells (DCs)1 are highly specialized APCs that possess unique immunostimulatory properties and function as the principal activators of quiescent T cells, and thus cellular immune responses in vivo. (1). These bone marrow–derived leukocytes express a unique repertoire of cell-surface molecules including high levels of MHC class I and II, adhesion molecules, and costimulatory molecules, all of which assist in the activation of T cells. As motile cells with elaborate cytoplasmic processes and a unique veiled morphology, DCs are specialized for antigen capture and transport from the periphery to T cell–dependent areas of lymphoid organs.
The key role of DCs in the initiation of immune responses has focused the attention of many investigators on the potential efficacy of these cells in tumor immunotherapy. Several groups have demonstrated that DCs pulsed with peptides from tumor associated antigens (TAA) can induce antigen-specific antitumor responses in vivo in a variety of murine tumor models (2–7). The successes of TAA-pulsed DCs in murine models has supported the use of autologous, peptide-pulsed DCs in recent clinical trials (8).
In developing strategies to optimize the use of DCs in tumor immunotherapy, retroviral transduction of DCs with TAA genes may offer important advantages over peptide-pulsed DCs and other methods of immunization currently in use. The efficacy of peptide-pulsed DCs might be limited in vivo, because peptides pulsed onto DCs stay bound to the MHC molecules only transiently due to variation in peptide binding affinities, peptide–MHC complex dissociation, and MHC turnover (9). Additionally, the use of peptide-pulsed DCs is dependent on the knowledge of the HLA haplotype of the patient, as well as the restriction element of the peptide epitopes for any particular antigen.
However, retroviral transduction of DCs with TAA genes may allow for constitutive expression of the full-length protein leading to prolonged antigen presentation in vivo, and presentation of multiple or unidentified antigen epitopes in the context of MHC class I, and possibly class II, molecules. Additionally, retrovirally transduced DCs are entirely autologous, thus abrogating the potential for development of neutralizing antibodies with repeated treatments, as can occur with recombinant viral immunization modalities. TAA-transduced DCs might also be given repeatedly and/or combined with other viral or peptide-based immunization strategies.
As nonreplicating, terminally differentiated cells, mature DCs are poor candidates for retroviral gene modification. However, dividing bone marrow progenitor cells can be efficiently transduced with retroviral vectors (10–12). Because DCs can successfully be generated in vitro from bone marrow cells in the presence of GM-CSF–containing cytokine combinations (13–16), we used a method by which bone marrow cells were retrovirally transduced by coculture with irradiated producer lines and then differentiated in vitro to DCs. This method has previously been shown to be effective in human DC by retroviral transduction of CD34+ hematopoietic progenitor cells (HPCs) and differentiation of transduced cells in vitro to mature DC (17, 18).
In this study, we demonstrate that murine DCs retrovirally transduced with the gene encoding β-galactosidase (β-gal) stably express, process, and present the gene in the context of MHC class I molecules, and that treatment with β-gal–transduced DCs is capable of mediating effective antitumor activity against established pulmonary metastases of a murine tumor expressing β-gal.
Materials and Methods
Cell Lines.
CT26.CL25, a subclone of CT26.WT, is a BALB/c (H-2d) carcinogen-induced, undifferentiated colon carcinoma stably transduced with a retrovirus encoding the lacZ gene driven by the Moloney murine leukemia virus long terminal repeat (LTR) promoter (19). Tumor cell lines were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/liter glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from Biofluids, Rockville, MD), 1.25 mg/ml amphotericin B (Fungizone; GIBCO BRL, Gaithersburg, MD), and 50 μg/ml gentamicin sulfate (GIBCO BRL). The CTL line (CTLx) which recognizes the naturally processed, H-2Ld–restricted β-gal epitope p876 to 884, has previously been described (20).
Retroviral Transduction and Differentiation of DCs from Bone Marrow Cells.
