

Abstract
MHC class I molecules load antigenic peptides in the endoplasmic reticulum and present them at the cell surface. Efficiency of peptide loading depends on the class I allele and can involve interaction with tapasin and other proteins of the loading complex. Allele HLA-B*4402 (Asp at position 116) depends on tapasin for efficient peptide loading, whereas HLA-B*4405 (identical to B*4402 except for Tyr116) can efficiently load peptides in the absence of tapasin. Both alleles adopt very similar structures in the presence of the same peptide. Comparative unrestrained molecular dynamics simulations on the α1/α2 peptide binding domains performed in the presence of bound peptides resulted in structures in close agreement with experiments for both alleles. In the absence of peptides, allele-specific conformational changes occurred in the first segment of the α2-helix that flanks the peptide C-terminal binding region (F-pocket) and contacts residue 116. This segment is also close to the proposed tapasin contact region. For B*4402, a shift toward an altered F-pocket structure deviating significantly from the bound form was observed. Subsequent free energy simulations on induced F-pocket opening in B*4402 confirmed a conformation that deviated significantly from the bound structure. For B*4405, a free energy minimum close to the bound structure was found. The simulations suggest that B*4405 has a greater tendency to adopt a peptide receptive conformation in the absence of peptide, allowing tapasin-independent peptide loading. A possible role of tapasin could be the stabilization of a peptide-receptive class I conformation for HLA-B*4402 and other tapasin-dependent alleles.
Keywords: MHC class I flexibility, empty class I structure, peptide loading, tapasin class I interaction, class I, protein dynamics, molecular simulation
Major histocompatibility complex (MHC) class I glycoproteins play a key role in the recognition of pathogens by presenting antigenic peptides at the cell surface. Class I molecules consist of three domains (α1, α2, α3) as well as a C-terminal membrane anchor region. Before they appear on the outer site of the cell membrane, MHC class I molecules associate with β2-microglobulin and an antigenic peptide consisting of 8–14 amino acids (Bouvier 2003; Ellgaard and Helenius 2003; Wright et al. 2004). These peptides are bound in a cleft formed by two α-helices (α1 and α2) on top of an extended, anti-parallel β-sheet at the α1/α2 domain (Madden 1995). Among the three class I subtypes in humans, HLA-A and HLA-B alleles play the largest role in presenting antigenic peptides at the cell surface and show the greatest polymorphic variation. The polymorphic variance is needed to enable the binding of HLA-A and HLA-B molecules to a large number of different peptides (every subtype binds to a specific set; Zhang et al. 1998). Despite the large polymorphic variance among HLA-A/B molecules, the basic concept of peptide binding is well conserved (Madden 1995). Side chains of specific residues point directly toward the β-sheet at the bottom of the α1/α2 domain and establish hydrogen bonds to residues within the β-sheet as well as the surrounding α-helices. These specific residues are always the C-terminal amino acid and additionally the residue either at position two or five of the peptide. These anchor residues are mainly responsible for a stable binding of the peptide to the class I binding cleft. Therefore, they are used to define binding motifs that are specific for particular MHC class I alleles. The formation of stable peptide-bound MHC class I complexes occurs in the endoplasmic reticulum (ER) within the peptide loading complex (PLC). This process consists of several steps that involve the interaction with several chaperone-like proteins: calnexin, calreticulin, tapasin, the protein disulfide isomerase ERp57, and the peptide transporter TAP (Transporter associated with Antigen Processing) (for reviews, see Bouvier 2003; Ellgaard and Helenius 2003; Wright et al. 2004). Tapasin plays a key role in the PLC by acting as a link between class I molecules and TAP (Lehner et al. 1998). Tapasin is also involved in stabilizing TAP, and appears to facilitate peptide binding to MHC molecules and to edit bound peptides (Williams et al. 2002; Zarling et al. 2003; Howarth et al. 2004). Editing refers to the observation that in the presence of tapasin the peptide cargo of class I molecules can shift to binding of higher-affinity peptides (Williams et al. 2002). The structural details of class I molecules interacting with tapasin have been investigated in experiments using point-mutated MHC proteins and tapasin or truncated tapasin molecules. Still, there are no X-ray crystallography data of MHC class I–tapasin complexes available, but experiments showed that residues in a loop below the α2-helix of the class I molecule consisting of amino acids 128–134 are crucial for tapasin binding (Lewis et al. 1996; Zernich et al. 2004) Another important region for forming a stable complex with tapasin is located in a loop consisting of residues 222–229 in the α3-domain of the MHC protein heavy chain (Suh et al. 1998).
Although most HLA molecules benefit from the presence of tapasin in the ER, there are some alleles that show reduced dependence or even complete tapasin independence (Williams et al. 2002; Park et al. 2003; Zernich et al. 2004). Interestingly, single amino acid differences between two alleles can have a major impact on the tapasin dependence of class I alleles (Williams et al. 2002). A particularly interesting example is the allele HLA-B*4405, which was shown to form stable complexes with antigenic peptides and exhibits cell surface presentation in cells that lack tapasin (Williams et al. 2002; Zernich et al. 2004). In contrast, under the same conditions cells with the closely related allele HLA-B*4402 showed strongly reduced presentation of peptides at the cell surface (in the absence of tapasin). Both alleles differ only by a single residue at position 116 (a tyrosine in case of HLA-B*4405 and an aspartate in case of HLA-B*4402) at the bottom of the region that binds the peptide C terminus (F-pocket; Garrett et al. 1989) and located distant from the proposed tapasin-binding region of class I. The crystal structure of both alleles in complex with the same high-affinity peptide has been determined (MacDonald et al. 2003; Zernich et al. 2004). In both structures, residue 116 is buried at the interface to the peptide, and there are no structural differences between the two alleles that could explain differences in tapasin dependence (MacDonald et al. 2003; Zernich et al. 2004). However, tapasin may interact and stabilize the unbound form of class I molecules, and residue 116 may have an influence on the structure and dynamics of the class I molecules in the absence of peptides.
