

Abstract
The pores of glutamate receptors and K+ channels share sequence homology, suggesting a conserved secondary structure. Scanning mutagenesis with substitution of alanine and tryptophan in GluR6 channels was performed based on the structure of KcsA. Our assay used disruption of voltage-dependent polyamine block to test for changes in the packing of pore-forming regions. Alanine scanning from D567 to R603 revealed reduced rectification resulting from channel block in two regions. A periodic pattern from F575 to M589 aligned with the pore helix in KcsA, whereas a cluster of sensitive positions around Q590, a site regulated by RNA editing, mapped to the selectivity filter in KcsA. Tryptophan scanning from D567 to R603 revealed similar patterns, but with a complete disruption of spermine block for 7 out of the 37 positions and a pM dissociation constant for Q590W. Molecular modeling with KcsA coordinates showed that GluR6 pore helix mutants disrupting polyamine block pack against M1 and M2, and are not exposed in the ion channel pore. In the selectivity filter, tryptophan creates an aromatic cage consistent with the pM dissociation constant for Q590W. A scan with glutamate substitution was used to map the cytoplasmic entrance to the pore based on charge neutralization experiments, which established that E594 was uniquely required for high affinity polyamine block. In E594Q mutants, introduction of glutamate at positions S593–L600 restored polyamine block at positions corresponding to surface-exposed residues in KcsA. Our results reinforce proposals that the pore region of glutamate receptors contains a helix and pore loop analogous to that found in K+ channels. At the cytoplasmic entrance of the channel, a negatively charged amino acid, located in an extended loop with solvent-exposed side chains, is required for high affinity polyamine block and probably attracts cations via a through space electrostatic mechanism.
Keywords: AMPA receptor, kainate receptor, polyamines, pore helix, ion channel block
INTRODUCTION
An accumulating body of evidence suggests that the pore region of glutamate receptor ion channels (GluRs) has a similar architecture to that found in K+ channels, sodium channels, calcium channels, hyperpolarization-activated channels, and cyclic nucleotide–gated channels (MacKinnon 1995). This large, and functionally diverse, family of proteins are believed to have in common a structural motif in which a variable number of membrane spanning helices surround a short α-helical segment (the pore helix), which acts both as a dielectric focusing device for cations as well as holding in place a short loop which in K+ channels forms ion binding sites and acts as the selectivity filter (Doyle et al. 1998; Yellen 1999). A pair of linkers connect two of the membrane spanning helices with the pore helix and selectivity filter. Because of their different transmembrane orientations, these linkers form the extracellular surface of K+ channels and the cytoplasmic surface of glutamate receptors.
Definitive evidence for this architecture is available only for one member of the large gene family of K+ channels, the KcsA channel from Streptomyces lividans, for which a structure was solved by X-ray diffraction to 3.2 Å (Doyle et al. 1998). For other channels, the argument for a conserved pore structure comes from multiple but indirect lines of evidence. First, amino acid sequence alignments reveal conserved residues in sequences corresponding to the pore helix, selectivity filter, and the two membrane spanning helices of KcsA (Wo and Oswald 1995; Wood et al. 1995; Yellen 1999). This is particularly striking in the case of GluR0, a prokaryotic glutamate receptor (Chen et al. 1999). For voltage-gated Na+ and Ca2+ channels, hydropathy analysis in addition reveals four repeating domains of additional membrane spanning segments (Catterall 1995), suggesting gene duplication of the core elements that form the channel and gating apparatus of tetrameric voltage-gated K+ channels. Related arguments based on sequence conservation and hydropathy analysis can be made for structural conservation in the pore regions of glutamate receptors, hyperpolarization-activated channels and cyclic nucleotide-gated channels (Chen et al. 1999; Santoro and Tibbs 1999; Wo and Oswald 1995; Wood et al. 1995; Zagotta and Siegelbaum 1996). Because glutamate receptors are ligand-gated and not voltage-gated channels they contain in addition structures which bind glutamate (Armstrong et al. 1998; Armstrong and Gouaux 2000; Chen et al. 1999). Despite increasing acceptance of this framework and the key role of glutamate receptors in central synaptic transmission, considerably less experimental work has been performed to substantiate this vision of pore structure for glutamate receptor ion channels compared with studies on voltage-gated and cyclic nucleotide–gated channels.