CreLacZ and CreNeu are fibroblast-derived, ecotropic-packaging cell lines encoding the lacZ and rat HER-2/neu genes, respectively, in the MFG retroviral vector backbone. CreLacZ was provided by Richard Mulligan (Children's Hospital, Boston, MA) and has previously been described (21). CreNeu was provided by Elizabeth Jaffee (Johns Hopkins University, Baltimore, MD). Bone marrow was flushed from the long bones of the hind limbs of female 8–12-wk-old BALB/c mice and depleted of erythrocytes with ACK lysing buffer (Biofluids). Bone marrow cells were depleted of lymphocytes and Ia+ cells by incubation at 4°C in an antibody cocktail (RA3-3A1/6.1, anti–B cell surface glycoprotein; HO-2.2, anti-Lyt 2.2; B21-2, anti-I-Ab,d; and GK1.5, anti-L3T4 T cell surface antigen; all from American Type Culture Collection, Rockville, MD) and cytotoxicity medium (CM; Cedarlane Labs. Ltd., Hornby, Canada). Antibody-bound cells were removed by incubation at 37°C with rabbit complement (Accurate Chemical and Scientific Corp., Westbury, NY). Bone marrow cells (7 × 105 cells/ml) were plated on irradiated (5,000 rads) Cre producer lines (5 × 103 cells/well) in DC CM and cocultured for 2 d at 37°C, 5% CO2 in 6-well plates. Cells were cultured in DC complete medium (DC CM), which is RPMI-1640 supplemented with 5% heat-inactivated fetal calf serum, 2 mmol/liter glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1.25 μg/ml amphotericin B, 50 μg/ml gentamicin sulfate, 5 × 10−5 μM 2-mercaptoethanol (2-ME; GIBCO BRL), 20 ng/ml recombinant murine GM-CSF (rmGM-CSF), and 100 ng/ml recombinant murine IL-4 (rmIL-4) (both from Peprotech, Rocky Hill, NJ). On day 2, nonadherent cells were carefully removed from adherent packaging cell lines and replated at 7 × 105 cells/ml in DC CM. On day 4, 10 ng/ml rmGM-CSF and 50 ng/ml rmIL-4 were added to each well. Cells were harvested on day 6 by gentle pipetting and replated in 100 mm tissue culture dishes at 106 cells/ml with DC CM supplemented with 20 ng/ml rmGM-CSF and 100 ng/ml rmIL-4.
Cell Surface Phenotype.
Selected monoclonal antibodies against the murine molecules (B7-1, B7-2, Iad, CD11c, B220/CD45R, CD3, Thy1.2, Mac-1, GR-1, and appropriate isotype controls; all from PharMingen, San Diego, CA) were obtained directly labeled with phycoerythrin or fluorescein isothiocyanate. For phenotypic analysis, 106 day 7 DCs or fresh syngeneic splenocytes were first incubated with 2.4G2, an antibody directed against the FcRIIγ receptor, and then double stained with the indicated directly labeled antibodies. Propidium iodide was used to exclude dead cells from the analysis.
Mixed Leukocyte Reaction.
The ability of DCs to stimulate quiescent T cells was assessed by the MLR. Bone marrow–derived DCs (transduced and nontransduced) or freshly prepared splenocytes were irradiated (2,000 rads) and plated in graded doses with 2 × 105 allogeneic T cells in 200 μl of RPMI-1640 media supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/liter glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1× nonessential amino acids, 1 mM sodium pyruvate (all from Biofluids), 1.25 μg/ml amphotericin B (Fungizone; GIBCO BRL), and 50 μg/ml gentamicin sulfate (GIBCO BRL) (mCM) in flat-bottomed 96-well tissue culture plates and incubated at 37°C, 5% CO2 for 3.5 d. Allogeneic T cells were prepared from C57BL/6 mice by passing RBC-depleted splenocytes through an immunoaffinity column (R&D Systems, Minneapolis, MN). During the last 6 h of incubation, cultures were pulsed with 1.0 μCi/well [3H]thymidine (New England Nuclear, Boston, MA). [3H]thymidine incorporation was measured using a β scintillation counter (Beta Plate; LKB, Gaithersburg, MD).
X-Gal Staining.
To detect β-gal expression in transduced DCs, 5 × 105 day 7 DCs were washed once with PBS (Biofluids) and fixed with 1 ml 0.5% glutaraldehyde for 10 min at room temperature. Cells were centrifuged, washed in PBS, and incubated in 0.5 ml X-gal solution (330 mM K3Fe(CN)6, 330 mM K4Fe(CN)6, 1 M MgCl2, 10% Triton X-100, 67 mg/ml X-gal, PBS) for 3–5 h at 37°C, 5% CO2. Bright blue cells in each sample were counted blindly and expressed as a percentage of the total cells.
In Vitro Cytokine Release Assays.
To determine whether endogenously expressed β-gal was processed and presented in the context of MHC class I molecules, β-gal–specific T cells (CTLx; 105) were cultured with day 7 bone marrow–derived DCs or peptide-pulsed splenocytes (105) in 200 μl mCM containing 60 IU/ml IL-2 (Chiron, Emeryville, CA) in flat-bottomed 96-well tissue culture plates and incubated at 37°C in a 5% CO2 humidified incubator. After 24 h, the supernatant was collected and analyzed for IFN-γ content with an ELISA assay (R&D Systems).
Peptides.
