

Abstract
The c-rel protooncogene encodes a member of the Rel/nuclear factor (NF)-κB family of transcriptional factors. To assess the role of the transcriptional activation domain of c-Rel in vivo, we generated mice expressing a truncated c-Rel (Δc-Rel) that lacks the COOH-terminal region, but retains a functional Rel homology domain. Mice with an homozygous mutation in the c-rel region encoding the COOH terminus of c-Rel (c-relΔCT/ΔCT) display marked defects in proliferative and immune functions. c-relΔCT/ΔCT animals present histopathological alterations of hemopoietic tissues, such as an enlarged spleen due to lymphoid hyperplasia, extramedullary hematopoiesis, and bone marrow hypoplasia. In older c-relΔCT/ΔCT mice, lymphoid hyperplasia was also detected in lymph nodes, liver, lung, and stomach. These animals present a more severe phenotype than mice lacking the entire c-Rel protein. Thus, in c-relΔCT/ΔCT mice, the lack of c-Rel activity is less efficiently compensated by other NF-κB proteins.
The c-rel protooncogene encodes a transcription factor that belongs to the family of Rel/nuclear factor (NF)1-κB proteins that play an important role in the expression of genes involved in immune and inflammatory responses (1–10). Rel/NF-κB proteins represent a group of homo- and heterodimeric complexes that are related through a common NH2-terminal domain known as the Rel homology domain (RHD), which consists of ∼300 amino acids and contains sequences important for protein dimerization, DNA binding, nuclear localization, and association with inhibitors of the IκB family. The COOH termini of Rel proteins have little sequence similarity and have been used to distinguish two classes of Rel proteins. One class includes NF-κB1 (protein [p]105/p50) and NF-κB2 (p100/p52), which, by proteolytic processing, generates the mature DNA-binding subunits p50 and p52, respectively. The second class includes c-Rel, RelA (p65), and RelB, which contain transcriptional activation domains in their COOH termini. The genes of the Rel/NF-κB family are differentially expressed in lymphoid tissues (11–12) and studies with mice lacking either p50, RelB, RelA, or c-Rel demonstrate that individual members of this family have distinct functions in vivo (for review see reference 13).
Activation NF-κB is regulated by posttranslational modification and degradation of IκB proteins that interact with the Rel/NF-κB complexes and sequester them in the cytoplasm by masking their nuclear localization signal. Members of the IκB family include IκBα, IκBβ, IκBγ, IκBε, Bcl-3, p105, and p100, which share conserved ankyrin-like repeats responsible for interaction with the Rel/NF-κB complexes. In the case of IκBα, phosphorylation and subsequent degradation of the inhibitor releases the active Rel/NF-κB complexes allowing their nuclear translocation. Degradation of IκBα is mediated by the ubiquitin– proteasome pathway, and phosphorylation of IκBα involves a ubiquitin-dependent protein kinase (4, 7, 8, 10, 14–16).
The mammalian c-rel gene was first identified as the cellular homologue of v-rel, the oncogene carried by Rev-T, an acutely transforming avian retrovirus that induces a variety of neoplastic diseases in chickens. Rearrangements in the c-rel gene have been associated with human lymphoid malignancies. Similar to v-rel, the altered c-rel genes lack sequences encoding the transcriptional activation domain (17–20). The in vivo roles of the c-rel gene have been recently addressed by gene targeting. Mice lacking the c-Rel protein (c-rel −/−) exhibit defects in lymphocyte proliferation, humoral immunity, and cytokine production (21–23). To understand the in vivo role of c-Rel in greater detail, we have generated mice lacking only the COOH-terminal transcriptional activation domain of c-Rel (c-relΔCT/ΔCT). This transcriptionally inactive molecule retains an intact RHD, is able to bind DNA, and to interact with other Rel/NF-κB family members and the IκB family of inhibitory molecules. Therefore, in c-relΔCT/ΔCTmice, other Rel/ NF-κB family members do not have the possibility of taking the function of c-Rel, as in the case of c-rel −/− mice. In addition, this approach allows us to address the functional significance of the different transcriptional activation domains present in c-Rel, RelA, and RelB, and the role of c-Rel COOH-terminal truncations in the generation of lymphoid malignancies.
Materials and Methods
Targeting Vector and Generation of Mutant Mice.
A genomic library (cloned in lambda DashII; Stratagene Corp., La Jolla, CA) prepared from D3 embryonic stem (ES) cell DNA was screened with the mouse c-rel cDNA probe (24). Two overlapping phages containing a total of 25 kbp of the c-rel gene were isolated and the fragments were subcloned into pBluescript KS+ (Stratagene Corp.). A 0.9-kbp fragment containing the SV40 polyadenylation sequence [p(A)] and a termination codon was prepared by PCR mutagenesis using the pMSG vector (Pharmacia, Piscataway, NJ) and subcloned in the plasmid PGK promoter neomycin thymidine kinase (pPNT) vector (25). A 4.7-kbp c-rel genomic DNA fragment containing exons 7–9 and the first portion of exon 10 (until the XhoI site) was inserted upstream of the stop codon and between the phosphoglycerate kinase (PGK)-neo cassette and the PGK promoter driving the herpes simplex virus thymidine kinase gene (PGK–tk cassette) of a pPNT vector containing the termination codon. A 8.5-kbp fragment from c-rel genomic DNA extending from the XhoI site of exon 10 to the flanking 3′ genomic sequences was cloned upstream and in opposite direction to the PGK–neo cassette of the pPNT vector. In this way, the genomic c-rel gene was interrupted by the neo selection marker in exon 10. Introduction of the stop codon 3′ to the XhoI restriction site produces a truncated c-rel messenger RNA that lacks the region encoding the truncated c-Rel (Δc-Rel) transactivation domain (pPNT/Δc-Rel).
CJ7 ES cells were electroporated with NotI-linearized pPNT/ Δc-Rel and grown under double selection conditions using G418 and fialuridine (FIAU). Homologous recombination events were screened by Southern blot analysis using a 5′ external probe, and additional random integrations were excluded with a neo probe (Fig. 1 A, and data not shown). Homozygous mutant animals were prepared as described (26).
Figure 1.