Before X-ray crystallographic analysis of KcsA the “M2” segment (see Fig. 1) in glutamate receptors was thought to form a hairpin motif (Wo and Oswald 1995; Wood et al. 1995). Studies in which cysteine substitution accessibility analysis were used to define solvent-exposed side chains in NMDA and AMPA receptors revealed patterns of labeling consistent with an α-helical structure lining the ion channel (Kuner et al. 1996, Kuner et al. 1999). However, these studies, which were performed before the crystallographic analysis of KcsA, revealed labeling of residues, which based on the structure of KcsA, are unlikely to be exposed to the lumen of the pore. Substituted cysteine labeling studies on AMPA receptors also revealed disruption of polyamine block via mechanisms difficult to interpret in the absence of a structural model. Based on our earlier finding that the removal of even a single methylene group from amino acid side chains in the pore region of homomeric GluR6 subtype kainate receptors had effects on polyamine block that exhibited strong positional effects, we decided to systematically test the effects of changes in side chain volume using alanine and tryptophan substitution. For example, the mutant Q590N produced a sevenfold reduction in affinity for spermine while E594D had no effect (Panchenko et al. 1999). The region studied was chosen based on the structure of KcsA and starts in the “turret” that links the first membrane spanning helix with the pore helix (Doyle et al. 1998; MacKinnon et al. 1998); proceeds through the pore helix and selectivity filter and a site at which RNA editing in GluRs regulates sensitivity to polyamine block and permeability to Ca2+, as well as discrimination between anions and cations (Bowie and Mayer 1995; Burnashev et al. 1995, Burnashev et al. 1996); and terminates in residues which align with the second membrane spanning segment of KcsA. The approach used is related to recent work on voltage-gated K+ channels that used alanine and tryptophan substitution and shifts in the voltage dependence of gating to identify the secondary structure and packing of transmembrane helices (Hong and Miller 2000; Li-Smerin et al. 2000a,Li-Smerin et al. 2000b; Monks et al. 1999). Such an approach obtains information from the pattern but not the amplitude or sign of the free energy change produced by individual mutations. Common to these studies is the assumption that disruption of normal channel function is produced by mutants that alter packing at protein–protein but not protein–lipid or protein–solvent interfaces. In addition, we performed a glutamate scan in the linker between the pore helix and the second membrane spanning segment to test the hypothesis that in GluRs this loop forms a pore surface with multiple amino acid side chains exposed to the cytoplasm.
Figure 1.

Homology between glutamate receptors and K+ channels. (A) Amino acid sequence alignment of pore region residues for GluR6, GluRD, GluR0, and KcsA. Residues common to KcsA and any of the three glutamate receptors are shown in bold; yellow shaded boxes indicate conservative substitutions; a red arrow indicates the conserved negative charge at E594. The original transmembrane helix assignments for GluR6 that were based on hydropathy analysis are shown as the top set of shaded boxes labeled “M1,” “M2,” and “M3” (Egebjerg et al. 1991; Lomeli et al. 1992). The numbering for GluR6 assumes processing of a 31–amino acid signal peptide. The dashed line below the GluR6 sequence indicates the extent of the alanine and tryptophan scans performed in the present study. Deletions in the sequence for GluR6, GluRD, and GluR0 are indicated by an asterisk. Below the sequence of KcsA shaded boxes indicate α-helical regions in the X-ray diffraction structure (Doyle et al. 1998); the numbering is for full-length protein. B and C show the secondary structure of KcsA with the sequence corresponding to that used for the GluR6 alanine and tryptophan scans shaded lighter than the surrounding structure; black and red spheres indicate the Cα positions of residues T75 and D80 in KcsA that align with Q590 and E594 in GluR6. The pore helix is indicated by P, and the membrane spanning helices by M1 and M2; this and subsequent figures were drawn using Molscript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997). In B, the orientation with respect to the membrane is that expected for glutamate receptors and is rotated by 180° with respect to that for K+ channels. In C, rotation by 90° shows the cytoplasmic surface and entrance to the pore for the orientation found in glutamate receptors.
The results of our experiments are consistent with a remarkably similar secondary structure of the pore regions of KcsA and GluR6. We also show that a negative charge near the cytoplasmic entrance is an essential requirement for high affinity block by polyamines, and that within an extended loop connecting the pore and inner membrane helices, the position of the charge can be moved over a distance exceeding 17 Å without disrupting block.
MATERIALS AND METHODS
Molecular Biology
Site-directed mutagenesis by PCR, preparation of RNA for injection in Xenopus oocytes, and preparation of DNA for transfection of HEK293 cells were performed as described previously (Panchenko et al. 1999). Silent NheI, MluI and BglII restriction sites in wild-type GluR6(Q) were designed to define the boundaries of 196 and 193 base pair cassettes surrounding the pore region. After generation of individual mutants, these cassettes were sequenced in their entirety and subcloned before use in electrophysiological experiments. Alanine (Ala), Tryptophan (Trp), glutamate (Glu), and truncation mutants were generated by introducing the required codons as appropriate, with valine introduced at positions with a wild-type alanine, and phenylalanine at positions with a wild-type tryptophan.
Oocyte and HEK Cell Preparation
Oocytes were surgically removed from Xenopus laevis (Xenopus One) using aseptic techniques after induction of anesthesia by immersion for 15 min in water containing 3 g/liter tricaine, a protocol approved by the NICHD Animal Care and Use Committee. Animals were killed after a maximum of up to six surgeries. Oocytes were defolliculated by incubation of ovarian fragments for 60–90 min with 1.5 mg/ml collagenase dissolved in Ca2+-free solution containing (in mM): 83 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.5. The preparation was thoroughly rinsed with solution containing (in mM): 88 NaCl, 2.5 NaHCO3, 1.1 KCl, 0.4 CaCl2, 0.3 Ca(NO3)2, 0.8 MgCl2, 2.5 mM sodium pyruvate, 10 HEPES, pH 7.3, and 5 μg/ml gentamicin; and stored overnight. Dumont stage V–VI oocytes were individually selected and injected with 40 pg to 50 ng of cRNA, as required, and stored at 18°C for 2–3 d before recording. Intervals of up to 10 d between mRNA injection and recording were used only for mutants that were nonfunctional or expressed low amplitude responses after an initial assay at 2–3 d.
HEK 293 cells (CRL 1573; American Type Culture Collection) were maintained at a confluency of 70–80% in MEM with Earle's salts, 2 mM glutamine, and 10% fetal bovine serum. 24 h after plating at low density (2 × 104 cells/ml) onto the center of 35-mm petri dishes, cells were transfected using the calcium phosphate technique; cotransfection with the cDNA for green fluorescent protein (S65T mutation) helped to identify transfected cells during experiments as described previously (Bowie and Mayer 1995). Cells were washed with PBS and then MEM 12–18 h after transfection and used for electrophysiological recordings after another 24–48 h.