The peptide (βgP), TPHPARIGL, representing the naturally processed H-2 Ld-restricted epitope spanning amino acids 876–884 of β-gal (22) and the P1A35–43 peptide LPYLGWLVF, presented by H-2 Ld, were synthesized by Peptide Technologies (Washington, DC) to a purity >99% as assessed by HPLC and amino acid analysis.
Antigen Pulsing of DCs.
Day 7 bone marrow–derived DC from BALB/c mice were resuspended in reduced serum media (Optimem; GIBCO BRL) at 2 × 106 cells/ml and pulsed with peptide (10 μg/ml) plus human β2-microglobulin (β2m; 10 μg/ ml; Intergen Co., Purchase, NY) for 4 h at room temperature with gentle mixing. Cells were then washed twice in HBSS (Biofluids) and DCs (4 × 105) in 0.5 ml HBSS were injected intravenously in the tail vein. Freshly isolated syngeneic splenocytes (5 × 106 cells/ml) were pulsed with peptide in an identical manner as DCs.
In Vivo Treatment Studies.
BALB/c mice were challenged with CT26.CL25 tumor cells (3 × 105) intravenously to establish pulmonary metastases. On days 3 and 6 after tumor challenge, mice were treated with day 7 bone marrow–derived transduced or peptide-pulsed DCs or peptide-pulsed splenocytes intravenously. All mice were randomized before receiving transduced or peptide-pulsed DCs or splenocytes. Mice were killed on day 12 and metastatic lung nodules were enumerated in a randomized and blinded manner. Numbers presented are the mean numbers of pulmonary metastases ± SEM. The significance of differences between the groups was determined with the Wilcoxon rank Sums test. All P values are two-tailed.
In Vitro Antigen Restimulation.
For CTL generation, naive mice were immunized with transduced or peptide-pulsed DCs or splenocytes (4 × 105 cells) on days 0 and 3. 3–4 wk after the second immunization, splenocytes from animals immunized with transduced or peptide-pulsed DCs or splenocytes were pooled and restimulated in vitro with the Ld-restricted β-gal peptide for 6 d in EHAA medium (Biofluids) supplemented with 0.5% heat-inactivated mouse serum (Sigma Chemical Co., St. Louis, MO) and 5 × 10−5 μM 2-mercaptoethanol (2-ME; GIBCO BRL). On days 2 and 4, 1 ml of fresh media with 60 IU/ml IL-2 (Chiron, Emeryville, CA) was added to each well of cells. On day 7, restimulated splenocytes were tested for antigen-specific cytolytic activity and cytokine release. Restimulated splenocytes (105) were plated in flat-bottomed 96-well tissue culture plates with 105 peptide-pulsed tumor targets in 200 μl media. After 24 h, the supernatant was collected and analyzed for IFN-γ content with an ELISA assay (R&D Systems).
Cytotoxicity Assays.
Target cells (CT26 and CT26.CL25 tumor cells) were harvested and labeled with 200 μCi Na2 51CrO4 (Amersham, Arlington Heights, IL). Restimulated splenocytes from mice immunized with transduced or peptide-pulsed DCs or splenocytes were harvested and mixed in graduated doses with 5 × 103 labeled target cells in 150 μl mCM in round-bottomed 96-well tissue culture plates. Cells were incubated for 4 h at 37°C in a 5% CO2 humidified incubator, the 51Cr released from the target cells was measured with a γ counter (Wallac, Gaithersburg, MD), and the percent specific lysis was calculated as follows: 100 × ([experimental release − spontaneous release]) / ([maximal release − spontaneous release]). Spontaneous and maximal release were determined in the presence of either medium or 2% SDS, respectively.
Results
Retroviral Transduction of Bone Marrow Cells and Differentiation into DCs.
Bone marrow cells from BALB/c mice were retrovirally transduced by coculture with the irradiated, ecotropic packaging cell lines, CreLacZ and CreNeu, for 2 d. This method of gene modification capitalizes on the suitability of rapidly dividing bone marrow cells as targets for retroviral transduction (10–12). Transduced bone marrow cells were then differentiated into DCs in vitro for an additional 5 d in the presence of rmGM-CSF and rmIL-4. This method of retroviral transduction consistently generated a cell population composed of 45–78% DCs as determined by the percentage of cells double positive for characteristic DC surface molecules (B7-2, I-Ad, B7-1, CD11c) (Fig. 1).
Figure 1.