In a previous comparative molecular dynamics (MD) simulation study on a HLA-A allele, we found that especially the C-terminal part of the α2-helix (termed α2–1-helix) showed enhanced flexibility in the absence of a bound peptide (Zacharias and Springer 2004). This helical segment was also found to be the most mobile segment in a comparative analysis of several crystal structures of class I molecules (Elliott 1997). In addition, in a recent crystal structure analysis of a class I homolog in the empty state (without a bound ligand), electron density for only part of the α2–1-helix was found (Olson et al. 2005), indicating either multiple conformations or increased flexibility of the segment. On one side the α2–1-helix contacts residue 116, whereas the other side is close to or may even overlap with the proposed tapasin binding site (Lewis et al. 1996). In the present study, we have employed MD simulations on the two alleles (HLA-B*4402 and HLA-B*4405), both in the absence and presence of bound peptides, starting from X-ray structures to characterize possible structural and dynamic differences. Simulations were restricted to the α1/α2 domain that has been shown to bind peptides (Rigney et al. 1998). This restriction was necessary in order to perform several fairly long and independent simulations and to calculate the free energy of opening the F-pocket binding region for both alleles. It is assumed that although the α3 domain and β2-microglobulin influence the flexibility of class I molecules and peptide binding properties, this influence is the same for the two alleles since both alleles differ only in one residue in the α1/α2 domain that is far from any interface to the α3 domain or β2-microglobulin.
No significant differences in the average structure and dynamics between the two alleles in the peptide bound forms were found. However, in the absence of peptide, significant differences in the dynamics of the F-pocket region were observed, with a greater tendency of the B*4402 allele to adopt a more open F-pocket structure that significantly differed from the “bound” form. In contrast, the B*4405 allele stayed close to the conformation in the “bound” structure even in the absence of a peptide. The results of independent unrestrained MD simulations were confirmed using free energy simulations to enforce opening of the F-pocket. The simulation result indicates that tapasin-independent class I alleles may have a greater tendency to adopt a conformation close to the peptide-bound state (receptive form) already, in the absence of peptides. This may have significant implications for understanding peptide binding to class I molecules in the absence of tapasin and for understanding the mechanism of peptide loading to class I molecules.
Results
Deviation of simulated structures from the experimental start structure
Comparative molecular dynamics simulations on the two MHC class I alleles HLA-B*4402 and HLA-B*4405 (Fig. 1) in the absence and presence of bound peptides were performed. The two alleles differ only in residue 116 (Asp in B*4402 and Tyr in B*4405) and show significant differences in the dependence on tapasin for effective peptide loading (Williams et al. 2002). For both alleles, high-resolution crystal structures in complex with the same antigenic peptide (sequence: EEFGRAFSF) are available (MacDonald et al. 2003; Zernich et al. 2004) and served as start structures for the simulations with and without bound peptides (see Materials and Methods). Simulations were restricted to the α1/α2 peptide binding domain in order to perform independent simulations with different initial conditions. For each molecule and each state, two independent simulations at 300K starting from different starting velocities were conducted up to a simulation time of 21 nsec. In the presence of bound peptide, the B*4402 and B*4405 alleles reached a stable main chain RMSD (root-mean-square deviation) of 1.7–2.3 Å, respectively, after ∼3 nsec with respect to the start structure (Fig. 2). The simulations on allele B*4402 (with peptide) showed on average a slightly larger RMSD deviation (∼0.3 Å) from the start structure than allele B*4405. Neither dissociation of bound peptides nor significant structural differences near residue 116 between both alleles were observed in the presence of the peptides. A superposition of average structures of the simulations in the presence of peptides with respect to the start structure indicated stable binding of the anchor residues at positions 2 and 9 with little deviation from the start conformation (Fig. 3) and supported the quality of the force field simulation approach. The average RMSD of the structures without peptides reached in both cases higher but also stable levels (up to 2.7–3.0 Å; Fig. 2). Nonuniform conformational fluctuations along the protein chain were observed (Fig. 4). The β-sheet regions that form the floor of the peptide binding cleft showed the smallest conformational fluctuations and little difference between peptide-bound and free forms (Fig. 4). Also, the region that forms the interface to the β2-microglobulin subunit underwent only small fluctuations (Fig. 4). The α-helical regions showed larger fluctuations, in particular during the simulations in the absence of peptide. The short α2–2-segment transiently lost its α-helical structure (not shown), and for the region around the α2–1-helix, but also around the N terminus of the α1–helix, an enhanced flexibility and drift from the start structure toward a more open structure was observed (Figs. 4, 5). The residues that contact the peptide N terminus and peptide C terminus, respectively, stayed close to the arrangement observed in the X-ray start structures during the simulations in the presence of peptides (Table 1). In the absence of peptides, an increased deviation from the start structure was observed (for both pocket residues), but a larger deviation of the F-pocket-forming residues from the X-ray structure was seen in the B*4402 allele compared with the B*4405 allele. In both cases, the F-pocket stayed in an open and accessible conformation; however, on average, the B*4402 allele adopted a more open structure around the F-pocket compared with the B*4405 allele (Fig. 5).
Figure 1.