Recording Conditions and Solutions
Two-electrode voltage clamp recording for Xenopus oocytes was performed using 3 M KCl filled agarose cushion microelectrodes of resistance 0.5–1.2 MΩ (Schreibmayer et al. 1994) and an amplifier (Axoclamp-2B; Axon Instruments) with an extracellular microelectrode used as the input for a virtual ground bath clamp. The recording chamber had a volume of 5 μl; solutions were applied at 250 μl min−1. The extracellular solution contained (in mM): 100 NaCl, 1 KCl, 0.7 BaCl2, 0.8 MgCl2, and 5 HEPES, pH 7.3 with NaOH. GluR6 responses were activated using 100 μM kainate, a concentration >100 times the EC50. 0.3–1 mg ml−1 concanavalin A (Sigma type IV) was applied for 4–16 min to reduce desensitization (Partin et al. 1993; Everts et al. 1999). The length of application of concanavalin A was adjusted for individual recordings to give current amplitudes of 1–5 μA at −60 mV. Current-voltage (I-V) plots from −100 to 100 mV were generated with ramp protocols (0.11–2.7 Vs−1). For mutants that did not give functional responses at −60 mV after application of 1 mg ml−1 concanavalin A for 8–10 min the voltage range was extended to −200 mV to test for high affinity polyamine block (Panchenko et al. 1999).
Outside-out patches were excised from HEK cells using fire-polished, thin-walled borosilicate glass pipets (2–5 MΩ) coated with dental wax to reduce electrical noise. Experiments were performed in an external solution containing (in mM): 150 NaCl, 1 KCl, 0.7 BaCl2, 0.8 MgCl2, and 5 HEPES, pH 7.3; and the osmolarity was adjusted to 295 mOsm with sucrose. GluR6 responses were evoked using 50 μM domoic acid, a weakly desensitizing agonist, applied via a stepper motor-based fast perfusion system (Vyklicky et al. 1990). In some experiments, patches were treated with concanavalin A (0.3 mg/ml for 1–1.5 min) to further reduce desensitization. The internal solution contained (mM): 110 NaCl, 10 NaF, 10 mM Na2ATP, 5 HEPES, 0.5 CaCl2, and 5 Na4BAPTA. Currents were recorded with an amplifier (Axopatch-200; Axon Instruments), filtered with an 8-pole Bessel filter, and stored on a Power Macintosh G3 computer using a 16-bit A/D converter (model ITC-16; Instrutech) under control of the program Synapse (Synergy Research Incorporated).
Data Analysis
Procedures in the Igor program (Wavemetrics) were used to generate and analyze conductance-voltage (G-V) plots. First, the reversal potential (Vrev) was estimated using a fifth order polynomial fit to I-V plots for either single (oocytes) or the average of five (HEK cells) leak-subtracted responses. G-V plots were generated from the relationship G = I/(V − Vrev) and used to measure the conductance at +80 and −80 mV. A fit of the following Boltzmann function over the range −100 to +20 mV was used to obtain an initial estimate of the voltage dependence of block by polyamines
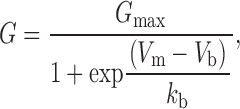 |
where G max is the conductance at a sufficiently hyperpolarized potential to produce full relief from block by polyamines, Vm is the membrane potential, Vb the potential at which 50% block occurs, and k b is a slope factor describing the voltage dependence of block. To analyze block over a wider range of membrane potentials for mutants with a low affinity for polyamines, it was necessary to correct for rectification that occurs in polyamine-free conditions (Bähring et al. 1997; Bowie et al. 1998). To do this, pooled responses recorded in separate experiments in the presence and absence of polyamines were normalized to have a value of 1 at −100 mV. Corrected G-V plots were generated by dividing responses recorded in the presence of polyamines by those recorded in polyamine-free conditions. The resulting plots were fit with a function describing the behavior of the two barrier one- site (2B1S) model for a permeant blocker:
 |
where G max, Vm, Vb, and kb have the same meaning as described above, Vp is the half unblock potential for the second Boltzmann function describing the permeation of polyamines on strong depolarization, and kp is the slope factor describing the voltage dependence of relief from block. A voltage-independent dissociation constant for polyamine block, K d(0), was calculated from the following relationship:
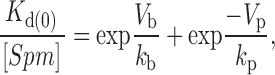 |
assuming a cytoplasmic concentration of spermine, spermidine, and possibly other polyamines functionally equivalent to the block produced by 20 μM spermine [Spm] alone (Panchenko et al. 1999).
Numerical values in the text and error bars in graphs indicate mean ± SD, unless noted differently. Unpaired t tests were used to test for significant differences where required.
Immunohistochemistry
4 d after injection with 10 ng cRNA, oocytes were fixed overnight by immersion in 100 mM PBS, pH 7.4, containing 4% paraformaldehyde and 15% of a saturated solution of picric acid; uninjected oocytes from the same preparations were processed identically. The oocytes were cryoprotected by overnight immersion in a solution containing 10 mM PBS, 10% glycerol, 0.008% NaN3, and 25 g/100 ml sucrose. 100-μM sections were cut on a freezing microtome, incubated with 2% Triton in PBS, blocked with 10% goat serum in 0.2% Triton for 2 h, and reacted overnight at 4°C with 1 μg/ml rabbit anti–GluR6 antibody (Wenthold et al. 1994) in PBS containing 10% goat serum and 0.2% Triton. Sections were incubated at room temperature for 60 min with FITC-conjugated goat anti–rabbit serum (1/200). An Olympus AX70 microscope and Optronics Magnafire digital camera was used for imaging.