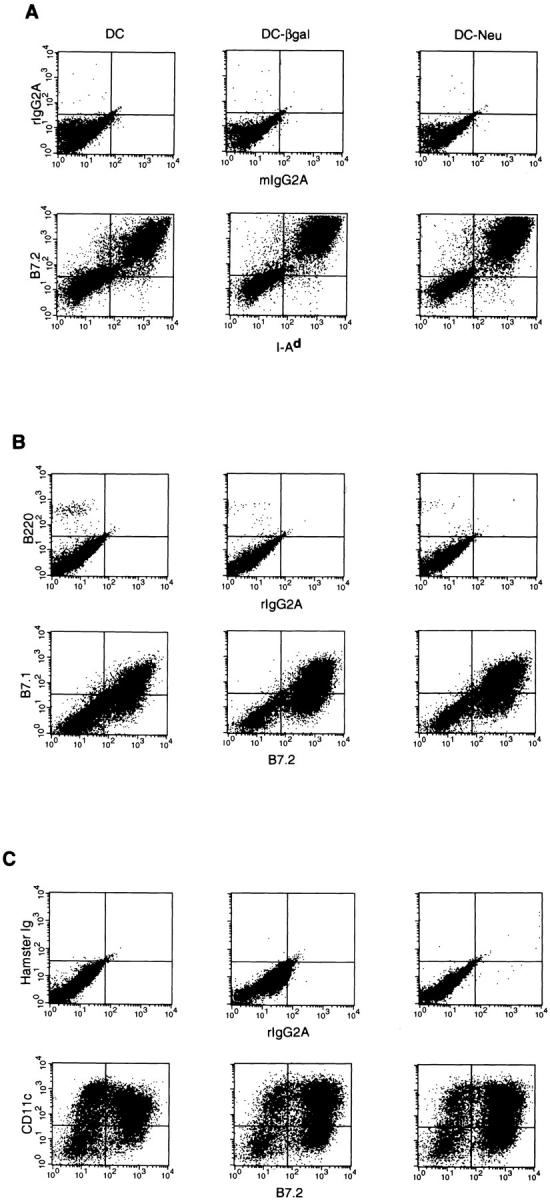
Cell surface phenotype of bone marrow–derived DCs as determined by FACS® analysis. For phenotypic analysis, 106 DCs on day 7 of culture or fresh syngeneic splenocytes were incubated with 2.4G2, an antibody directed against the FcRIIγ receptor, and then double stained with the indicated directly labeled antibodies. Propidium iodide was used to exclude dead cells from the analysis. (A) B7-2 and I-Ad with isotype controls, (B) B7-1 and B7-2 with isotype controls, and (C) CD11c and B7-2 with isotype controls.
To determine the percent of cells expressing β-gal in the transduced cell population, day 7 DCs were X-gal stained. Transduction efficiencies between 41–72% were consistently observed by this method with very low levels of background staining in the nontransduced or HER-2/neu– transduced controls (Table 1). Both transduced and nontransduced DCs displayed a characteristic DC morphology with prominent cytoplasmic processes (Fig. 2).
Table 1.
Expression of β-gal in Retrovirally Transduced DCs
| Percent of cells expressing β-gal | ||||||
|---|---|---|---|---|---|---|
| Experiment | DC (nontransduced) | DC–β-gal | DC–Neu | |||
| I | 0 | 54 | ND | |||
| II | 2 | 72 | 3 | |||
| III | 1 | 65 | 0 | |||
| IV | 4 | 41 | 1 | |||
| V | 3 | 48 | 0 | |||
On day 7 of in vitro culture, DCs were X-gal stained as described in Materials and Methods. Bright blue cells in each sample were counted blindly and expressed as a percentage of the total cells.
Figure 2.

Expression of β-gal as determined by X-gal staining. On day 7 of in vitro culture, nontransduced DCs (A) and β-gal–transduced DCs (B) were fixed and stained as described in Materials and Methods. Phase contrast photomicrographs were taken at 100×.
Bone Marrow–derived, Retrovirally Transduced DCs Function as Potent Stimulators in an Allogeneic Mixed Leukocyte Reaction.
The allostimulatory capacity of the DCs were assessed by mixed leukocyte reaction (MLR) (Fig. 3). Both transduced and nontransduced DCs were potent stimulators of quiescent, allogeneic T cells in repeated experiments. DCs were greater than 1,000-fold stronger than fresh, bulk splenocytes in stimulating proliferation of allogeneic T cells. No significant difference was observed among the nontransduced, β-gal–transduced, or Neu-transduced DCs.
Figure 3.

Allogeneic MLR of bone marrow–derived DCs and splenocytes. Bone marrow cells were transduced by coculture with retroviral producer lines, CreLacZ (DC–β-gal) and CreNeu (DC–Neu), and differentiated into DCs in vitro. DCs were cocultured with allogeneic, C57BL/6 T cells, isolated from bulk splenocytes by passing cells through an immunoaffinity column. After 3.5 d in culture, cells were pulsed with [3H]thymidine as described in Materials and Methods. Results from triplicate wells were corrected for [3H]thymidine incorporation by irradiated stimulators and T cells alone, and are plotted as the mean ± SEM.