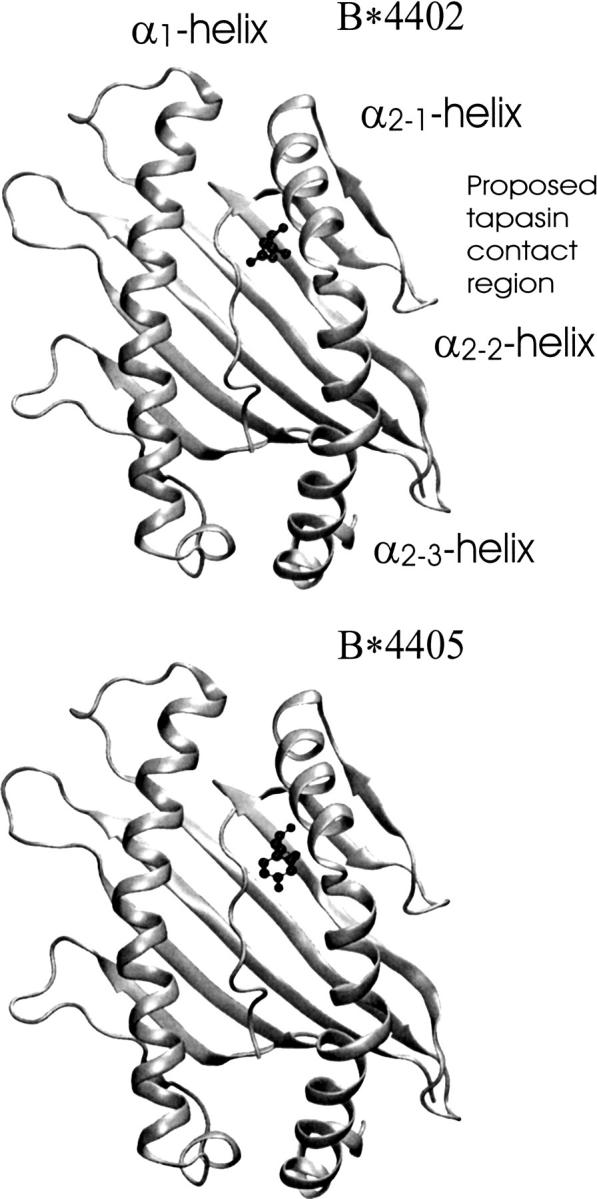
Top view of the α1/α2 peptide binding domain of MHC class I molecules HLA-B*4402 (PDB entry 1M6O) and HLA-B*4405 (PDB entry 1SYV). Both molecules are complexed with an antigenic peptide consisting of nine residues (peptide sequence: EEFGRAFSF). The two alleles differ only at residue 116, which is given in ball-and-stick representation (B*4402, aspartate; B*4405, tyrosine). The proposed tapasin contact region and the α-helical segments are indicated (for B*4402).
Figure 2.

RMSD time course (moving average) for backbone atoms of the α1/α2-domains of B*4402 and B*4405 with respect to the X-ray start structure. Time courses are shown over 21-nsec trajectories (two independent simulations) for the peptide-bound (black) and ligand-free forms (gray), respectively. (Thick lines) Average of two independent simulations.
Figure 3.

Superposition of average peptide-bound structures (approximately the same point of view) obtained during the MD simulations (gray) on the crystal start structure (black) of B*4402 and B*4405.
Figure 4.

Conformational flexibility of the α1/α2-domain of HLA-B*4402 and HLA-B*4405 in complex with an antigenic peptide (peptide bound) and in the absence of peptide (peptide free). Averages of mean square fluctuations per residue are shown for the two independent simulations at 300 K.
Figure 5.

Superposition of the average structure obtained from the simulation of HLA B*4402 in the absence of a peptide (gray) on the X-ray start structure (black). (Straight light-gray lines) Helical segments that flank the F-pocket (binding region for the peptide C-terminal anchor) and segments used to monitor the F-pocket opening and to drive F-pocket opening during free energy simulations (residues 74–85 and 138–149, respectively); (light-gray double arrows) increased distance between the two segments observed during the unrestrained simulations.
Table 1.
Average root mean square deviation (RMSD, in Å) of class I residues that contact the peptide C-terminal and N-terminal anchor residues, respectively

Distance distribution of the α-helical segments surrounding the F-pocket
The mobility of the α2–1-helix and the conformational changes of the F-pocket during the simulations in the absence of peptide were further analyzed by monitoring the distance distribution of the center-of-mass of the α2–1-helix (residues 138–149) and the α1-helix segment on the opposite side of the F-pocket (residues 74–85). These two segments form the “walls” of the F-pocket region. Enhanced mobility of the α2–1-helix in the absence of peptide was observed already in a previous study on a HLA-A allele (Zacharias and Springer 2004). For both alleles, in the peptide-bound states the distance probability during the simulations adopted very similar nearly Gaussian distributions centered around a distance of 14 Å (in both independent simulations; Fig. 6). In simulations in the absence of peptides, significantly different distance distributions between the two α-helical segments were obtained (Fig. 6). Both simulations of HLA-B*4402 without peptide showed that this molecule favors a conformation where the binding cleft around the F-pocket is widened by ∼3 Å compared with the state with bound peptide (Fig. 5). The widening F-pocket does not provide the same contact surface seen in the bound state. Unlike B*4402, the simulations of B*4405 did not show a significant further opening of the binding cleft that was stable for more than a few nanoseconds. Instead, a still open and ligand-accessible conformation of the α-helical elements closer to the complexed state was favored (Fig. 6). Thus, only a smaller proportion of the observed conformations presented an opening of the binding cleft similar to B*4402, as indicated by the small local maximum at ∼16 Å. Direct comparison of the conformational changes of both simulated MHC class I molecules indicated that both proteins can conduct the same movements of the last part of the α1-helix and the α2–1-helix, but the probability of observing B*4405 in a conformation closer to the bound state was much higher than for B*4402.
Figure 6.

Distribution of the center-of-mass distance between the two α-helical segments (residues 74–85 of the α1-helix and residues 138–149 of the α2–1-helix) that flank the class I F-pocket observed during unrestrained MD simulations. Two independent simulations (thin continuous and dashed lines) were performed (each 21 nsec) for peptide-bound (black) and peptide-free (gray) class I molecules. (Thick lines) Sum of the distance distributions from the two independent simulations.