Online Supplemental Material
A PDB file for the model shown in Fig. 8 (below) is available online at: http://www.jgp.org/cgi/content/full/117/4/345/DC1.
Figure 8.

Mapping GluR6 Q/R site and glutamate scan mutations to KcsA secondary structure. Two complete KcsA subunits plus the pore helix and surrounding linkers to M1 and M2 for a third subunit are shown and labeled as for Fig. 1. The side chain for T75 that maps to Q590 in an alignment with GluR6 (Fig. 1) was mutated to tryptophan. The side chain bond angles between the Cα–Cβ–Cγ atoms were adjusted to rotate the indole ring into the cavity of KcsA; the resulting aromatic cage has minimal bad VDW contacts with surrounding residues. The Cα positions of residues in the pore surface that, when mutated to glutamate, restored high affinity polyamine block in the E594Q background are shown as red spheres. The two positions for which mutation to glutamate abolished polyamine block in the wild-type background are shown as black spheres.
RESULTS
We began our experiments by performing sequence alignments for GluR6 (Egebjerg et al. 1991) and GluRD (Keinänen et al. 1990), polyamine-sensitive glutamate receptor ion channels (GluRs) from the kainate and AMPA receptor gene families; for GluR0, a prokaryotic glutamate receptor K+ channel (Chen et al. 1999); and for KcsA, a prokaryotic K+ channel of known structure (Doyle et al. 1998; Schrempf et al. 1995). This revealed conservation of amino acid sequence between KcsA and both eukaryotic and prokaryotic GluRs in regions corresponding to the pore helix, pore loop, and the inner and outer membrane crossing helices (Fig. 1).
GluR6 Sequence Mapped onto the KcsA Crystal Structure
The amino acid sequence conservation shown in Fig. 1 suggests that GluRs and KcsA might have a similar structure in the pore helix and pore loop (Wo and Oswald 1995; Wood et al. 1995), a hypothesis reinforced by the recent discovery of a K+ selective prokaryotic glutamate receptor ion channel GluR0 (Chen et al. 1999). To test whether KcsA can be used to accurately predict secondary structure in the pore of GluRs we performed scanning mutagenesis for GluR6 with substitution of small and large side chains to look for patterns in changes of polyamine block. We limited our scan to the pore helix and pore loop because eukaryotic GluRs have an additional membrane spanning segment (M3) that most likely packs against M1 and M2. As a result, in eukaryotic GluRs, the outer faces of the helices equivalent to M1 and M2 in KcsA will most likely make both protein–protein contacts with M3 as well as protein–lipid interactions and require a different amino acid side chain chemistry from that in KcsA. To allow for uncertainty in the length of the pore helix in GluR6 resulting from variability in the number of residues between the end of M1 and the predicted start of the pore helix, a region which is also of variable length in K+ channels and which in KcsA forms turret protruding beyond the mouth of the pore, we designed our Ala scan to start at D567 well before the likely start of a pore loop in GluR6 (Fig. 1).
Alanine Scanning Mutagenesis in the Pore Region of GluR6
Wild-type GluR6 responses show such strong biphasic rectification due to permeable block by cytoplasmic polyamines that there is nearly complete attenuation of outward current flow between 10 and 50 mV (Bowie and Mayer 1995). In previous experiments for which wild-type GluR6 responses were recorded from >100 oocytes, we estimated that cytoplasmic polyamine concentrations varied at most twofold between oocytes (Panchenko et al. 1999). At 0 mV membrane potential K d(0), the equilibrium dissociation constant of wild-type GluR6 for spermine, was 1.25 μM at 100 mM [Na+]o and decreased e-fold per 15-mV depolarization over the range −100 to +50 mV (Panchenko et al. 1999). GluR6 mutants with alanine substituted from D567 to R603 showed a complete loss of polyamine block for 3 out of the 37 positions tested, F583A, G592A, and E594A. For the other 34 mutants tested, we recorded biphasic rectification with strong voltage dependence, and for some mutants, shifts in the half block potential. Analysis of I-V and G-V plots for two typical examples (Q590A and M589A) are shown in Fig. 2. For Q590A, the block of outward current by polyamines was reduced compared with wild type (Fig. 2 A). Analysis of G-V plots revealed a rightward parallel shift compared with responses for wild-type GluR6 with Vb and k b values of −12.0 ± 4.1 mV and 13.4 ± 0.9 mV−1 (n = 6) and a K d(0) of 8.3 ± 2.9 μM (Fig. 2 B). For wild-type GluR6, the corresponding values for Vb and k b were −43.6 ± 3.1 mV and 15 ± 1.0 mV−1 (n = 9). In contrast, responses for M589A were indistinguishable from those for wild-type GluR6. I-V plots for M589A showed nearly complete attenuation of outward current flow between 10 and 50 mV (Fig. 2 A), and analysis of G-V plots gave values of −44.6 ± 1.6 mV and 15.9 ± 0.8 mV−1 (n = 6) for Vb and k b, respectively, with a K d(0) for spermine of 1.2 ± 0.1 μM (Fig. 2 B).