Retrovirally Transduced DCs Process and Present Endogenously Expressed β-gal.
To determine whether the β-gal transduced DCs were capable of processing and presenting β-gal peptides, transduced and peptide-pulsed DCs were cocultured with an Ld-restricted β-gal–specific T cell line, CTLx (20). After 24 h, IFN-γ was measured in the culture supernatant (Table 2). DCs transduced with β-gal induced antigen-specific cytokine release from activated T cells indicating that the endogenously expressed transgene was processed and presented in an MHC-restricted manner. DCs as well as fresh, syngeneic splenocytes pulsed with the Ld-restricted βgP, also induced IFN-γ release from CTLx as positive controls. DC transduced with HER-2/neu or pulsed with an irrelevant peptide, P1A, failed to cause cytokine release, demonstrating the antigen specificity of the response.
Table 2.
Cytokine Release from Ld-restricted, β-gal–specific T Cell Line (CTLx) in Response to Bone Marrow–derived DCs
| Responder (CTLx) | ||
|---|---|---|
| Stimulator | mIFN-γ (pg/ml/24 h) | |
| HBSS | 73 | |
| DC–β-gal transduced | 20,890 | |
| DC–Neu transduced | 169 | |
| DC–βgP pulsed | 46,140 | |
| DC–P1A pulsed | 139 | |
| Splen–βgP pulsed | 68,000 | |
| CT26 | 616 | |
| CT25.CL26 | 113,730 |
Nontransduced, bone marrow–derived DCs and fresh, syngeneic splenocytes were pulsed with either βgP or P1A plus β2m for 4 h in reduced-serum medium. Pulsed cells or transduced DCs (105) were incubated with 105 CTLx for 24 h, and the supernatant was analyzed for murine IFN-γ content. Tumor lines CT26 (parental, wild type) and CT26.CL25 (which stably expresses β-gal) are included as negative and positive controls, respectively.
Retrovirally Transduced DCs Are Therapeutically Effective against Established Pulmonary Metastases.
To determine the therapeutic efficacy of retrovirally transduced and peptide-pulsed DCs against established pulmonary metastases, BALB/c mice were challenged with 3 × 105 CT26.CL25 tumor cells intravenously and treated intravenously with either 4 × 105 β-gal or HER-2/neu–transduced DCs, 4 × 105 peptide-pulsed DCs, 4 × 105 βgP-pulsed splenocytes, or HBSS on days 3 and 6 after tumor challenge. On day 12, the number of pulmonary metastastic nodules were enumerated in a blinded fashion. Mice treated with DCs transduced with β-gal (DC–β-gal) or pulsed with βgP (DC– βgP) showed a significant reduction in the number of pulmonary metastases compared with mice treated with HER-2/neu–transduced (DC–Neu) or P1A peptide-pulsed DCs (DC–P1A) (Fig. 4 A). The unique role of DCs in this response is evidenced by the lack of treatment effect seen in mice treated with βgP-pulsed splenocytes (splen– βgP). These results are representative of data obtained from four independent experiments.
Figure 4.


Active immunotherapy with retrovirally transduced or peptide-pulsed DCs significantly reduces the number of established pulmonary metastases in this 3-d tumor model. (A) 8–10 BALB/c mice/group were injected intravenously with 3 × 105 CT26.CL25 tumor cells. On days 3 and 6 after tumor challenge, mice received intravenous injections of 4 × 105 transduced or peptide-pulsed DCs, fresh peptide-pulsed splenocytes, or HBSS alone. On day 12 after tumor challenge, lungs were harvested and pulmonary nodules were enumerated in a blinded fashion. (B) BALB/c mice were challenged with 3 × 105 CT26.CL25 tumor cells. On days 3 and 6 after tumor challenge, mice received intravenous injections of transduced DCs or fresh peptide-pulsed splenocytes at varying doses (4–16 × 105). On day 12 after tumor challenge, lungs were harvested and pulmonary nodules were enumerated in a blinded fashion.
To assess further the efficacy of retrovirally transduced DCs in this tumor model, BALB/c mice bearing CT26.CL25 pulmonary metastases (3 × 105 tumor cells) were treated with varying doses of retrovirally transduced DCs or βgP-pulsed splenocytes (4 × 105–1.6 × 106) on days 3 and 6 after tumor challenge (Fig. 4 B). Mice receiving β-gal–transduced DCs showed a significant reduction in pulmonary metastases compared with mice receiving HER-2/neu–transduced DCs or βgP-pulsed splenocytes at all doses tested. Increasing the dose of β-gal–transduced DCs from 4 × 105 to 1.6 × 106 did not significantly enhance therapeutic efficacy. Identical studies with Ld-restricted βgP-pulsed DCs also failed to show a significant correlation between antitumor activity and increased DC dose in this model (data not shown). These studies demonstrate that in this rapidly lethal tumor model, β-gal–transduced DCs (DC–β-gal), and βgP-pulsed DCs (DC–βgP) were both capable of mediating significant, specific antitumor activity.