Umbrella sampling of F-pocket-opening
The distance distributions between the α1-helix and the α2–1-helix obtained from unrestrained MD simulations gave a qualitative indication of the opening tendency of the F-pocket for the two class I alleles. If one defines a state with a distance of ∼17 Å between the helices as open and with largely disrupted peptide binding interface (nonreceptive state) and a state with ∼14 Å as a receptive (“bound”) state, the distance distribution curves can be used to estimate a free energy difference between the two states for the two alleles. The comparison of the probabilities at the two states translates to an estimate for the free energy difference of ∼1–1.5 kcal mol−1 in favor of the “bound” state in the case of B*4405, and approximately the same in favor of the “unbound” state in the case of B*4402. It has been shown that folding of peptides or small proteins that involve several transitions in the peptide backbone can occur on the nanosecond timescale (Lapidus et al. 2002; Kubelka et al. 2006). The conformational shift of the α2–1-helical segment does not involve any major changes in the peptide backbone structure. However, due to the limited time scale of the simulations it is possible that a transition to a more open form seen in B*4402 may also occur in B*4405, but it has not been sufficiently sampled on the current nanosecond time scale. To investigate the conformational preference of helices that surround the F-pocket more quantitatively, umbrella sampling MD simulations were performed. In such a simulation, a transition between two states can be enforced along a reaction coordinate and the associated work or free energy change can be recorded. As a reaction coordinate, the same center-of-mass distance between the helical segments was employed that has already been used for the analysis of the unrestrained simulations. A quadratic distance-dependent restraining potential was used, and the reference distance between the centers of the helical segments was changed in 0.5-Å steps (see Materials and Methods for details). Employing the weighted histogram analysis method, it was possible to extract a free energy change along the reaction coordinate. In the case of the peptide-bound simulations, a free energy minimum at ∼14 Å was found for both alleles close to the distance that showed the highest probability during the unrestrained MD simulations (data not shown). In contrast to the peptide-bound forms, significant differences in the potentials of mean force for F-pocket opening of the peptide free states for the two alleles were observed (Fig. 7). In the case of B*4405, the free energy minimum was located at a distance of ∼14 Å between the helical segments (close to the distance with the maximum probability found in the case of the unrestrained simulations; Fig. 6). This distance is also close to the equilibrium distance observed during the simulations in the presence of peptide. Further closing of the F-pocket as well as opening are accompanied by an increase in free energy. The calculated free energy change for increasing the distance between the helical segments that flank the F-pocket in the case of B*4405 to ∼17 Å amounted to ∼2 kcal mol−1. In contrast, the calculations on B*4402 predicted an unfavorable “closed” state of the F-pocket with a distance between the helical segments close to the peptide-bound form. In this case a conformation of the F-pocket with favorable helical segment distances of 15–17 Å was favored (nonreceptive form). Simulations using different force constants in the umbrella sampling runs showed no quantitative agreement but gave qualitatively similar results (Fig. 7). This indicates that complete convergence of the free energy calculations may not be achieved on the present nanosecond time scale. However, all three simulations resulted in almost the same free energy differences between open (∼15–18 Å) and receptive (∼14 Å) states, and a very similar overall shape of the free energy curves. The results of the free energy simulations also compare well qualitatively with the histogram analysis of the α-helical elements surrounding the F-pocket during the unrestrained simulations. The positions of the probability maxima observed in the histograms (Fig. 6) coincide very well with the free energy minima obtained from the potential of mean force calculations (Fig. 7).
Figure 7.

Free energy vs. distance between helical segments that flank the F-pocket HLA-B*4402 (black) and HLA-B*4405 (gray) obtained during umbrella sampling simulations in the absence of peptides. Free energy changes were extracted from simulations with different reference distances (ranging from 12–18 Å in steps of 0.5 Å: 13 reference distances). Data gathering at each reference distance was performed for 3 nsec. (Continuous, dashed, and dotted lines) Free energy results were extracted from different intervals of the data-gathering simulation time at each reference distance (see graph legend).