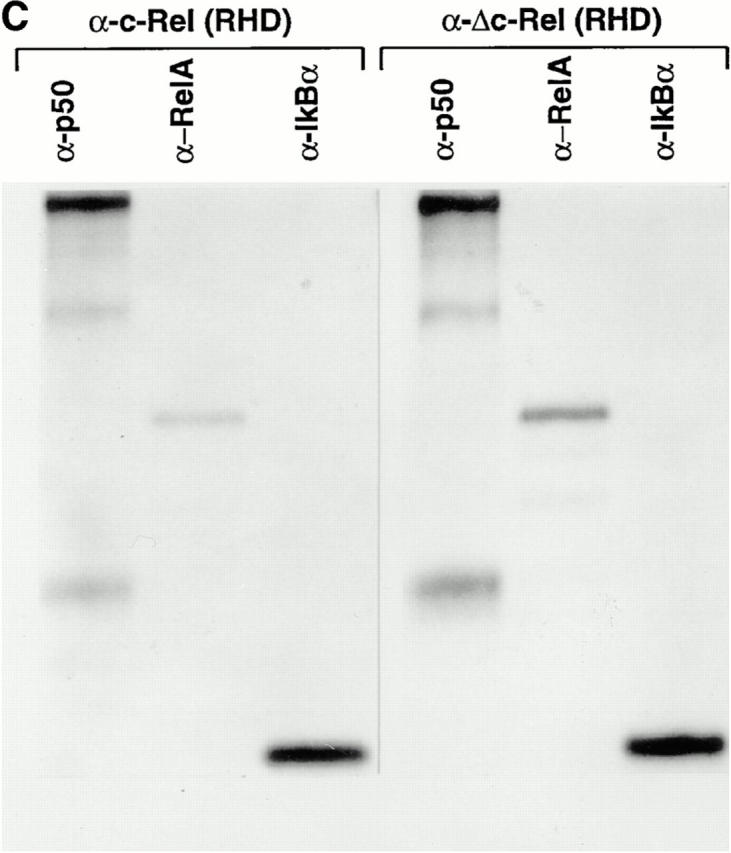
Generation of mice lacking the transcriptional activation domain of c-Rel. (A) The targeting vector for positive– negative selection, pPNT/Δc-Rel is shown at the top. Open boxes, the SV40 p(A), the PGK– neo, and the PGK–tk cassettes. Homologous recombination of the targeting vector with wild-type genomic DNA introduces the neo selection marker at the beginning of exon 10 (bottom). The targeted allele and the length of diagnostic restriction fragments used for Southern blot analyses and the probe fragment are indicated. (B) Screening of ES cells and mice generated from heterozygote intercrosses and expression of Δc-Rel protein. (B a) ES cell genomic DNAs were digested with XbaI and subjected to Southern blot analysis using the 5′ external probe. The 25-kbp band (wt) indicates the wild-type allele, whereas the 18.5-kbp band (ko) represents the targeted allele. (B b) Tail DNAs were prepared and subjected to PCR analyses using an appropriate pair of c-rel and neo oligonucleotides. Wild-type mice presented a single band, c-rel; heterozygous mice showed two bands, c-rel and neo; and the homozygous mice showed a single band, neo. (B c) Total protein extract from wild-type (+/+), heterozygous (+/ΔCT), and homozygous (ΔCT/ΔCT) mutant splenocytes labeled with [35S]methionine were immunoprecipitated with an anti–c-Rel/ RHD antiserum. Specific signals for c-Rel and Δc-Rel are indicated. (C) Δc-Rel interacts with other Rel/NF-κB proteins. Total protein extract from wild-type (left) and homozygous (right) mutant splenocytes labeled with [35S]methionine were first immunoprecipitated with an anti–c-Rel/RHD antiserum, and the complexes were denatured and reprecipitated with the indicated specific antisera.
Histology, Immunofluorescence, Immunohistochemistry, and Flow Cytometry.
Tissues were immersion fixed in 10% buffered formalin and embedded in paraffin blocks. Sections were stained with hematoxylin and eosin. Apoptotic cells were detected with the Apoptag assay and stained with methyl green according to Oncor, Inc. (Gaithersburg, MD). For detection of germinal centers (GCs), mice were immunized with SRBCs, and frozen sections of spleen were prepared and stained as described (27). Flow cytometry analysis was performed as described (26, 28). Anti–mouse macrophage 1 (Mac-1), granulocyte 1 (Gr-1), and total erythroid cells (Ter) 119 were obtained from GIBCO BRL (Gaithersburg, MD).
In Vitro Proliferation, Listeria Monocytogenes, and Lymphocytic Choriomeningitis Virus Infections.
B cell proliferation assays were performed as described (29). Lymph node T cells were purified by murine T cell enrichment columns (R&D Systems, Inc., Minneapolis, MN) and (105) cells in 96-well plates were stimulated with IL-2 or with cluster of diffentiation (CD)3 plus CD28 coated antibodies (PharMingen, San Diego, CA) in 200 μl medium for 48 h. Cell proliferation was measured after 12 h of culture by [3H]thymidine incorporation in a scintillation spectroscopy. Results are expressed as the arithmetic mean ± SD of triplicate cultures.
Adult mice were injected intraperitoneally with 2,500 CFU of L. monocytogenes and killed after 5 d. Animals were killed after 5 d or on day 4 of infection when moribund. Numbers of viable L. monocytogenes in lung, liver, and spleen of infected animals were determined by plating serial dilutions of organ homogenates in PBS on sheep blood agar. Other mice were injected intraperitoneally with 1.2 × 105 PFU (Armstrong strain) of Lymphocytic Choriomeningitis Virus (LCMV) and killed 3 or 7 d later. Infectious LCMV titers of lung, liver, and spleen (PFU/g tissue) were quantitated by plaque assay using Vero cell monolayers as described (30). Data from six to nine animals per genotype were recorded for each experiment.
Thioglycollate-induced Peritonitis, Granuloma Formation, and Nitric Oxide Production.
Peritoneal macrophages were prepared 5 d after intraperitoneal injection with 1.5 ml sterile Brewers thioglycollate broth (3%) as described (31). Three cell preparations from the various genotypes were used in in vitro experiments. Lung granuloma formation was induced by glucan tail injection (32). For nitric oxide synthase (NOS) assays, resting peritoneal macrophages (5 × 105/0.5 ml DMEM in a 24-well plate) were treated for 72 h with LPS (1 μg/ml) alone or in combination with IFN-γ (100 U/ml). NO was measured as nitrite with Greiss reagent (33).
ELISA.
Purified lymph node T cells (5 × 105/ml) isolated from 6-wk-old mice were incubated with or without coated anti-CD3 and anti-CD28 antibodies for 72 h. Macrophages were stimulated for 2 h with media alone or in the presence of a combination of LPS (1 μg/ml) and IFN-γ (100 U/ml). Cytokine levels in supernatants were determined by ELISA (R&D Systems, Inc.). All cultures were done in triplicate.
Immunoprecipitation and Electrophoretic Mobility Shift Assays.
Splenocytes from 6-wk-old animals were isolated as described (28) and labeled with 800 μCi/ml of [35S]methionine (Amersham Corp., Arlington Heights, IL) for 6 h. Cells were lysed directly in 1× RIPA buffer, followed by immunoprecipitation as described (34). For electrophoretic mobility shift assays (EMSAs), nuclear extracts were prepared and incubated with a palindromic κB site as described (35). Protein loading was normalized by comparing Oct 1 DNA-binding activity.
Expression Vectors, Cell Culture, and Transfections.
Expression vectors for p50, RelA, c-Rel, and Δc-Rel were constructed by cloning the corresponding coding regions into the mammalian expression vector pMexneo as described (36). S107 cells were cotransfected using the standard calcium phosphate coprecipitation method (37). Typically, 2 μg of reporter vector and 0.5 μg of each of the expression vectors were used. The reporter vector, 2× κB-tk-CAT (38; CAT, chloramphenicol acetyl transferase), containing two κB-binding sites from the mouse immunoglobulin κB enhancer, was used in all experiments. The standard reaction for CAT enzyme assay was performed according to manufacturer's recommendations (Promega Corp., Madison, WI). Reactions were normalized for luciferase levels and protein concentration.
Results
Generation of Mice Lacking the Transcriptional Activation Domain of c-Rel.
A targeted disruption of the transcriptional activation domain of c-rel was created by introducing a termination signal at codon 366 followed by SV40 p(A) and a PGK–neo cassette (Fig. 1 A). After electroporation and selection, 200 double-resistant CJ7 ES cell clones were picked and screened by Southern blot analysis (Fig. 1 B). Homologous recombination in 11 clones was identified by the appearance of a 18.5-kbp recombinant band in addition to the 25-kbp wild-type band in XbaI-digested DNA (Fig. 1 B a).
Eight chimeric males transmitted the ES cell–derived agouti coat and the targeted c-rel gene to their offspring (data not shown). Intercrosses between heterozygous animals produced progeny with normal Mendelian transmission of the disrupted c-rel allele according to genotypic PCR analysis (Fig. 1 B b).
Expression of c-Rel Lacking the Transcriptional Activation Domain in Splenocytes.