Generation of β-gal–specific CTLs by In Vivo Immunization with Bone Marrow–derived DCs.
To determine whether antigen-specific CTLs were generated in vivo after DC administration, naive, nontumor-bearing mice were immunized on day 0 and day 3 with 4 × 105 peptide-pulsed DCs, 4 × 105 β-gal–transduced DCs, or βgP-pulsed splenocytes intravenously. 3–4 wk after the second immunization, splenocytes from immunized mice were restimulated in vitro with βgP for 6 d. On day 7, restimulated splenocytes were cocultured with tumor targets, and 24-h IFN-γ release was measured (Table 3).
Table 3.
Cytokine Release From CTL Generated In Vivo by Immunization with Bone Marrow–derived DCs
| Splenocytes from mice immunized with: | Stimulators | |||||
|---|---|---|---|---|---|---|
| mIFN-γ (pg/ml/24 h) | ||||||
| CT26/P1A* | CT26/βgP‡ | CT26.CL25 | ||||
| HBSS | 0 | 0 | 0 | |||
| DC–β-gal transduced | 266 | 216,700 | 151,100 | |||
| DC–Neu transduced | 164 | 210 | 372 | |||
| Splen–βgP pulsed | 0 | 0 | 0 | |||
| DC–βgP pulsed | 5 | 1197 | 1436 | |||
| DC–P1A pulsed | 420 | 351 | 477 | |||
CT26 tumor cells pulsed with P1A peptide (10 μg/ml).
CT26 tumor cells pulsed with βgP (β-gal Ld peptide) (10 μg/ml). Splenocytes from animals immunized with transduced or peptide-pulsed DCs or peptide-pulsed splenocytes were pooled and restimulated in vitro with the Ld-restricted β-gal peptide (βgP) for 6 d in EHAA media with 30 IU/ml IL-2 (as described in Materials and Methods). On day 7, restimulated splenocytes (1 × 105) were plated in flat-bottomed 96-well tissue culture plates with 105 peptide-pulsed tumor targets. After 24 h, the supernatant was collected and analyzed for IFN-γ content.
Splenocytes from mice immunized with either β-gal– transduced or βgP-pulsed DCs induced antigen-specific cytokine release; however, the T cells generated in mice immunized with β-gal–transduced DCs produced significantly more IFN-γ than T cells from mice immunized with βgP-pulsed DCs. Immunization with control DCs or peptide-pulsed splenocytes failed to generate detectable CTL activity. These results were observed in three independent experiments.
The ability of the CTL to lyse relevant tumor targets was also assessed (Fig. 5). CTLs grown from mice immunized with β-gal–transduced DCs demonstrated antigen-specific lytic activity, whereas CTLs from mice immunized with βgP-pulsed DCs were minimally reactive. Immunization with control DCs or βgP-pulsed splenocytes failed to generate lytic CTLs.
Figure 5.

Lysis of tumor targets by CTLs from mice immunized with transduced (DC–β-gal and DC–Neu) or peptide-pulsed DCs (DC–βgP and DC–P1A) or peptide-pulsed splenocytes (splen–βgP). Splenocytes from immunized animals were harvested, restimulated in vitro for 7 d with the Ld-restricted β-gal peptide876–884, and mixed in graduated doses with 5 × 103 51Cr-labeled target cells. Cells were incubated for 4 h at 37°C in a 5% CO2 humidified incubator. 51Cr released from the lysed target cells was measured. Results are plotted as the mean percent specific lysis ± SEM.
Discussion
The identification of tumor rejection antigens recognized by cytotoxic T cells has led to the development of vaccination strategies aimed at generating an immune response capable of mediating tumor regression in cancer patients (23). As potent APCs capable of initiating immune responses from quiescent T cells, DCs pulsed with peptides from TAA have been successful in murine models in generating antitumor immunity and treating established tumor (2–7). Retroviral gene modification of DCs may offer important advantages over other methods of immunization; perhaps the most important of these being stable, prolonged expression of the full-length antigen leading to presentation of multiple epitopes in the context of MHC class I molecules. To examine the potential therapeutic efficacy of such gene-modified DCs, we used the well-characterized murine β-gal tumor model (24, 25).