Discussion
A single amino acid polymorphism that distinguishes HLA-B*4405 (Tyr116) from B*4402 (Asp116) permits B*4405 to acquire peptides in the ER largely independent of tapasin and the peptide-loading complex (Williams et al. 2002). Position 116 is located at the floor of the peptide binding site and is inaccessible in the peptide-bound state. The crystal structures of alleles HLA-B*4405 and B*4402 bound to the same peptide do not reveal any obvious structural differences that could explain the differences in tapasin dependence as well as the differences in association with the PLC of the two alleles (MacDonald et al. 2003; Zernich et al. 2004). Also, the proposed binding site of class I molecules to tapasin is located on the opposite side of the peptide binding groove and does not overlap with residue 116 (Lewis et al. 1996; Bouvier 2003). However, residues at position 116 can modulate the structure and dynamics of the class I structure in the absence of bound peptide, and this may in turn alter the association with the PLC. The aim of the present simulation study was to compare the dynamics of the peptide bound and free forms of the two alleles on the nanosecond time scale. In previous studies, the molecular dynamics method has been used to study the interaction between peptides and MHC class I molecules (Rognan et al. 1992a,b; Pohlmann et al. 2004) and the role of water molecules and water structure located at the class I–peptide interface (Meng et al. 1997, 2000; Petrone and Garcia 2004). The study by Petrone and Garcia (2004) also indicated a larger mobility of the HLA-A2 molecule in the absence of a peptide compared with simulations in the presence of a peptide from the HIV reverse transcriptase on the 5-nsec time scale. A previous comparative MD simulation study on the α1/α2 peptide binding domain of a HLA-A allele (Zacharias and Springer 2004) indicated that a helical region that forms part of the wall of the F-pocket (α2–1 segment) showed enhanced mobility in the absence of a bound peptide. The present simulation studies were also restricted to the α1/α2 peptide binding domain, which allowed us to perform several independent simulations and calculate a potential of mean force for an opening of the class I region responsible for binding the peptide C terminus (F-pocket). It is assumed that other parts of the class I molecule can modulate the mobility of the α1/α2 peptide binding domain, but the influence is the same for the two alleles since there are no sequence differences between the two alleles outside of the α1/α2 peptide domain. On the time scale of the current simulations, the secondary structure of the class I molecules was largely preserved during the simulations in the bound as well as peptide-free states without significant changes at the interface to β2-microglobulin. Overall modest conformational changes and fluctuations even in the unbound state, and no unfolding of the empty class I molecule during the simulations, were observed. This agrees with recent crystal structures on a class I molecule complexed with a partial peptide epitope (missing the anchor region at the peptide; Glithero et al. 2006) and with a crystal structure of an empty class I homolog (Olson et al. 2005). In both crystal structures, the overall fold of the class I molecules is preserved. It also agrees with experimental studies on refolded “empty” class I molecules that showed a circular dichroism (CD) spectrum similar to the bound form, indicating that both empty as well as bound class I structures have similar secondary structure content (Bouvier and Wiley 1998). The largest dynamical and structural differences between the two alleles and between peptide-bound and free states during the MD simulations were found for loop and α-helical segments. The floor of the peptide binding groove showed the smallest conformational fluctuations both in the absence and presence of peptides. The analysis was focused on the region near residue 116, which is part of the F-pocket, contacts the mobile α2–1 segment, and is the only residue that differs between the two alleles. The side of this segment pointing away from the peptide binding groove is close to the proposed region involved in interaction with tapasin (Fig. 1).
In two independent sets of simulations on the two alleles in the presence of bound peptide, the α2–1-segment stayed close to the start structure and showed similar mobility. However, significant differences were observed during independent simulations in the absence of peptide. In none of the simulations without peptide was a collapse or closing of the binding cleft observed. Instead, the α2–1-segment of the B*4402 allele preferred a more open conformation at the peptide C-terminal binding region compared with the simulation in the presence of the bound peptide. In this more open conformation, the arrangement of several residues responsible for anchoring the peptide C terminus deviates from the configuration in the bound (nonreceptive) form. In contrast, during the two independent simulations on B*4405, the peptide C-terminal binding region remained close to the conformation seen in the bound structure (receptive form). Since the α2–1-helix remained in a helical conformation even in the simulation of the unbound class I structures, the distance between the α2–1-helix and the α1-helix on the opposite side of the peptide binding groove could be used as a simple coordinate to monitor opening and closing of the peptide C-terminal binding region. The corresponding distance of the most populated states in the case of the bound structures and the B*4405 allele without peptide was ∼14 Å, compared with ∼17 Å in case of the B*4402 allele without peptide. Since the distance distribution curves obtained from unrestrained MD simulations can suffer from limited sampling on the nanosecond time scale, we used the same centers-of-mass distance between the two helical segments as the reaction coordinate to enforce the transition during umbrella sampling simulations (for a recent review on umbrella sampling and related free energy simulation methods, see Adock and McCammon 2006). The resulting free energy curves largely confirmed the probability distributions obtained during unrestrained MD simulations. The calculations predict that B*4405 prefers a conformation close to the bound form (peptide receptive form) over a more open form by ∼2 kcal mol−1. It should be emphasized that this form is also an “open” F-pocket structure accessible for ligand binding. Conversely, in the case of B*4402, a further opened conformation at the F-pocket is preferred by about the same free energy difference. In this form, however, the peptide contact interface as seen in the bound form is largely disrupted (nonreceptive). A differential behavior of the two alleles with respect to the stability of a peptide receptive form is not unexpected since the receptive form partially buries the Asp116 in the case of the B*4402 allele. In the “open” conformation, Asp116 is much more solvent-exposed, which contributes favorably to the stability of “open” B*4402 structures. In the case of B*4405, partial burying of Tyr116 (in the receptive form) is more favorable due to the hydrophobic aromatic ring of the Tyr side chain. The greater solvent exposure of the Tyr116 upon “opening” of the F-pocket adds an unfavorable contribution to F-pocket opening in the case of B*4405.
Additional experimental evidence that the region at the α2–1-helix is a mobile segment and may play a key role during peptide loading comes from the comparison of various peptide-bound class I structures (Madden 1995; Elliott 1997; Wright et al. 2004). The comparison of crystal structures indicated that the α2–1-segment can undergo conformational shifts similar to those seen in the present simulations as a response to allelic differences or different bound peptides (Elliott 1997). Also, in a recent crystal structure analysis of a class I homolog in the empty state (Olson et al. 2005), the α2–1-helix could not be fully defined due to the lack of electron density in this region, also indicating structural disorder or flexibility of the segment. Interestingly, a crystal structure of a mouse class I molecule with a partial peptide epitope that lacks the N-terminal anchor residues recently has been solved (Glithero et al. 2006). The crystal structure was found to be overall very similar to the structure with a full bound peptide. In class I refolding experiments, the presence of the partial epitope lacking the N-terminal anchor led to stabilization of the folded structure, whereas the corresponding partial peptide lacking the C-terminal anchor region had no effect. In addition, it was not possible to crystallize the class I molecule in the presence of the partial epitope that contained only the peptide N-terminal binding region (Glithero et al. 2006). These results can again be interpreted by an enhanced conformational flexibility of the F-pocket region in the absence of peptide (or a partial peptide that lacks the C-terminal anchor), which interferes with ordered crystallization.