Whole cell lysates from mouse splenocytes labeled with [35S]methionine were immunoprecipitated with a c-Rel antiserum raised against the RHD. The results demonstrated that homozygous mutant mice (c-rel ΔCT/ΔCT) lacked c-Rel, but expressed the truncated c-Rel protein, Δc-Rel (Fig. 1 B c). In heterozygous mice (c-rel +/ΔCT), both the normal c-Rel and the truncated Δc-Rel proteins are detected. We further studied selected protein interactions of Δc-Rel (Fig. 1 C). After immunoprecipitation with c-Rel antibodies under nondenaturing conditions, the immunocomplexes were dissociated and then sequentially reprecipitated with p50, RelA, and IκBα antibodies. The immunoprecipitation patterns observed in control (Fig. 1 C, left) and in Δc-Rel (Fig. 1 C, right) cells were similar, indicating that Δc-Rel, by virtue of its intact RHD, maintains normal interactions with other Rel/NF-κB family members and IκBα.
Histopathological Alterations in c-relΔCT/ΔCT Mutant Mice.
Young c-rel ΔCT/ΔCTmice, between 3 and 8 wk old, appeared normal as assessed by habit, weight, posture, and histologic and flow cytometric analysis of lymphoid cells (data not shown). However, after 5 to 7 mo of age, an increasing number of animals began to develop exzematoid skin lesions around the nose, eyes, ears, tail, and foreskin. These lesions were not related to infection, according to microbiologic testing and serologic analysis (not shown). The disease progressed slowly, without severely compromising the health status and survival of c-rel ΔCT/ΔCT mice if the animals were kept in microisolators. However, when c-rel ΔCT/ΔCT mice were left in a non–pathogen-free environment, their survival was reduced compared with control littermates (data not shown). Apparently, healthy and sick c-rel ΔCT/ΔCT mice were systematically analyzed by histopathology. A constant observation was the presence of enlarged spleens, lymph nodes, and in 30% of the cases analyzed, the presence of pale bones and changes in the color and consistency of the stomach, liver, and lung (Fig. 2, a and b, and data not shown). The TUNEL assay revealed increased numbers of apoptotic nuclei inside of macrophages in medullary areas of the thymus and within the marginal zone of the spleen in c- rel ΔCT/ΔCT mice (Fig. 2, c and d, and data not shown). This result is in agreement with recent reports documenting a role of Rel/NF-κB in preventing apoptosis (39–41).
Figure 2.
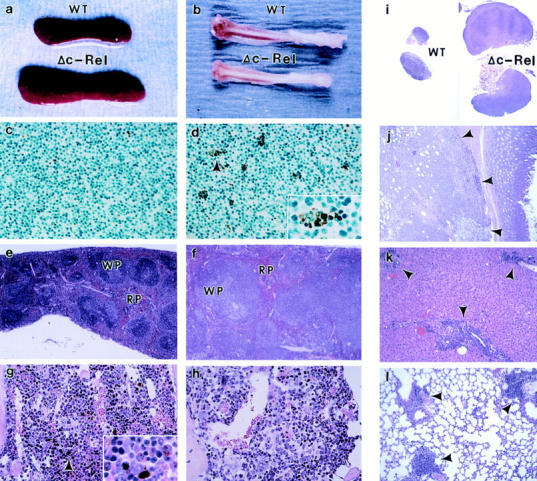
Histopathology of c-relΔCT/ΔCT mice. Necropsies of 5–7-mo-old c-relΔCT/ΔCT mice reveals splenomegaly (a) and bone marrow hypoplasia (b). Thymus tissue sections from 4-wk-old control (c) and c-relΔCT/ΔCT (d) mice were stained with the TUNEL procedure. (d, inset) Apoptotic nucleus. Spleen (e and f ) and bone marrow (g and h) sections from 5-mo-old control (e and g) and c-relΔCT/ΔCT (f and h) mice were stained with hematoxylin and eosin. (g, inset) Macrophages containing iron particles. Photomicrographs were taken at magnifications of 100 (c, d, g, and h), 12.5 (e and f ), and 250 (d and g, insets). WT, wild type; WP, white pulp; RP, red pulp. Tissue sections from lymph nodes (i), stomach (j), liver (k), and lung (l) from 5-mo-old c-relΔCT/ΔCT mice revealed lymphoid hyperplasia (arrows). Photomicrographs were taken at magnifications of 6.25 (i) and 25 (j, k, and l).
Staining of mutant spleen sections revealed increased white and red pulp areas when compared with control tissue (Fig. 2, e and f ). Analysis at higher magnification revealed mild to moderate increase of normoblast and megakaryocytes and a reduced number of metachromatic macrophages in red pulp areas of spleens from c-rel ΔCT/ΔCT mice (data not shown). Reduced cellularity and increased empty spaces were observed in the bone marrow of c-rel ΔCT/ΔCT mice that presented macroscopic alterations in the bones (Fig. 2, g and h). Lymphoid cellular infiltration was observed in tissues of the mutant mice with enlarged lymph nodes and macroscopic alterations in the stomach, liver, and lung (Fig. 2, i–l).
The previous observations suggests an essential role of c-Rel in normal bone marrow hematopoiesis and lymphoid development. Since these changes were not observed in wild-type and heterozygous c-rel +/ΔCTmice, Δc-Rel does not behave as a trans-dominant mutant of c-Rel.
Bone Marrow Hypoplasia, Extramedullary Hematopoiesis, and Lymphoid Hyperplasia in c-relΔCT/ΔCT Mice.
Flow cytometric analysis of hemopoietic cells from young c-rel ΔCT/ΔCTmice did not show major alterations in the expression of the surface markers in cells derived from thymus (CD4, CD8, and TCR-α/β), spleen (CD4, CD8, TCR-α/β, CD25, Mac-1, Gr-1, B220, IgM, IgK, and Ter 119), and bone marrow (Ter 119, IgM, IgK, and B220) (data not shown). However, after 5 mo of age, alterations began to be detected in c-rel ΔCT/ΔCTmice (Fig. 3, A and B). For instance, in agreement with the hypocellularity observed in bone marrow at histopathology, a reduced percentage of macrophages (Fig. 3 A, a and b) and erythroid precursors (Fig. 3 A, c and d) was detected in bone marrow–derived cells from c-rel ΔCT/ΔCT mice, but not in control littermates. In addition, a concomitant and graded increase in the number of erythroid precursors (Fig. 3 B, a and b) and granulocytes (Fig. 3 B, c and d) was observed in the enlarged spleens from c-rel ΔCT/ΔCTmice, reflecting the presence of extramedullary hematopoiesis. When B cell markers were used in flow cytometric analysis, a 1.5–2.5-fold increase in the total number and percentage of B cells was observed in the enlarged spleens of c-rel ΔCT/ΔCTmice, correlating with the enlarged white pulp areas (Fig. 3 C, and data not shown). Immunostaining of control (Fig. 3 C a) and c-rel ΔCT/ΔCT (Fig. 3 C b) splenic tissue sections with anti–Mac-1 revealed a dramatic enlargement of the lymphatic follicles with compression and displacement of the red pulp to the periphery of the organ. A concomitant reduction of Mac-1 positive cells in the red pulp between the follicles was also observed. Labeling with anti-B220 antibodies revealed diffusely enlarged white pulp areas with poorly demarcated white/red pulp boundaries, marginal zones, and periarterial lymphatic sheaths (Fig. 3 C, c and d). In addition, a decrease in the intensity of the B220-stained cells was also evident in spleens from c-rel ΔCT/ΔCT mice compared with control littermates. In contrast to the B cell areas, the T cell areas (periarterial lymphatic sheaths), were not enlarged in spleens from mutant mice, as revealed by the immunostaining with anti–TCR-α/β monoclonal antibodies (Fig. 3 C, e and f ).
Figure 3.