In generating retrovirally transduced DCs, we took advantage of the suitability of dividing bone marrow cells as targets for retroviral gene modification. Gene-modified cells were then differentiated in vitro to DCs with cytokine support. This approach for the transduction of DCs has been used successfully in human CD34+ hematopoietic progenitor cells to generate DCs that stably express the MART-1 melanoma antigen (17). The data presented here provide evidence that primary murine DCs can be stably gene modified by retroviral transduction to express a model TAA gene.
The ability of the transduced DCs to process and present endogenously expressed β-gal was evidenced by recognition and specific cytokine release by β-gal–specific T cells. The difference observed in IFN-γ release in response to β-gal–transduced DCs and βgP-pulsed DCs is attributable to the high concentration of peptide pulsed onto the DCs compared with the transduced DCs, where the antigen is processed and presented at physiologic levels. A similar effect is also observed with the peptide-pulsed splenocytes in this assay.
Although we saw no difference in specific antitumor activity between β-gal–transduced and β-gal peptide-pulsed DCs under the conditions tested, it is possible that further titrations of DCs and tumor dose might reveal a difference in antitumor response between these two treatments. In fact, immunization with β-gal–transduced DCs generated CTLs that were significantly more reactive in vitro than CTLs grown from mice immunized with β-gal peptide-pulsed DCs. The expression and presentation of multiple epitopes in the β-gal–transduced DCs might lead to the generation of CTLs with multiple specificities. The presence of such additional CTL populations in the restimulated T cells might have contributed to the enhanced reactivity observed in cytolytic activity. In addition, the difference in CTL reactivity may be due to the prolonged presentation of β-gal in vivo by β-gal–transduced DCs compared with peptide-pulsed DCs. An increased duration of antigen presentation could lead to the generation of a greater number of CTLs, the expansion of which may require a longer period of time than the rapidly lethal, 12-d pulmonary metastases model will allow. We are currently attempting development of longer tumor models to determine whether transduced DCs might have enhanced in vivo activity compared with peptide-pulsed DCs.
Our current and future efforts are focused on the development of tumor models that more closely approximate the characteristics of the immune response to tumor in patients. The cloning and characterization of several shared, human, melanoma-associated antigens that are recognized by T cells, including tyrosinase, tyrosinase-related protein-1 (TRP-1), MART-1, and gp100, provide opportunities for studying the ability of retrovirally transduced DCs to induce an immune response against nonmutated, self-antigens (26–29). In the mouse, TRP-2 has recently been identified as a nonmutated, melanoma-associated antigen in B16 melanoma (30). We have begun construction of retroviral vectors encoding the murine melanoma-associated antigens, TRP-2 and gp100, expressed in B16 melanoma. The ability of transduced DCs to immunize against and/or treat this nonimmunogenic tumor may be a useful predictor of the potential efficacy of retrovirally transduced DCs in humans.
Approaches designed to enhance the immunostimulatory function of DCs are also being explored. These include transduction of DCs with cytokine genes in an attempt to enhance DC immunogenicity. Activation of DCs by CD40–CD40 ligand association, as well as the use of adjuvant cytokines such as GM-CSF and IL-12, in vivo are also being investigated.
The ability to transduce a primary DC retrovirally to express stably a foreign gene also introduces a novel, potentially powerful, approach to studying the mechanisms of DC activation and differentiation as well as protein trafficking and antigen processing in primary DCs. Retroviral transduction of genes encoding proteins containing intracellular targeting sequences, such as the hexapeptide present in melanosomal membrane proteins gp75 and gp100 (31), might allow efficient trafficking of protein antigens to the MHC class II endosomal pathway, resulting in presentation of antigen epitopes in the context of MHC class I and II molecules. The efficient presentation of antigen in association with both class I and class II may allow the initiation of a more potent immune response.
In summary, we present here evidence that murine DCs can be retrovirally transduced to express stably a model TAA gene. TAA-transduced DCs generated by this method express the transgene at high levels, and are capable of processing and presenting the antigen in the context of MHC class I molecules. Immunization with gene-modified DCs results in the production of highly reactive, antigen-specific CTLs. Finally, treatment with TAA-transduced DCs is capable of mediating effective antitumor activity against established pulmonary metastases. These results suggest that TAA-transduced DCs may be a promising treatment modality in tumor immunotherapy.
Acknowledgments
The authors thank A. Mixon and E. Fitzgerald for assistance in FACS® analyses, and P. Spiess and D. Jones for their assistance with animal experiments.
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; βgP, β-gal peptide; β2m, β2-microglobulin; CM, cytotoxicity medium; DC, dendritic cell; rm, recombinant murine; TAA, tumor-associated antigen.