Figure 2.

Alanine scanning mutagenesis disrupts polyamine block with characteristic patterns separating sensitive and insensitive residues. (A) Leak-subtracted I-V plots reveal reduced rectification and increased outward currents for Q590A but not M589A. (B) Mean G-V plots for these mutants with the SD plotted at 20-mV intervals. Responses for wild-type GluR6 (dashed line) nearly overlie responses for M589A. (C) K d(0) values for alanine-substituted GluR6 mutants from D567 to R603 measured using Boltzmann fits to G-V plots like those shown in B. Shaded boxes indicate responses for which G-V plots did not show the biphasic rectification characteristic of polyamine block; a conservative lower estimate of K d(0) for these positions is >100 μM. The shaded horizontal box indicates a plus/minus twofold change from the K d(0) value for wild-type GluR6. α-Helical regions in the secondary structure of KcsA mapped onto the GluR6 sequence as shown in Fig. 1 are shown above the bar plot.
For the 34 Ala mutants with measurable biphasic rectification because of polyamine block, the mean value for k b was 16.4 ± 1.94 mV−1, with values for Vb ranging from −47.9 to 3.9 mV and K d(0) values from 0.8 to 26 μM (Table ). The similar values for kb for these mutants indicate changes in affinity for polyamines but not the electrical location of the binding site(s) (Panchenko et al. 1999). We did not attempt to analyze the weak rectification observed for F583A, G592A, and E594A, for which polyamines bind with too low an affinity to allow accurate estimation of K d(0) at the concentrations present in the cytoplasm of Xenopus oocytes. Because we were interested primarily in the pattern of changes produced by scanning mutagenesis, we simply note for these mutants that K d(0) is likely to be >100 μM. Plots of the value for K d(0) versus amino acid sequence for GluR6 Ala scan mutants revealed a cluster of amino acid residues sensitive to alanine substitution located close to position 590, the Q/R site at which RNA editing regulates polyamine block (Fig. 2 C). When mapped to the sequence alignment with KcsA, these positions correspond to the selectivity filter (Fig. 1). Alanine substitutions within the preceding region corresponding to the pore helix produced a periodic pattern of changes in polyamine affinity which, as discussed later, map to those faces of the pore helix in KcsA that interact with the surrounding pore loop and membrane spanning helices. Ala mutations in the sequence preceding the pore helix, corresponding to the turret in KcsA, and mutations corresponding to the start of M2 (the inner helix) had no effect. The reduction of polyamine block by mutation of amino acid side chains to alanine most likely results from either a change in structure of the pore region due to altered packing of the pore-forming segments, or possibly a specific contribution of individual solvent accessible amino acid side chains to polyamine binding sites. To distinguish between these possibilities, we performed a 37-residue tryptophan scan over the same region (from D567 to R603), allowing us to compare the effects of introducing small and large hydrophobic chains at the same positions. Our expectation was that introduction of tryptophan would produce large structural perturbations in the pore and, hence, change polyamine block for residues making protein–protein contacts, whereas for solvent-exposed residues, an increase in affinity was possible because of a larger surface for hydrophobic interactions. Such a mechanism has been shown to play a major role in the mechanism of binding of polyamines to both GluR6 and K+ channels (Cui et al. 1998; Guo and Lu 2000b; Pearson and Nichols 1998).
Table 1.