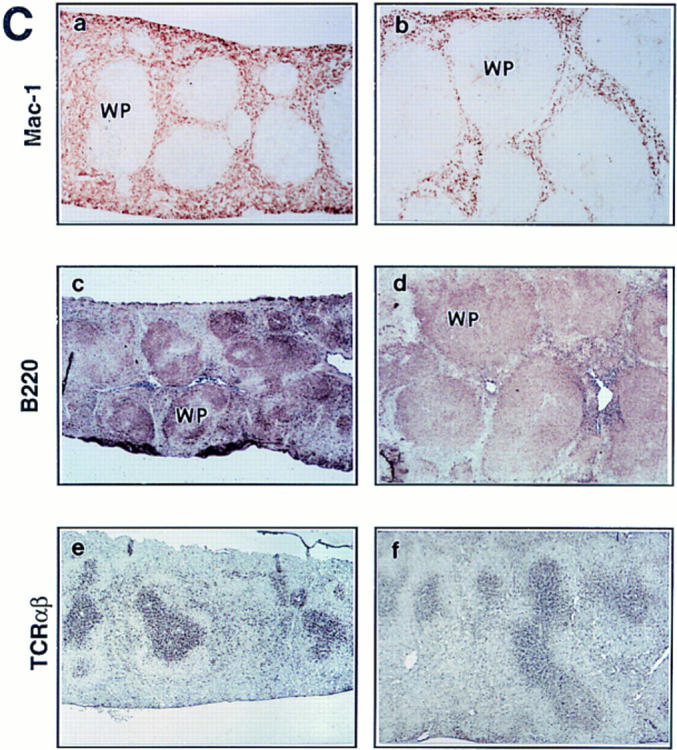

Bone marrow hypoplasia, extramedullary hematopoiesis, and lymphoid hyperplasia in c-relΔCT/ΔCT mice. Single cell suspensions were treated with LCK lysis buffer and an equivalent number of bone marrow (A) and splenic (B) cells from c-relΔCT/ΔCT and control littermates were stained with anti–Mac-1 (A, a and b), anti–Ter 119 (A, c and d; and B, a and b), and anti–Gr-1 (B, c and d) antibodies and analyzed by flow cytometry. Results show a representative profile from quadruplicates of several independent analyses. (C) Spleen sections from 5-mo-old control (a, c, and e) or c-relΔCT/ΔCT mice (b, d, and f) immunostaining with anti–Mac-1 (a and b), anti-B220 (c and d), or anti–TCR-α/β (e and f) mouse monoclonal antibodies. Photomicrographs were taken at a magnification of 12.5. WP, white pulp.
Defective Clearance of L. Monocytogenes in c-relΔCT/ΔCT Mice.
Although infectious disease as a primary cause of the c-rel ΔCT/ΔCT phenotype was ruled out (data not shown), opportunistic bacterial infections were observed in older c-rel ΔCT/ΔCT mice. To examine whether c-rel ΔCT/ΔCT mice might fail to eliminate bacterial pathogens, we used the L. monocytogenes model (42). Groups of wild-type and homozygous mutant mice were infected intraperitoneally with L. monocytogenes, killed at day 5, and bacterial CFU were determined in spleen, liver, and lung. As shown in Fig. 4 A, c-rel ΔCT/ΔCT mice had >20-fold higher listerial titers in all the tissues examined compared with control animals. This result defined an impaired capability of c-rel ΔCT/ΔCT mice in handling bacterial infections and explained the increased susceptibility to bacterial infections of these mice when kept in a non–pathogen-free environment.
Figure 4.

Macrophage alterations in c-relΔCT/ΔCT mice. (A) Defective clearance of L. monocytogenes. Mice were injected intraperitoneally with 2,500 CFU of L. monocytogenes and killed 5 d later. Each solid square (control mice) or empty circle (c-relΔCT/ΔCT mice) represents the bacterial CFU isolated from spleen, liver, or lung homogenates of individual animals. (B) Normal numbers of total inflammatory cells (B a), but reduced numbers of peritoneal macrophages (b and c) in c-relΔCT/ΔCT mice. Representative cytospin preparation from thioglycollate-elicited macrophages from control (B b) and c-relΔCT/ΔCT mice (B c). (C) NO production from unstimulated (solid bars) or LPS/IFN-γ–stimulated resting peritoneal macrophages (hatched bars). (D) Cytokine production from unstimulated (solid bars) or LPS/IFN-γ–stimulated peritoneal macrophages (hatched bars). Results represent absolute amounts (mean ± SD). (E) Reduced granuloma formation in c-relΔCT/ ΔCT mice. Control (a and b) and c-relΔCT/ΔCT (c and d) mice were injected intravenously with glucan, killed 24 h later, and lung tissue sections were stained with hematoxylin and eosin. Photomicrographs were taken at magnifications of 250 (a and b), 25 (c and d), and 100 (e and f). M, macrophage; G, granulocyte. Arrowheads, granulomatous infiltration.
Reduced Production of NO and Cytokines in c-relΔCT/ΔCT Macrophages.
To investigate whether migration is impaired in Δc-Rel macrophages, mice were injected with thioglycollate. The total number of peritoneal inflammatory cells elicited was comparable in control and c-rel ΔCT/ΔCT mice (Fig. 4 B a). However, the differential cell count revealed a twofold reduction in the number of macrophages and a relative increase in the number of polymorphonuclear leukocytes in c-rel ΔCT/ΔCT mice, indicating that recruitment of macrophages is impaired in the mutant animals (Fig. 4 B, b and c). The altered clearance of the facultative intracellular bacterium L. monocytogenes in c-rel ΔCT/ΔCT mice may be due to reduced bactericidal activity of the macrophage, which is known to be mediated by release of toxic nitrogen intermediates (43). Resident c-rel ΔCT/ΔCT peritoneal macrophages after in vitro stimulation with LPS and IFN-γ had a significantly lower NO production compared with control macrophages (Fig. 4 C).
Cytokines synthesized by activated macrophages include, among others, IL-1β, TNF-α, IL-6, and GM-CSF (44). The production of these cytokines was compared in nonstimulated and LPS/IFN-γ–activated control and Δc-Rel resident peritoneal macrophages (Fig. 4 D). Basal levels of IL-1β (Fig. 4 D a), GM-CSF (Fig. 4 D b), TNF-α (Fig. 4 D c), and IL-6 (Fig. 4 D d) were reduced in nonstimulated Δc-Rel resident peritoneal macrophages; however, upon LPS and IFN-γ activation, TNF-α and IL-6 production was almost completely reestablished. This finding contrasts with the production of IL-1β and GM-CSF by activated macrophages, whereas cytokine levels secreted by Δc-Relmacrophages were significantly reduced.
Impaired Granuloma Formation in c-relΔCT/ΔCT Mice.
The intravenous administration of glucan to a variety of experimental animals results in a marked proliferation of macrophages and granuloma formation (32). Control and c-rel ΔCT/ΔCT mice were injected intravenously with glucan and after 24 h, lung tissue sections were prepared for analysis of granuloma formation (Fig. 4 E). In control lungs, multiple and massive angiocentric granulomas were observed (Fig. 4 E a). Analysis at higher magnification revealed that the granulomas were composed primarily of granulocytes and, to a lesser extent, by macrophages (Fig. 4 E b). In contrast, in c-rel ΔCT/ΔCT mice, lung compromise was minimal with small scattered granulomas (Fig. 4 E c), composed primarily of macrophages and rare granulocytes (Fig. 4 E d). This result indicates that macrophage proliferation and granulocyte recruitment in response to glucan injection is impaired in c-rel ΔCT/ΔCT mice. Since we have not detected c-Rel expression in granulocytes (12), the impairment in granulocyte recruitment observed after glucan injection is most probably due to defective cytokine production by alveolar macrophages. Collectively, these results define an essential need of a transcriptionally active c-Rel in macrophages.
c-relΔCT/ΔCT Mice Display Impaired Antibody Production, B Cell Proliferation, and Germinal Center Formation.
To evaluate the humoral immunity of c-rel ΔCT/ΔCT mice, we measured the basal production of immunoglobulins in naive animals. Serum isotype levels from nonimmunized c-rel ΔCT/ΔCT mice and control littermates are shown in Fig. 5 A. Although the levels of IgG1 (threefold), IgG2b (twofold), and IgG3 (fourfold) in c-rel ΔCT/ΔCT mice were reduced compared with wild-type littermates (Fig. 5 A a), those of IgG2a (twofold), IgM (threefold), and IgA (twofold) were increased (Fig. 5 A b).