The greater preference of B*4405 for a peptide-receptive conformation in the peptide-free state close to the bound class I structure offers a structural explanation for the striking functional difference of this allele compared with B*4402. Our calculations predict that the B*4405 allele has an intrinsic preference for a peptide-receptive structure already in the unbound state. During the relatively short time the class I molecules stay in the ER (∼20–60 min; Jackson et al. 1994), such a preference still allows peptides to load even without the assistance of the PLC (in particular, tapasin). In contrast, in the case of B*4402, a transition from an open state to a receptive state is necessary before peptide binding can occur (this means that the peptide-loading kinetics of HLA-B*4402 should significantly differ from those of HLA-B*4405 even if peptides are bound with the same affinity). A possible role of tapasin is to stabilize a peptide-receptive class I conformation in the case of B*4402 such that rapid peptide association becomes possible. In essence, the role of tapasin in such a model would be to shift the control of peptide association from kinetic to thermodynamic control. A kinetic control of peptide loading would mean that low- and high-affinity peptides have similar chances to fill the binding pocket, and since the majority of peptides in the ER are low-affinity peptides, this kind of control would increase the proportion of low-affinity peptides bound to class I molecules compared with a thermodynamic control. A prerequisite of thermodynamic control is high association (and dissociation) rates, such that peptide selection during the short time in the ER is mainly determined by the relative peptide binding affinity. In this model of tapasin function, the intrinsic preference for a peptide receptive conformation in the case of B*4405 would allow a more thermodynamic control of peptide loading in the absence of tapasin compared with B*4402 and hence would explain the tapasin-independence of this allele.
The tapasin dependence of only a few class I alleles has been investigated in detail so far. An interesting allele is B*2705, which also has an aspartate at position 116 and shows a reduced tapasin dependence (Park et al. 2003; Fiorillo et al. 2005; Goodall et al. 2006). However, in this case the neighboring residue (position 114) at the floor of the peptide binding groove is a His (Asp114 in B*4402 and B*4405). The His114 residue may form a salt bridge with Asp116 in the case of B*2705. Such a salt bridge is neutral overall, and the desolvation penalty for partial burying of a neutral structural element is much smaller than for a charged element (in the case of B*4402, both residues 114 and 116 are Asp). This hypothesis is further supported by the mutagenesis studies of Park et al. (2003). These investigators found a correlation of tapasin dependence with the nature of the amino acid present at the naturally polymorphic and also partially buried position 114. A substitution to a histidine at position 114 in the B*4402 allele (creating a His114/Asp116 pair) changed the phenotype and allowed loading of high-affinity peptides in the absence of tapasin as well as cell surface expression levels similar to the levels seen in the presence of tapasin (Park et al. 2003). Conversely, the opposite substitution (a His to Glu) at position 114 of allele B*2705 created a tapasin-dependent phenotype with a similar amino acid pattern as in the tapasin-dependent allele B*4402 (a Glu114/Asp116 pair). In line with the hypothesis outlined above, the two acidic residues in this mutant result in a much larger desolvation penalty to partially burying both residues in the peptide receptive form compared with a His-Asp/Glu pair and hence lead to the stabilization of a more open structure around the F-pocket in the unbound state.
Conclusions
The comparative MD simulation studies on a tapasin-dependent and a tapasin-independent allele indicate that differences in the structure and flexibility of class I molecules in the absence of bound peptide may play a key role in determining the dependence on tapasin during peptide loading. In particular, differential dynamics of α-helical regions that flank the F-pocket and contact the only residue that differs in the two alleles (residue 116) were observed. The tapasin-independent allele (HLA-B*4405) showed a greater tendency to adopt a conformation close to the peptide-bound state (receptive state) during both unrestrained and umbrella sampling free energy simulations compared with the tapasin-dependent allele (HLA-B*4402). The simulation results indicate the presence of a significant portion of HLA-B*4405 molecules in the receptive state in the absence of tapasin that could allow this allele to load peptides without association to the PLC or interaction with tapasin. In this model, a possible role for tapasin would be to stabilize a conformation close to the receptive structure in the case of HLA-B*4402. The basis of the model can be further investigated by future simulation studies on other class I alleles that differ in tapasin dependence as well as experimental studies on the peptide loading kinetics of class I alleles.
Materials and methods
Molecular dynamics simulations
Comparative molecular dynamics simulations were performed on the crystal structures of α1/α2 domains (residues 1–182) of human class I alleles HLA-B*4402 (MacDonald et al. 2003; protein databank entry: pdb1M6O) and HLA-B*4405 (Zernich et al. 2004; pdb1SYV; Fig. 1). Both class I molecules differ only at position 116 (HLA-B*4402: Asp, HLA-B*4405: Tyr) and have been crystallized in the presence of the same bound peptide (sequence: EEFGRAFSF). The bound peptide represents a high-affinity ligand derived from HLA-DPA*0201, residues 46–54 (MacDonald et al. 2003). Simulations were performed either in the presence or the absence of the antigenic peptide using the sander module of the Amber8 software package (Case et al. 2004) in combination with the parm03 force field (Duan et al. 2003). The proteins were placed into rectangular boxes with periodic boundary conditions together with 24 additional sodium and chloride counter-ions and roughly 9000 TIP3 water molecules (Jorgensen et al. 1983). Short-range nonbonded interactions were taken into account up to a cut-off value of 9 Å. Long-range electrostatic interactions were treated employing the particle mesh Ewald option (Darden et al. 1993) using a grid spacing of ∼0.9 Å. The structures were energy minimized and heated up from 50 to 300K within 0.1 nsec using positional restraints on solute atoms (force constant: 50 kcal mol−1 Å−2) followed by a step-wise removal of the positional restraints on the solute within another 0.1 nsec. Subsequently, all systems consisting of HLA-B*4402 and HLA-B*4405, respectively, in bound and peptide-free states were simulated for 21 nsec. During all simulations temperature and pressure (1 bar) were controlled using a relaxation time of 5 psec. Coordinates were stored every 2 psec for further analysis. In order to control the dependence of the results on the starting conditions, in each case two separate simulations with different initial velocities were performed. Visualization of trajectories and preparation of figures was performed using VMD (Visual molecular dynamics; Humphrey et al. 1996).