The Effect of Amino Acid Substitutions on Polyamine Block in Homomeric GluR6
| Alanine scan | Tryptophan scan | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GluR6 | GR ± 80 | Vb | k b | K d(0) | GR ± 80 | Vb | k b | K d(0) | KcsA | ||
| mV | μM | mV | μM | ||||||||
| D567 | 17.1 ± 5.0 | −42.5 ± 3.5 | 15.2 ± 1.1 | 1.2 ± 0.1 | 19.1 ± 1.6 | −44.2 ± 1.3 | 14.1 ± 0.3 | 1.0 ± 0.01 | — | — | — |
| S568 | 14.1 ± 4.4 | −42.9 ± 6.0 | 15.7 ± 0.8 | 1.3 ± 0.2 | 15.4 ± 2.1 | −41.6 ± 1.2 | 14.4 ± 0.6 | 1.3 ± 0.01 | G53 | Turret | Solvent |
| D569 | 13.3 ± 4.2 | −41.9 ± 5.4 | 14.7 ± 0.7 | 1.2 ± 0.2 | 14.7 ± 2.7 | −41.8 ± 3.0 | 14.3 ± 0.3 | 1.1 ± 0.01 | A54 | Turret | Solvent |
| V570 | 11.6 ± 2.1 | −43.8 ± 5.6 | 14.1 ± 0.3 | 0.9 ± 0.1 | 12.6 ± 0.7 | −40.9 ± 1.0 | 14.5 ± 0.4 | 1.3 ± 0.01 | P55 | Turret | Solvent |
| V571 | 13.0 ± 3.1 | −47.3 ± 3.0 | 15.0 ± 0.4 | 0.9 ± 0.1 | 11.4 ± 0.6 | −40.9 ± 0.8 | 14.2 ± 0.1 | 1.3 ± 0.01 | G56 | Turret | Solvent |
| E572 | 19.0 ± 3.2 | −37.2 ± 2.7 | 15.1 ± 0.4 | 1.7 ± 0.1 | 29.8 ± 3.0 | −42.0 ± 1.9 | 15.3 ± 0.2 | 1.3 ± 0.01 | A57 | Turret | Solvent |
| N573 | 6.8 ± 1.7 | −47.9 ± 1.7 | 15.0 ± 0.8 | 0.8 ± 0.1 | 12.0 ± 0.3 | −48.2 ± 0.7 | 15.5 ± 0.4 | 1.2 ± 0.01 | Q58 | Turret | Solvent |
| N574 | 14.0 ± 2.7 | −36.8 ± 2.0 | 15.2 ± 0.9 | 1.8 ± 0.1 | 14.2 ± 0.8 | −40.7 ± 0.3 | 15.5 ± 0.2 | 1.8 ± 0.02 | L59 | Turret | Protein |
| F575 | 56.7 ± 6.1 | −18.1 ± 1.4 | 18.5 ± 1.1 | 7.6 ± 0.7 | 10.0 ± 2.1 | −54.5 ± 2.1 | 14.4 ± 0.6 | 0.4 ± 0.01 | I60 | Turret | Solvent |
| T576 | 29.8 ± 3.7 | −31.3 ± 1.3 | 21.4 ± 1.8 | 4.7 ± 0.4 | 96.5 ± 4.2 | ND | ND | >100 | T61 | Turret | Solvent |
| L577 | 10.9 ± 1.7 | −38.1 ± 3.0 | 16.1 ± 0.6 | 1.9 ± 0.2 | 12.4 ± 2.1 | −41.1 ± 1.5 | 14.3 ± 0.4 | 1.4 ± 0.02 | Y62 | Helix | Lipid |
| L578 | 7.0 ± 1.5 | −39.4 ± 1.2 | 14.7 ± 0.6 | 1.4 ± 0.1 | 18.5 ± 1.2 | −31.8 ± 0.6 | 17.9 ± 0.5 | 3.7 ± 0.04 | P63 | Helix | Lipid |
| N579 | 17.4 ± 2.0 | −29.4 ± 1.1 | 17.1 ± 0.5 | 3.6 ± 0.2 | 75.3 ± 5.1 | ND | ND | >100 | R64 | Helix | Solvent |
| S580 | 8.4 ± 1.2 | −40.9 ± 0.6 | 14.4 ± 0.7 | 1.2 ± 0.1 | 87.9 ± 4.3 | ND | ND | >100 | A65 | Helix | Protein |
| F581 | 9.4 ± 1.9 | −40.6 ± 2.0 | 15.6 ± 1.3 | 1.5 ± 0.1 | 11.0 ± 1.8 | −43.6 ± 1.0 | 14.7 ± 0.4 | 1.3 ± 0.01 | L66 | Helix | Lipid |
| W582 | 50.5 ± 8.4 | −10.8 ± 3.2 | 20.7 ± 2.9 | 12.0 ± 3.9 | 41.9 ± 2.7 | −24.5 ± 1.2 | 17.0 ± 0.6 | 5.2 ± 0.05 | W67 | Helix | Lipid |
| F583 | 70.3 ± 1.5 | ND | ND | >100 | 9.1 ± 0.2 | −53.3 ± 3.0 | 15.5 ± 0.8 | 0.7 ± 0.01 | W68 | Helix | Protein |
| G584 | 8.8 ± 2.3 | −41.9 ± 1.2 | 14.7 ± 0.9 | 1.2 ± 0.1 | 327.4 ± 56.5 | ND | ND | >100 | S69 | Helix | Protein |
| V585 | 18.6 ± 2.5 | −27.1 ± 1.3 | 16.1 ± 0.7 | 3.8 ± 0.2 | 3.6 ± 1.2 | −50.5 ± 1.5 | 14.7 ± 1.3 | 0.7 ± 0.01 | V70 | Helix | Lipid |
| G586 | 54.1 ± 5.0 | −9.5 ± 4.7 | 18.3 ± 2.4 | 12.1 ± 6.2 | 109.3 ± 1.8 | ND | ND | >100 | E71 | Helix | Protein |
| A587 | 75.2 ± 9.3 | 3.9 ± 2.4 | 15.8 ± 1.4 | 26.0 ± 15.8 | 70.0 ± 5.8 | 3.1± 6.4 | 20.9 ± 1.0 | 23.3 ± 1.38 | T72 | Helix | Protein |
| L588 | 5.8 ± 1.3 | −45.1 ± 2.1 | 15.8 ± 0.6 | 1.2 ± 0.1 | 53.3 ± 5.2 | −7.6 ± 0.6 | 32.7 ± 5.8 | 17.2 ± 0.40 | A73 | Helix | Protein |
| M589 | 7.9 ± 1.6 | −44.6 ± 1.6 | 15.9 ± 0.8 | 1.2 ± 0.1 | 94.0 ± 5.9 | −8.4 ± 1.3 | 18.3 ± 0.5 | 12.2 ± 0.16 | T74 | Helix | Protein |
| Q590 | 74.7 ± 2.4 | −12.0 ± 4.1 | 13.4 ± 0.8 | 8.3 ± 2.9 | 0.0 ± 0.0 | −191.3 ± 4.7 | 17.1 ± 1.3 | 190 ± 29 pM | T75 | P–loop | Pore |
| Q591 | 77.6 ± 5.1 | −6.0 ± 2.5 | 19.8 ± 0.7 | 15.0 ± 6.2 | 70.4 ± 2.6 | −14.9 ± 2.4 | 23.7 ± 0.8 | 12.1 ± 0.12 | V76 | P–loop | Protein |
| G592 | 61.9 ± 2.2 | ND | ND | >100 | 195.8 ± 6.7 | ND | ND | >100 | G77 | P–loop | Protein |
| S593 | 18.9 ± 2.4 | −32.9 ± 1.4 | 16.7 ± 0.8 | 2.8 ± 0.2 | 62.1 ± 1.1 | 9.8 ± 1.5 | 29.9 ± 0.6 | 25.6 ± 0.98 | G79 | P–loop | Protein |