Figure 5.

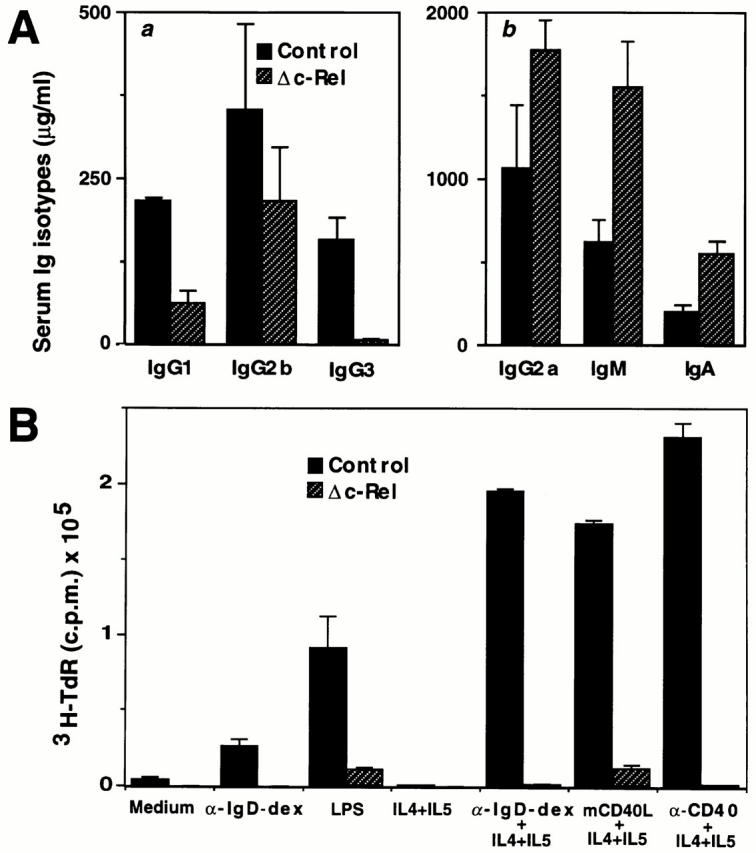
B cell alterations in c-relΔCT/ΔCT mice. (A) c-relΔCT/ΔCT mice display defects in antibody production. The levels of serum immunoglobulins (μg/ml) in naive 6–8-wk-old litter-matched mice were determined by isotype-specific ELISA in control and c-relΔCT/ΔCT mice. (B) Δc-Rel B cells show defects in DNA synthesis in vitro. Splenic B cells (103 cells/ml) isolated from control (solid bars) or c-relΔCT/ΔCT (hatched bars) mice were incubated for 72 h in various conditions: unstimulated, α-IgD-dex (3 ng/ ml), LPS (0.2 mg/ml), IL-4 (3,000 U/ml) + IL-5 (150 U/ml), α-IgD-dex (3 ng/ml) + IL-4 (3,000 U/ml) + IL-5 (150 U/ml), mCD40L (1/2,000 vol/vol) + IL-4 (3,000 U/ml) + IL-5 (150 U/ml), or anti-CD40 mAb (1.25 mg/ml). [3H]thymidine was added for the final 12 h, and cells were then harvested for determination of [3H]thymidine incorporation. Values represent the means in triplicate cultures ± SD. (C) Absence of GCs in spleen from immunized c-relΔCT/ΔCT mice. Serial spleen sections from control (a and b) and c-relΔCT/ΔCT mice (c and d) were stained with peanut agglutinin (a and c) or anti-IgD antibodies (b and d). Photomicrographs were taken at a magnification of 50 (a–d).
Splenic B cells from control and c-rel ΔCT/ΔCT mice were stimulated with different agents over a 72-h period and proliferation was monitored by [3H]thymidine incorporation. Control B cells stimulated with α-IgD-dex, LPS, ligand for the mouse CD40 (mCD40L), or α-CD40 showed proliferation, and these responses were further augmented by IL-4 and IL-5 addition. Δc-Rel B cells showed only minimal response to LPS and mCD40L and IL-4 plus IL-5, but did not proliferate under other stimuli (Fig. 5 B). However, when unpurified Δc-Rel B cells were used for in vitro proliferation assays, significant levels of [3H]thymidine incorporation were observed (data not shown). These results indicate that there are two different pathways leading to B cell proliferation, one intrinsic and c-Rel dependent, and another extrinsic, c-Rel independent, which requires additional exogenous stimuli.
The abnormal architecture of the spleen in c-rel ΔCT/ΔCT mice suggests that the immune responses dependent on cellular interactions in the lymphatic follicles may not be fully functional. Therefore, we immunized mice with SRBCs and evaluated the formation of GCs 7 d later (Fig. 5 C). The spleen of control mice had numerous GCs as defined by central areas of cells that bind peanut agglutinin (PNA; Fig. 5 C a) surrounded by IgD+ cells (Fig. 5 C b). In contrast, spleens of c-rel ΔCT/ΔCT mice had reduced numbers and poorly defined GCs (Fig. 5 C c and d). These results indicate that clonal expansion of B cells in response to the T cell–dependent antigen (SRBC) is impaired in c-rel ΔCT/ΔCT mice.
Normal Clearance of LCMV in c-relΔCT/ΔCT Mice.
Acute infection of adult mice with LCMV induces a protective immunity and it has been shown that virus-specific CTLs play an essential role in virus elimination from the infected host (45). To examine whether a transcriptionally active c-Rel is required to induce a CTL response, control and c-relΔCT/ΔCT mice were infected with LCMV and their capacity to clear virus loads from various organs was determined at days 3 and 7 after virus inoculation. As shown in Fig. 6 A, c-relΔCT/ΔCT and control mice exhibited equivalent viral titers in all organs tested at 3 (Fig. 6 A a) or 7 (Fig. 6 A b) d after inoculation. This result demonstrates that c-relΔCT/ΔCT mice can mount a protective CTL response against LCMV.
Figure 6.

Normal T cell functions in c-relΔCT/ΔCT mice. (A) c-relΔCT/ΔCT mice clear LCMV infection. Control and c-relΔCT/ΔCT mice were inoculated with 1.2 × 105 PFU LCMV by intraperitoneal injection, killed 3 (A a) or 7 (A b) D after LCMV infection, and virus titers of spleen, liver, and lung were determined. Bars represent data (mean ± SD) from six to nine mice per genotype. (B) Normal T cell proliferation of Δc-Rel T cells. Control or Δc-Rel lymph node–derived T cells were incubated with or without various stimuli, and cell proliferation was measured by [3H]thymidine incorporation. (C) Cytokine production in control or Δc-Rel lymph node T cells. Cells were incubated without (solid bars) or with (hatched bars) αCD3 and αCD28. Cytokine levels in supernatants were determined after 24 h. All cultures were performed in triplicate.
Normal Proliferative Responses and Cytokine Production in c-relΔCT/ΔCT–derived T Cells.
Proliferative response of T cells from c-relΔCT/ΔCT mice was comparable to that seen in control cells (Fig. 6 B). Equivalent levels of [3H]thymidine incorporation were observed in control and mutant T cells after stimulation of either IL-2 or with a combination of anti-CD3 plus anti-CD28.
The cytokine secretion profile of normal and Δc-Rel T cells in the absence of stimuli or after anti-CD3 plus anti-CD28 stimulation is shown in Fig. 6 C. In the absence of stimuli, cytokines were undetectable in either control or Δc-Rel T cells (Fig. 6 C, a–c). Anti-CD3 plus anti-CD28 stimulation increased production of GM-CSF, TNF-α, and IL-2 in both control and c-relΔCT/ΔCT T cells. However, quantitative differences were observed. For instance, the levels of GM-CSF and TNF-α were higher in c-relΔCT/ΔCT T cells, whereas the levels of IL-2 production were equivalent in control and c-relΔCT/ΔCT T cells.