References
- 1.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 2.Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CJ, Ildstad ST, Kast WM, DeLeo AB, Lotze MT. Bone marrow–derived dendritic cells pulsed with synthetic tumor peptides elicit protective and therapeutic antitumor immunity. Nat Med. 1995;1:1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 3.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD. Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med. 1996;183:283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med. 1996;183:317–322. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell. J Exp Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayordomo JI, Loftus DJ, Sakamoto H, De Cesare CM, Appasamy PM, Lotze MT, Storkus WJ, Appella E, DeLeo AB. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183:1357–1365. doi: 10.1084/jem.183.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pogador A, Snyder D, Gilboa E. Induction of antitumor immunity using bone marrow–generated dendritic cells. J Immunol. 1996;156:2918–2926. [PubMed] [Google Scholar]
- 8.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 9.Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 10.Dunbar CE, Seidel NE, Doren S, Sellers S, Cline AP, Metzger ME, Agricola BA, Donahue RE, Bodine DM. Improved retroviral gene transfer into murine and Rhesus peripheral blood or bone marrow repopulating cells primed in vivo with stem cell factor and granulocyte colony–stimulating factor. Proc Natl Acad Sci USA. 1996;21:11871–11876. doi: 10.1073/pnas.93.21.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nienhuis AW. Gene transfer into hematopoietic stem cells. Blood Cells. 1994;20:141–148. [PubMed] [Google Scholar]
- 12.Cassel A, Cottler-Fox M, Doren S, Dunbar CE. Retroviral-mediated gene transfer into CD 34-enriched human peripheral blood stem cells. Exp Hematol. 1993;21:585–591. [PubMed] [Google Scholar]
- 13.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witmer-Pack MD, Olivier W, Valinsky J, Schuler G, Steinman RM. Granulocyte/macrophage colony-stimulating factor is essential for the viability and function of cultured murine epidermal Langerhans cells. J Exp Med. 1987;166:1484–1498. doi: 10.1084/jem.166.5.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inaba K, Steinman RM, Witmer-Pack M, Aya K, Inaba M, Sudo T, Wolpe S, Schuler G. Identification of proliferating dendritic cell precursors in mouse blood. J Exp Med. 1992;175:1157–1167. doi: 10.1084/jem.175.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature (Lond) 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 17.Reeves ME, Royal RE, Lam JS, Rosenberg SA, Hwu P. Retroviral transduction of human dendritic cells with a tumor-associated antigen gene. Cancer Res. 1996;56:5672–5677. [PubMed] [Google Scholar]
- 18.Henderson RA, Nimgaonkar MT, Watkins SC, Robbins PD, Ball ED, Finn OJ. Human dendritic cells genetically engineered to express high levels of the human epithelial tumor antigen mucin (MUC-1) Cancer Res. 1996;56:3763–3770. [PubMed] [Google Scholar]
- 19.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 20.Wang M, Chen PW, Bronte V, Rosenberg SA, Restifo NP. Anti-tumor activity of cytotoxic T lymphocytes elicited with recombinant and synthetic forms of a model tumor-associated antigen. J Immunother Emphasis Tumor Immunol. 1995;18:139–146. doi: 10.1097/00002371-199510000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danos O, Mulligan RC. Safe and efficient generation of recombinant retroviruses with amphotropic and ecotropic host ranges. Proc Natl Acad Sci USA. 1988;85:6460–6464. doi: 10.1073/pnas.85.17.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1995;151:3971–3980. [PubMed] [Google Scholar]
- 23.Rosenberg SA. The development of new cancer therapies based on the molecular identification of cancer regression antigens. Cancer J Sci Am. 1995;1:90–100. [PubMed] [Google Scholar]
- 24.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA, Restifo NP. IL-2 enhances the function of recombinant poxvirus–based vaccines in the treatment of established pulmonary metastases. J Immunol. 1995;154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 25.Irvine KR, Rao JB, Rosenberg SA, Restifo NP. Cytokine enhancement of DNA immunization leads to effective treatment of established pulmonary metastases. J Immunol. 1996;156:238–245. [PMC free article] [PubMed] [Google Scholar]
- 26.Robbins PF, El-Gamil M, Kawakami Y, Stevens E, Yannelli JR, Rosenberg SA. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy (erratum published 54:3952) Cancer Res. 1994;54:3124–3126. [PubMed] [Google Scholar]
- 27.Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A3–restricted tumor–infiltrating lymphocytes (erratum published 181: 1261) J Exp Med. 1995;181:799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, Rosenberg SA. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6452. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, El-Gamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vijayasaradhi S, Bouchard B, Houghton AN. Intracellular sorting and targeting of melansomal membrane proteins: identification of signals for sorting of the human brown locus protein, gp75. J Cell Biol. 1995;130:807–820. doi: 10.1083/jcb.130.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]