| E594 | 216.8 ± 8.2 | ND | ND | >100 | 264.0 ± 6.6 | ND | ND | >100 | D80 | Surface | Solvent |
| L595 | 14.4 ± 1.9 | −35.9 ± 1.7 | 16.4 ± 0.9 | 2.3 ± 0.2 | 20.0 ± 3.3 | −40.7 ± 2.0 | 21.3 ± 1.9 | 3.2 ± 0.04 | L81 | Surface | Solvent |
| M596 | 20.6 ± 2.0 | −34.2 ± 1.5 | 16.1 ± 0.5 | 2.4 ± 0.1 | 18.9 ± 3.3 | −40.9 ± 0.6 | 15.9 ± 0.8 | 1.8 ± 0.02 | Y82 | Surface | Solvent |
| P597 | 42.9 ± 2.0 | −15.9 ± 2.3 | 18.7 ± 1.3 | 8.7 ± 1.4 | 48.6 ± 2.6 | −13.6 ± 3.8 | 30.2 ± 2.7 | 13.4 ± 0.20 | P83 | Surface | Protein |
| K598 | 7.0 ± 1.8 | −41.7 ± 2.4 | 16.5 ± 1.5 | 1.6 ± 0.2 | 50.1 ± 4.6 | −48.2 ± 7.9 | 28.0 ± 7.0 | 5.9 ± 0.08 | V84 | Surface | Solvent |
| A599 | 21 ± 4.1 | −38.6 ± 3.3 | 16.1 ± 0.3 | 1.8 ± 0.2 | NR | NR | NR | NR | T85 | Surface | Solvent |
| L600 | 7.7 ± 0.7 | −38.6 ± 1.2 | 15.5 ± 0.8 | 1.7 ± 0.1 | 16.7 ± 3.9 | −32.0 ± 4.3 | 18.2 ± 1.2 | 3.8 ± 0.04 | L86 | M2 | Lipid |
| S601 | 21.2 ± 2.0 | −41.7 ± 2.1 | 17.9 ± 1.2 | 2.0 ± 0.2 | NR | NR | NR | NR | W87 | M2 | Lipid |
| T602 | 8.5 ± 1.1 | −45.6 ± 1.6 | 15.0 ± 0.3 | 1.0 ± 0.0 | NR | NR | NR | NR | G88 | M2 | Protein |
| R603 | 24.8 ± 2.2 | −40.5 ± 0.6 | 20.2 ± 0.4 | 2.7 ± 0.1 | 2.8 ± 2.3 | −44.2 ± 1.8 | 16.5 ± 0.7 | 1.7 ± 0.02 | R89 | M2 | Solvent |
| WT | 9.0 ± 1.3 | −43.6 ± 3.1 | 15.1 ± 1 | 1.2 ± 0.02 | |||||||
Changes in voltage dependence for polyamine block of GluR6 alanine and tryptophan scan mutants were measured using Boltzmann analysis of G-V plots. Values indicate mean ± SD for three to seven oocytes per mutant. NR, nonfunctional mutants. Labels for KcsA indicate the projections and structural role of side chains for residues that align with GluR6, as shown in Fig. 1. The pore helix, selectivity filter, and inner membrane spanning helix in the KcsA crystal structure are indicated by Helix, P-loop, and M2, respectively.
Tryptophan Scanning Mutagenesis Reveals Disruption of a Pore Helix
Substitution of tryptophan from D567 to R603 produced both increases and decreases in polyamine block as well as three nonfunctional mutants. Changes in polyamine block were quantified by analysis of G-V plots as described for results of the alanine scan. Although tryptophan mutants showed much larger changes in polyamine block than observed for alanine substitution, these effects also showed strong positional dependence and, for 14 mutants, K d(0) values for spermine were within twofold of the wild type (Table ). For two positions, tryptophan mutants showed stronger block than for wild-type GluR6, an effect not observed for any of the alanine mutants. As reported previously, this effect was dramatic for the mutant Q590W for which the affinity for spermine increased 6.6 × 103-fold (Panchenko et al. 1999). In our previous study, which focused only on mutations at Q590 and E594, we did not identify the structural basis for this striking effect or determine whether introduction of tryptophan at positions other than Q590 or E594 would increase polyamine affinity. The Trp scan performed in the present study revealed that a high affinity for spermine, K d(0) = 190 ± 29 pM (n = 6), was unique for the Q590W mutant (Fig. 3). A much smaller threefold decrease in K d(0) to 0.42 ± 0.01 μM (n = 5) was observed for the mutant F575W, whereas for F583W and V585W, although K d(0) values were below 1 μM, they were less than twofold different from wild type (Table ). Since the F583W mutation slightly increased polyamine block, whereas the F583A mutation was highly disruptive (Fig. 2), we tested additional mutations with side chain volumes between those for Ala and Trp. This revealed intermediate effects on polyamine block for the F583L and F583H mutants for which we obtained K d(0) values of 18.9 ± 0.3 μM (n = 4) and 18.1 ± 0.3 μM (n = 4), respectively.