Differential Alteration of κB-binding Activity in Δc-Rel B and T Cells.
The functional alterations observed in some hematopoietic cell compartments but not in others in c-relΔCT/ΔCTmice could be due to differential alterations in the NF-κB activity. Thus, we determined the κB-binding activity of purified in vitro activated control and mutant T and B cells (Fig. 7 A). Nuclear protein extracts from B cells (Fig. 7 A a) and T cells (Fig. 7 A b) from control (lanes 1–6) and Δc-Rel (lanes 7–12) cells were analyzed by EMSA using a palindromic κB-binding site. Antibody challenge of the nuclear extracts demonstrate that the major κB-binding complexes in control B cells are p50/c-Rel heterodimers and p50/p50 homodimers, whereas in control T cells, p50/c-Rel and p50/ RelA heterodimers together with p50/p50 homodimers are the major κB-binding complexes (Fig. 7 A b). In Δc-RelB cells, the major κB-binding complex is a p50/Δc-Rel heterodimer (Fig. 7 A a), whereas in Δc-RelT cells, two major complexes containing Δc-Rel were identified, RelA/Δc-Rel and p50/Δc-Rel heterodimers (Fig. 7 A b). As expected, due to the COOH-terminal deletion present in Δc-Rel, the p50/Δc-Rel and RelA/Δc-Rel heterodimers migrate faster than p50/c-Rel and RelA/c-Rel heterodimers, respectively.
Figure 7.

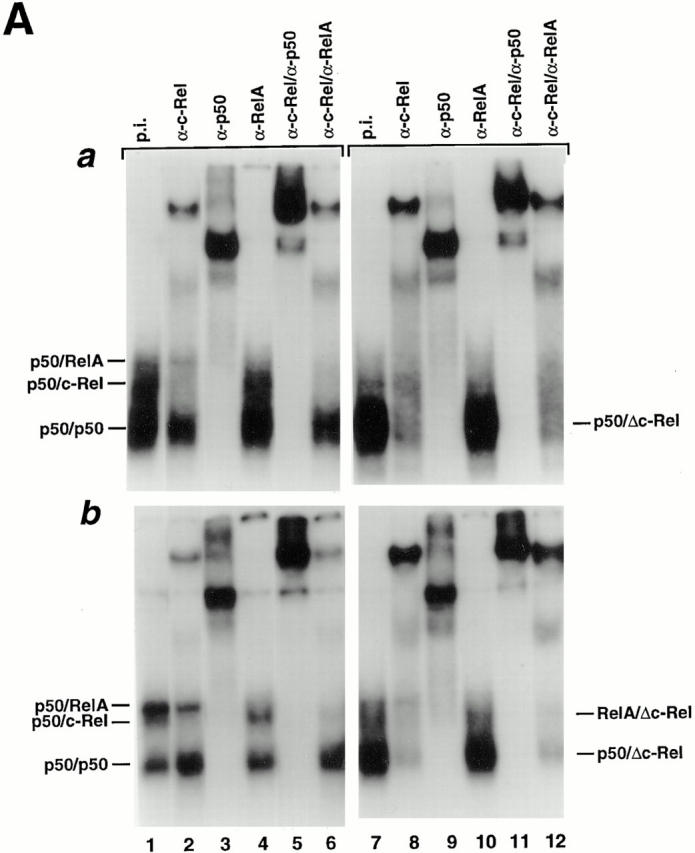
Differential κB-binding activity of Δc-Rel in T and B cells. (A) Control and c-relΔCT/ΔCT–derived B (A a) or T cells (A b) were incubated in the presence of LPS (10 μg/ml), or PMA (10 ng/ml) and PHA (5 μg/ml), respectively, for 3 h and nuclear protein extracts were prepared to analyze κB-binding activity by EMSA. Nuclear extracts were challenged with the indicated antibodies. p.i., preimmune. (B) Representative CAT assays of cotransfection experiments performed in the S107 cell line. The expression vectors for Δc-Rel and c-Rel were transfected either alone or in combination with p50 or RelA as described in Material and Methods. The basal levels of CAT activity in cells transfected with pMexneo vector have been subtracted before plotting.
The transcriptional activity of Δc-Rel complexes was studied in cotransfected S107 cells (Fig. 7 B). These studies demonstrate that the activity of the RelA/Δc-Rel heterodimer is similar to the wild-type RelA/c-Rel heterodimer (compare lanes 5 and 8), probably due to the presence of one transcriptional activation domain in RelA. The p50/Δc-Rel heterodimer is much less active than the wild-type p50/c-Rel heterodimer (compare lanes 7 and 9), most likely due to the absence of transcriptional activation domains in both p50 and Δc-Rel. In summary, these results indicate that the presence of p50/c-Rel activity is indispensable for normal B cell function, but not for T cell function.
Discussion
c-Rel was the first member of the Rel/NF-κB family of transcriptional factors demonstrated to be a protooncogene. v-Rel is the oncogene carried by the retrovirus of the reticuloendotheliosis virus strain T that produces lymphoproliferative disorders in birds (17–20). The chicken v-Rel has also been demonstrated to induce multicentric lymphoma/ leukemia in mammalian cells (34). Although all Rel/NF-κB members contain the highly conserved RHD, they have a highly divergent COOH-terminal end that in c-Rel, RelA, and RelB confer powerful transcriptional activity (24, 36, 46, 47). Interestingly, v-Rel represents a mutated version of c-Rel in which one of the major differences is a deletion of its COOH-terminal sequence (17–20). To better understand the functional consequences of the COOH-terminal truncation on the transforming capability of c-Rel and the functional role of the different transcriptional activation domains among different Rel/NF-κB members, we have generated mice in which the endogenous c-Rel protein has been replaced by a truncated version lacking the transcriptional activation domain (c-relΔCT/ΔCTmice). In this paper, the initial characterization of the phenotype of c-relΔCT/ΔCTmice is described.
Multiple Hematopoietic Abnormalities and Lymphoid Hyperplasia in c-relΔCT/ΔCT Mice.
In agreement with the broad expression pattern of c-Rel in hematopoietic cell lineages (12, 36), a complex spectrum of hematopoietic alterations was observed in mice lacking the transcriptional activation domain of c-Rel. c-relΔCT/ΔCTmice showed increased susceptibility to bacterial infection, impaired bone marrow hematopoiesis, and histopathologic alterations in hematopoietic tissues (including B cell hyperplasia). These alterations appear to represent defects in the cross-regulation between various hematopoietic cells, and for this reason, it is difficult to sort out secondary from primary defects. For instance, the fact that bone marrow hypoplasia was accompanied by compensatory extramedullary hematopoiesis in the spleen indicates that there are no primary abnormalities in hematopoietic precursors. Instead, there is an inappropriate microenvironment in the bone marrow of c-relΔCT/ΔCT mice, probably related to altered cytokine production by stromal cells, macrophages, B cells, or T cells. The defective GC formation in c-relΔCT/ΔCT mice is likely due to the structural alterations in the spleen, or due to functional alterations at some level in the network between macrophages, dendritic follicular cells, T cells, and B cells that are essential for the normal development of GCs (48, 49). Understanding the primary cellular alterations in c-relΔCT/ΔCT mice will require specific cellular assays in purified single cell populations. A successful example of this strategy is the recent observation that isolated c-relΔCT/ΔCT–derived B cells exhibit selective defects in germline transcription and Ig class switching (50).