Free energy simulations of F-pocket opening
Umbrella sampling simulations (for review, see Adock and McCammon 2006) were used in order to enforce opening of the binding region of the peptide C terminus (F-pocket). As a reaction coordinate for the umbrella sampling simulations a distance restraint between two groups of atoms was used. The two groups corresponded to the backbone (heavy) atoms of the two α-helical segments that flank the F-pocket (residues 74–85 and 138–149, respectively). Starting from the equilibrium distance of the two segments (∼15 Å), a quadratic restraining potential was used to enforce opening (and closing) of the F-pocket region. The reference distance in the restraining potential was changed in steps of 0.5 Å to cover a range of distances from 12–18 Å. A step in the restraining distance was taken every 1 nsec. Data gathering was performed during 3-nsec simulation for each restraining window. During the first 2 nsec a restraining force constant of 0.5 kcal mol−1 Å−2 was employed followed by 2.0 kcal mol−1 Å−1 to control the influence on the calculated free energy profile. The recorded distance probabilities for each simulation window were then used to create a free energy profile using the weighted histogram analysis method (WHAM) (Kumar et al. 1992; implemented by Grossfield, http://dasher.wustl.edu/alan/).
Acknowledgments
We thank A. May, C. Schneeweiß, and Drs. A Barthel, D. Roccatano, and T. Elliott for helpful discussions. This work was performed using the computational resources of the CLAMV (Computational Laboratories for Animation, Modeling and Visualization) at IUB and supercomputer resources of the EMSL (Environmental Molecular Science Laboratories) at the PNNL (Pacific Northwest National Laboratories; grant gc11-2002).
Footnotes
Reprint requests to: Martin Zacharias, International University Bremen, School of Engineering and Science, Campus Ring 6, D-28759 Bremen, Germany; e-mail: m.zacharias@iu-bremen.de; fax: +49-421-2003249.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062568407.
References
- Adock, S.A. and McCammon, J.A. 2006. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 106: 1589–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier, M. 2003. Accessory proteins and the assembly of human class I MHC molecules: A molecular and structural perspective. Mol. Immunol. 39: 697–706. [DOI] [PubMed] [Google Scholar]
- Bouvier, M. and Wiley, D.C. 1998. Structural characterization of a soluble and partial folded class I major histocompatibility chain/β2m heterodimer. Nat. Struct. Biol. 5: 377–384. [DOI] [PubMed] [Google Scholar]
- Case, D.A., Darden, T.A., Cheatham III, T.E., Simmerling, C.L., Wang, J., Duke, R.E., Luo, R., Merz, K.M., Wang, B., and Pearlman, D.A., et al. 2004. AMBER8. University of California, San Francisco.
- Darden, T., York, D., and Pedersen, L. 1993. Particle mesh Ewald: An NlogN method for Ewald sums in large systems. J. Chem. Phys. 98: 10089–10092. [Google Scholar]
- Duan, Y., Wu, C., Chowdhury, S., Lee, M.C., Xiong, G., Zhang, W., Yang, R., Cieplak, P., Luo, R., and Lee, T., et al. 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24: 1999–2012. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L. and Helenius, A. 2003. Quality control in the endoplasmic reticulum. Nature Reviews Mol. Cell. Biol. 4: 181–191. [DOI] [PubMed] [Google Scholar]
- Elliott, T. 1997. How does TAP associate with MHC class I molecules? Immunol. Today 18: 375–378. [DOI] [PubMed] [Google Scholar]
- Fiorillo, M.T., Ruckert, C., Hulsmeyer, M., Sorrentino, R., Saenger, W., Ziegler, A., and Uchanska-Ziegler, B. 2005. Allele-dependent similarity between viral and self-peptide presentation by HLA-B27 subtypes. J. Biol. Chem. 280: 2962–2971. [DOI] [PubMed] [Google Scholar]
- Garrett, T.P., Saper, M.A., Bjorkman, P.J., Strominger, J.L., and Wiley, D.C. 1989. Specificity pockets for the side chains of peptide antigens in HLA-Aw68. Nature 392: 692–696. [DOI] [PubMed] [Google Scholar]
- Glithero, A., Tormo, J., Doering, K., Kojima, M., Jones, E.Y., and Elliott, T. 2006. The crystal structure of H-2Db complexed with a partial peptide epitope suggests an MHC class I assembly-intermediate. J. Biol. Chem. 281: 12699–12704. [DOI] [PubMed] [Google Scholar]
- Goodall, J.C., Ellis, L., and Hill Gaston, J.S. 2006. Spondylarthritis-associated and non-spondylarthritis-associated B27 subtypes differ in their dependence upon tapasin for surface expression and their incorporation into the peptide loading complex. Arthritis Rheum. 54: 138–147. [DOI] [PubMed] [Google Scholar]
- Howarth, M., Williams, A., Tolstrup, A.B., and Elliott, T. 2004. Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc. Natl. Acad. Sci. 101: 11737–11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD—Visual Molecular Dynamics. J. Mol. Graph. 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Jackson, M.R., Cohen-Doyle, M.F., Peterson, P.A., and Williams, D.B. 1994. Regulation of MHC class I transport by the molecular chaperone, calnexin (p88, IP90). Science 263: 384–387. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandrasekhar, J., Madura, J., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79: 926–935. [Google Scholar]