The fact that c-relΔCT/ΔCTmice present lymphadenopathy and lymphoid hyperplasia in stomach, liver, and lungs is particularly interesting in light of the information that chromosomal translocations associated with structural alterations of the Rel/NF-κB family of proteins have been documented in several cases of human lymphomas (51–53). More specifically, c-rel rearrangements in several human non-Hodgkin's lymphomas have been found. In one cell line derived from a pre–T diffuse large cell human lymphoma, several abnormal c-rel cDNAs were isolated that encoded a large portion of the RHD that is fused to cellular sequences of unknown origin (53). The explanation of lymphoid hyperplasia found in older c-relΔCT/ΔCT mice is complex and may be related to a combination of different factors. The fact that in vitro [3H]thymidine incorporation was decreased in purified c-relΔCT/ΔCT B cells but not different from control unpurified B cells indicates that the truncated Δc-Rel protein does not affect the intrinsic mitogenic activity of B cells. Instead, it may affect the external signals regulating B cell proliferation. Cytokines are potent regulatory molecules secreted by cells of the immune system; deletion of certain cytokines in mouse mutants leads to immune dysregulation and expansion of some hematopoietic lineages (44). For instance, IL-8–deficient mice exhibit lymphoadenopathy, which results from an increase in B cells, and splenomegaly, which results from an increased number of metamyelocytes, band cells, and mature neutrophils (54). GM-CSF null mice present extensive lymphoid hyperplasia associated with lung airways and blood vessels (55). Interestingly, disruption of cytokine production and myeloid hyperplasia has been observed in mice deficient in RelB (26), and in mice lacking the COOH-terminal ankyrin domain of NF-κB2 (56).
The fact that B cell hyperplasia appears at later times, and in older animals, indicates that the lymphoid expansion may be related to a reduced clearance of aged cells by the reticuloendothelial system. This possibility is particularly attractive in light of the observation of reduced total number and functional alterations of macrophages in c-relΔCT/ΔCT mice. It is also possible that a secondary event may lead to an increase in B cell proliferation in older c-relΔCT/ΔCTmice. In this sense, it is important to mention that there are some biochemical similarities between Δc-Rel in c-relΔCT/ΔCT cells and v-Rel in transformed T cells (34). For example, a significant amount of Δc-Rel is found in the nucleus (data not shown) and increased κB-binding activity, mainly composed of p50/Δc-Rel, is observed in Δc-Rel cells, as in v-Rel–transformed T cells. In addition, similar to v-Rel transgenic mice, lymphoid hyperplasia in c-relΔCT/ΔCT animals was evident after a long period of latency, suggesting that a secondary event is required for the appearance of this phenotype. We have previously expressed Δc-Rel in transgenic thymocytes by using the lck T cell–specific promoter (34). In this model, Δc-Rel did not produce T cell abnormalities, suggesting that the use of a rearranged c-rel gene under the control of its own regulatory elements, as in c-relΔCT/ΔCT mice, is required to recapitulate the situation found in lymphoproliferative disorders.
Molecular Mechanisms Underlying c-Rel and Δc-Rel Function.
The phenotype of c-relΔCT/ΔCT mice has quantitative and qualitative differences from the phenotype described previously for mice lacking the entire c-Rel molecule (21– 23) with c-relΔCT/ΔCTmice showing a more severe phenotype. Since the same strain of mice were used to generate both c-rel −/− and c-relΔCT/ΔCTmice, these differences must be attributable to other factors. In c-relΔCT/ΔCTmice, a transcriptionally inactive molecule has been generated that retains its capability to bind DNA and interact with other Rel/ NF-κB members. The absence of phenotypic alterations in heterozygous c-rel+/ΔCTmice indicate that Δc-Rel does not behave as a transdominant mutant protein of c-Rel. We speculate that by keeping the original niche of c-Rel, Δc-Rel may prevent partial compensation of its function by other Rel/NF-κB members in some cell lineages, which may be the case in c-rel −/− mice. The degree of compensation in c-rel −/− mice depends on the cell type, its physiologic state, and the level of expression of other Rel/NF-κB members. In the case of Δc-Rel, its degree of inactivation will also depend on similar parameters, because of the possibility of forming either transcriptionally inactive or transcriptionally active heterodimers. For instance, in c-rel −/−, but not in c-relΔCT/ΔCTmice, defective proliferation of T cells and cytokine production by T cells was identified (22). Band shift assays performed with nuclear protein extracts from control T cells showed p50/RelA heterodimers as one of the major κB-binding components, whereas in Δc-RelT cells, a significant amount of complexes containing Δc-Rel were detected, including Δc-Rel/RelA heterodimers (Fig. 7 A a). Since cotransfection transcriptional assays demonstrated that Δc-Rel/RelA heterodimers are as active as p50/RelA heterodimers (Fig. 7 B, lanes 6 and 8), it is possible that T cell proliferation and cytokine production are normal in c-relΔCT/ΔCTmice due to cellular-specific compensation by Δc-Rel/RelA heterodimers. On the other hand, in c-rel −/− mice, T cell proliferation is altered because other Rel family proteins in the κB-binding complexes do not compensate for the loss of c-Rel in T cells (22). Alteration in B cell function in c-relΔCT/ΔCTmice is likely related to the fact that the major κB-binding activity in normal B cells is composed of p50/c-Rel heterodimers (Fig. 7 A a), which cannot be substituted by the transcriptional inactive p50/Δc-Rel heterodimer present in Δc-RelB cells (Fig. 7 B, lanes 7 and 9). Quantitative differences in the level of cytokine production in resting macrophages have also been observed between c-rel −/− and c-relΔCT/ΔCTmice. For instance, GM-CSF and IL-6 production are decreased in c-relΔCT/ΔCTmice, but increased in c-rel −/− mice. Since p50/c-Rel is the major κB-binding activity in resting macrophages (data not shown and reference 23), replacement with the inactive p50/Δc-Rel heterodimer in Δc-Rel macrophages results in decreased expression of GM-CSF and IL-6 (Fig. 7 B, lanes 7 and 9). In c-rel −/− macrophages, the p50/c-Rel heterodimer is substituted by the p50/RelA heterodimer that is a more powerful transcriptional activator (Fig. 7 B, lanes 6 and 7). In this way, the expression of GM-CSF and IL-6 genes will likely increase, instead of decreasing as in resting c-rel −/− macrophages.
The function of individual Rel/NF-κB family members has been studied by a gene targeting approach (13). The phenotypic differences between c-rel −/− and c-relΔCT/ΔCTmice suggest the existence of partial compensation of c-Rel function by other Rel/NF-κB members. Molecular compensation has also been suggested in mice deficient for other members of the Rel/NF-κB family. For instance, mice lacking only NF-κB1 or NF-κB2 do not show alterations in bone development. However, mice lacking both NF-κB1 and NF-κB2 develop osteopetrosis due to a defect in osteoclast differentiation (57).
The c-relΔCT/ΔCT mice are a useful model system to study in more detail the role of NF-κB factors in cells of the immune system. The pathological changes observed in c-relΔCT/ΔCT mice may help to understand the pathogenesis of human immune and lymphoproliferative disorders.
Acknowledgments
We are grateful to S. Lira, M. Swerdel, and L. Chen for generating mutant mice; R.-P. Ryseck, J. Caamaño, E. Claudio, and J. Cates for critical discussion; C. Reventos and K. Class for FACS® analysis; T. Gridley (The Jackson Laboratory, Bar Harbor, ME) for CJ7 ES cells; and the staff of Veterinary Sciences at Bristol-Myers Squibb (Princeton, NJ) for their excellent support.
Footnotes
1 Abbreviations used in this paper: Δc-Rel, truncated c-Rel; CAT, chloramphenicol acetyl transferase; CD, cluster of differentiation; EMSA, electrophoretic mobility shift assay; ES, embryonic stem; GC, germinal center; Gr-1, granulocyte 1; LCMV, lymphocytic choriomeningitis virus; Mac-1, macrophage 1; mCD40L, ligand for the mouse CD40; NF, nuclear factor; p, protein; p(A), polyadenylation sequence; PGK, phosphoglycerate kinase; pPNT, plasmid PGK promoter neomycin thymidine kinase; RHD, Rel homology domain; Ter, total erythroid cells.
D. Carrasco's present address is Department of Pathology, Massachusetts General Hospital, Boston, MA 02114.
References
- 1.Liou H-C, Baltimore D. Regulation of the NF-κB/Rel transcription factor and κB inhibitor system. Curr Opin Cell Biol. 1993;5:477–487. doi: 10.1016/0955-0674(93)90014-h. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 3.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 4.Finco TS, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 5.Kopp EB, Ghosh S. NF-κB and Rel proteins in innate immunity. Adv Immunol. 1995;58:1–27. doi: 10.1016/s0065-2776(08)60618-5. [DOI] [PubMed] [Google Scholar]
- 6.Miyamoto S, Verma IM. Rel/NF-κB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 7.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 8.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 9.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin AS. The NF-κB and Iκβ proteins, new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 11.Carrasco D, Ryseck R-P, Bravo R. Expression of relB transcripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- 12.Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-relprotooncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- 13.Attar R, Caamaño J, Carrasco D, Iotsova V, Ishikawa H, Ryseck R-P, Weih F, Bravo R. Genetic approaches to study Rel/NF-κB/ IκB function in mice. Semin Cancer Biol. 1997;8:93–101. doi: 10.1006/scbi.1997.0060. [DOI] [PubMed] [Google Scholar]
- 14.Whiteside ST, Epinat J-C, Rice NR, Israël A. I kappa B epsilon, a novel member of the IκB family, controls RelA and cRel NF-κB activity. EMBO (Eur Mol Biol Organ) J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of the IκBα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 16.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bose HR. The Rel family: models for transcriptional regulation and oncogenic transformation. Biochim Biophys Acta. 1992;1114:1–17. doi: 10.1016/0304-419x(92)90002-g. [DOI] [PubMed] [Google Scholar]
- 18.Kabrun N, Enrietto PJ. The Rel family of proteins in oncogenesis and differentiation. Semin Cancer Biol. 1994;5:103–112. [PubMed] [Google Scholar]
- 19.Gilmore TD. Role of relfamily genes in normal and malignant lymphoid cell growth. Cancer Surv. 1992;15:69–87. [PubMed] [Google Scholar]
- 20.Gilmore TD, Koedood M, Piffat KA, White DW. Rel/NF-κB/IκB proteins and cancer. Oncogene. 1996;13:1367–1378. [PubMed] [Google Scholar]
- 21.Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 22.Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck J-PY, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grigoriadis G, Zhan Y, Grumont RJ, Metcalf D, Handman E, Cheers C, Gerondakis S. The Rel subunit of NF-κB–like transcription factors is a positive and negative regulator of macrophage gene expression: distinct roles for Rel in different macrophage populations. EMBO (Eur Mol Biol Organ) J. 1996;15:7099–7107. [PMC free article] [PubMed] [Google Scholar]
- 24.Bull P, Morley KL, Hoekstra MF, Hunter T, Verma IM. The mouse c-relprotein has an N-terminal regulatory domain and a C-terminal transcriptional transactivation domain. Mol Cell Biol. 1990;10:5473–5485. doi: 10.1128/mcb.10.10.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 26.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DC. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 28.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober. 1992. Isolation and fractionation of mononuclear cell populations. In Current Protocols in Immunology. J.W. Sons, editor. Greene Publishing Associates & Wiley-Interscience, New York. 3.0.1–3.0.5.
- 29.Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, Weih F. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. 1996;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MBA. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. J Exp Med. 1984;60:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grove R, Allegretto N, Kiener P, Warr G. Lipopolysaccharide alters phosphatidylcholine metabolism in elicited peritoneal macrophages. J Leukocyte Biol. 1990;48:38–42. doi: 10.1002/jlb.48.1.38. [DOI] [PubMed] [Google Scholar]
- 32.Johnson KJ, Glovsky M, Schrier D. Pulmonary granulomatous vasculitis: pulmonary granulomatous vasculitis induced in rats by treatment with glucan. Am J Pathol. 1984;114:515–516. [PMC free article] [PubMed] [Google Scholar]
- 33.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- 34.Carrasco D, Rizzo CA, Dorfman K, Bravo R. The v-reloncogene promotes malignant T-cell leukemia/ lymphoma in transgenic mice. EMBO (Eur Mol Biol Organ) J. 1996;15:3640–3650. [PMC free article] [PubMed] [Google Scholar]
- 35.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 36.Dobrzanski P, Ryseck R-P, Bravo R. Both N- and C-terminal domains of RelB are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol Cell Biol. 1993;13:1572–1582. doi: 10.1128/mcb.13.3.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorman C, Moffat LF, Howard B. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryseck R-P, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, Bravo R. RelB, a new Rel family transcription activator that can interact with p50-NF-κB. Mol Cell Biol. 1992;12:674–684. doi: 10.1128/mcb.12.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C-Y, Mayo MW, Baldwin AS., Jr TNF– and cancer therapy–induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 40.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 41.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α–induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann SHE. Immunity to intracellular bacteria. Annu Rev Immunol. 1993;11:129–163. doi: 10.1146/annurev.iy.11.040193.001021. [DOI] [PubMed] [Google Scholar]
- 43.Nathan CF, Hibbs JB., Jr Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Biol. 1991;3:65–70. doi: 10.1016/0952-7915(91)90079-g. [DOI] [PubMed] [Google Scholar]
- 44.Gordon S, Clarke S, Greaves D, Doyle A. Molecular immunobiology of macrophages: recent progress. Curr Opin Immunol. 1995;7:24–33. doi: 10.1016/0952-7915(95)80025-5. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann-Grube F, Moskophidis D, Löhler J. Recovery from acute virus infection. Role of cytotoxic T lymphocytes in the elimination of lymphocytic choriomeningitis virus from spleens of mice. Ann NY Acad Sci. 1988;532:238–256. doi: 10.1111/j.1749-6632.1988.tb36343.x. [DOI] [PubMed] [Google Scholar]
- 46.Richardson PM, Gilmore TD. vRel is an inactive member of the Rel family of transcriptional activating proteins. J Virol. 1991;65:3122–3130. doi: 10.1128/jvi.65.6.3122-3130.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO (Eur Mol Biol Organ) J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarlinton D. Germinal centers: a second childhood for lymphocytes. Curr Biol. 1997;7:R155–R159. doi: 10.1016/s0960-9822(97)70077-0. [DOI] [PubMed] [Google Scholar]
- 49.Thorbecke GJ, Amin AR, Tsiagbe VK. Biology of germinal centers in lymphoid tissue. FASEB J. 1994;8:832–840. doi: 10.1096/fasebj.8.11.8070632. [DOI] [PubMed] [Google Scholar]
- 50.Zelazowski P, Carrasco D, Rosas FR, Moorman MA, Bravo R, Snapper CM. B cells genetically deficient in the c-rel transactivation domain have selective defects in germline CHtranscription and Ig class switching. J Immunol. 1997;154:3133–3139. [PubMed] [Google Scholar]
- 51.Neri A, Fracciolla NS, Rosecetti E, Garatti S, Trecca D, Bolatini A, Perletti B, Bardini L, Maiolo AT, Berti E. Molecular analysis of cutaneous B- and T-cell lymphomas. Blood. 1995;86:3160–3172. [PubMed] [Google Scholar]
- 52.Chang C-C, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu D, Thompson JD, Gorski GK, Rice NR, Mayer MG, Yunis JJ. Alterations at the rellocus in human lymphoma. Oncogene. 1991;6:1235–1241. [PubMed] [Google Scholar]
- 54.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 55.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC. Involvement of granulocyte–macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 56.Ishikawa H, Carrasco D, Claudio E, Ryseck R-F, Bravo R. Gastric hyperplasia and increased proliferative response of lymphocytes in mice lacking the COOH-terminal ankirin domain of NF-κB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iotsova, V., J. Caamaño, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 1285–1289. [DOI] [PubMed]