- Kubelka, J., Chiu, T.K., Davies, D.R., Eaton, W.A., and Hofrichter, J. 2006. Sub-microsecond protein folding. J. Mol. Biol. 359: 546–553. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Rosenberg, J.M., Bouzida, D., Swendsen, R.H., and Kollman, P.A. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13: 1011–1021. [Google Scholar]
- Lapidus, L.J., Eaton, W.A., and Hofrichter, J. 2002. Measuring dynamic flexibility of the coil state of a helix forming peptide. J. Mol. Biol. 319: 19–25. [DOI] [PubMed] [Google Scholar]
- Lehner, P.J., Surman, M.J., and Cresswell, P. 1998. Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line.220. Immunity 8: 221–231. [DOI] [PubMed] [Google Scholar]
- Lewis, J.W., Neisig, A., Neefjes, J., and Elliott, T. 1996. Point mutations in the a2 domain of HLA-A2.1 define a functionally relevant interaction with TAP. Curr. Biol. 6: 873–883. [DOI] [PubMed] [Google Scholar]
- MacDonald, W.A., Purcell, A.W., Mifsud, N.A., Ely, L.K., Williams, D.S., Chang, L., Gorman, J.J., Clements, C.S., Kjer-Nielsen, L., and Koelle, D.M., et al. 2003. A naturally selected dimorphism within the Hla-B44 supertype alters class I structure, peptide repertoire, and T cell recognition. J. Exp. Med. 198: 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden, D.R. 1995. The three-dimensional structure of peptide-MHC-complexes. Annu. Rev. Immunol. 13: 587–622. [DOI] [PubMed] [Google Scholar]
- Meng, W.S., v. Grafenstein, H., and Haworth, I.S. 1997. A model of water structure inside the HLA-A2 peptide binding groove. Int. Immunol. 9: 1339–1346. [DOI] [PubMed] [Google Scholar]
- Meng, W.S., v. Grafenstein, H., and Haworth, I.S. 2000. Water dynamics at the binding interface of four different HLA-A2-peptide complexes. Int. Immunol. 12: 949–957. [DOI] [PubMed] [Google Scholar]
- Olson, R., Huey-Tubman, K.E., Dulac, C., and Bjorkman, P.J. 2005. Structure of a pheromone receptor-associated MHC molecule with an open and empty groove. PLoS Biol. 3: 1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, B., Lee, S., Kim, E., and Ahn, K. 2003. A single polymorphic residue within the peptide-binding cleft of MHC class I molecules determines spectrum of tapasin dependence. J. Immunol. 170: 961–968. [DOI] [PubMed] [Google Scholar]
- Petrone, P.M. and Garcia, A.E. 2004. MHC–peptide binding is assisted by bound water molecules. J. Mol. Biol. 338: 419–435. [DOI] [PubMed] [Google Scholar]
- Pohlmann, T., Bockmann, R.A., Grubmuller, H., Uchanska-Ziegler, B., Ziegler, A., and Alexiev, U. 2004. Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J. Biol. Chem. 279: 28197–28201. [DOI] [PubMed] [Google Scholar]
- Rigney, E., Kojima, M., Glithero, A., and Elliott, T. 1998. A soluble major histocompatibility complex class I peptide binding platform undergoes a conformational change in response to peptide epitopes. J. Biol. Chem. 273: 14200–14204. [DOI] [PubMed] [Google Scholar]
- Rognan, D., Zimmermann, N., Jung, G., and Folkers, G. 1992a. Molecular-dynamics study of a complex between the human histocompatibility antigen HLA-A2 and the Imp58-66 nona-peptide from influenza-virus matrix protein. Eur. J. Biochem. 208: 101–113. [DOI] [PubMed] [Google Scholar]
- Rognan, D., Scapozza, L., Folkers, G., and Daser, A. 1992b. Molecular-dynamics study of MHC-peptide complexes as a tool for predicting potential T-cell epitopes. Biochemistry 33: 11476–11485. [DOI] [PubMed] [Google Scholar]
- Suh, W.K., Derby, M.A., Cohen-Doyle, M.F., Schoenhals, G.J., Fruh, K., Berzofsky, J.A., and Williams, D.B. 1998. Interaction of murine MHC class I molecules with tapasin and TAP enhances peptide loading and involves the heavy chain α3 domain. J. Immunol. 162: 1530–1540. [PubMed] [Google Scholar]
- Williams, A.P., Peh, C.A., Purcell, A.W., McCluskey, J., and Elliott, T. 2002. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16: 509–520. [DOI] [PubMed] [Google Scholar]
- Wright, C., Kozik, P., Zacharias, M., and Springer, S. 2004. Tapasin and other chaperones: Models of the MHC class I loading complex. Biol. Chem. 385: 763–778. [DOI] [PubMed] [Google Scholar]
- Zacharias, M. and Springer, S. 2004. Conformational flexibility of the MHC class I α1/α2 domain in peptide bound and free states: A molecular dynamics simulation study. Biophys. J. 87: 2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling, A.L., Luckey, C.J., Marto, J.A., White, F.M., Brame, C.J., Evans, A.M., Lehner, P.J., Cresswell, P., Shabanowitz, J., and Hunt, D.F., et al. 2003. Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J. Immunol. 171: 5287–5295. [DOI] [PubMed] [Google Scholar]
- Zernich, D., Purcell, A.W., Macdonald, W.A., Kjer-Nielsen, L., Ely, L.K., Laham, N., Crockford, T., Mifsud, N.A., Bharadwaj, M., and Chang, L., et al. 2004. Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J. Exp. Med. 200: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C., Anderson, A., and DeLisi, C. 1998. Structural principles that govern the peptide-binding motifs of class I MHC molecules. J. Mol. Biol. 281: 929–947. [DOI] [PubMed] [Google Scholar]