Figure 3.

Tryptophan scanning mutagenesis strongly disrupts polyamine block. (A) Mean G-V plots for the adjacent pair of mutants M589W (n = 3) and Q590W (n = 6); dashed lines show the fit of one (Q590W) or two (M589W) Boltzmann functions; SDs are plotted every 20 mV. (B) Mean G-V plots for S580W and G584W, two examples of mutants for which introduction of tryptophan abolished the strongly voltage-dependent rectification resulting from permeable block by polyamines. (C) Changes in K d(0) for tryptophan-substituted GluR6 mutants from D567 to R603 measured from responses like those shown in A and B; shaded boxes indicate responses like those for G584W and S580W for which G-V plots did not show the biphasic rectification characteristic of polyamine block. Of the 37 positions studied, no response (NR) was recorded for 3 positions. The shaded horizontal box indicates a plus/minus twofold change from the K d(0) value for wild-type GluR6. α-Helical regions in the secondary structure of KcsA mapped onto the GluR6 sequence as shown in Fig. 1 are shown above the bar plot.
Similar to the results of the alanine scan, a reduction in polyamine block was the more typical effect recorded for 18 positions substituted with tryptophan. As for the Ala scan mutants, these responses fell into two classes. For 11 positions, there were parallel rightward shifts in G-V plots compared with responses for wild-type GluR6, reflecting a decrease in affinity for spermine with little change in the voltage dependence of block (Fig. 3). For these mutants, the mean value for k b was 23.5 ± 5.8 mV−1 with values for Vb ranging from −48.2 to 9.8 mV and K d(0) values of 3.2 to 25.6 μM (Table ). Responses for a typical example (M589W), with k b = 18.3 ± 0.5 mV−1, Vb = −8.4 ± 1.3 mV, and K d(0) = 12.2 ± 0.2 μM (n = 3), are shown in Fig. 3 A. For 7 other positions, polyamine block was much more strongly disrupted. This prevented an accurate estimation of K d(0) at the physiological concentrations of spermine present in the cytoplasm of Xenopus oocytes. G-V plots for mutants at these positions were either outward-rectifying with conductance ratios at +80/−80 mV of 327 ± 57% for G584W (n = 4), 195 ± 7% for G592W (n = 4), and 264 ± 7% for E594W (n = 5), or showed very weak voltage dependence with conductance ratios at +80/−80 mV of 96 ± 2% for T576W (n = 4), 75 ± 5% for N579W (n = 3), 88 ± 4% for S580W (n = 4), and 109 ± 2% for G586W (n = 3). Typical examples are shown in Fig. 3 B for S580W and G584W. For these and other mutants with low sensitivity to polyamines, we again note that the K d(0) is likely to be >100 μM. Since our goal was to measure the pattern of changes produced by scanning mutagenesis with tryptophan, rather than individual K d(0) values for mutants with very low affinity for polyamines, we did not attempt to characterize these mutants further.
For three mutants, A599W, S601W, and T602W, we were unable to evoke functional responses even though the corresponding Ala mutants for these positions expressed well and had K d(0) values within twofold of the wild type. In an attempt to maximize expression for these nonfunctional mutants, we used an interval of 10 d between recording and injection of 50 ng mRNA with incubation of 10 μM concanavalin A for 14–16 min before application of agonist. For wild-type GluR6, these conditions typically gave responses that were in excess of 20 μA at −60 mV after only 4-min application of concanavalin A and saturated the amplifier with longer applications of lectin. The threshold for detection of agonist responses was typically 1–2 nA at −60 mV. Ramps to −200 mV to check for high affinity polyamine block like that seen for Q590W also failed to reveal latent responses. To distinguish between the possibility that these nonfunctional responses were due to gating mutants, or simply reflected a lack of cell surface expression, oocytes were injected with mRNA for A599W and S601W (T602W was not tested) and stained by indirect immunofluorescence with an antibody directed against the COOH terminus of GluR6. Staining was indistinguishable from that for uninjected oocytes, whereas positive controls with wild-type GluR6 showed intense cell-surface expression as described previously (Panchenko et al. 1999). Thus, the lack of functional responses for these mutants reflects the absence of cell-surface expression due to either instability, misfolding, or accumulation of protein in intracellular compartments, and not a disruption of gating.
Ala and Trp Mutants that Reduce Polyamine Block Cluster on One Face of the KcsA Pore Helix
When the results of the Ala and Trp scans were compared (Fig. 2 and Fig. 3) it was obvious that the most sensitive regions aligned with the pore helix and selectivity filter of KcsA. When results for the GluR6 sequence from T576 to M589 (which aligns with the pore helix in KcsA) were mapped onto a helical wheel, we found that those mutants which disrupted polyamine block showed a clustered distribution (Fig. 4 A). Using this sequence alignment and the KcsA atomic coordinates, we asked where are residues that disrupted polyamine block in GluR6 located in the KcsA structure. This analysis showed that, in the pore helix, the side chains of residues for which Ala and Trp substitution disrupted polyamine block projected either towards the membrane spanning helices and selectivity filter within the same subunit, or towards the selectivity filter of an adjacent subunit (Fig. 4 B). In contrast, the amino acid side chains of positions insensitive to substitution with either Ala or Trp projected away from both the selectivity filter and M1 or M2 and likely face lipid. A detailed discussion of results for some individual positions is given later. The other mutants that strongly disrupted polyamine block were either within sequences corresponding to the selectivity filter in KcsA or neutralized the negative charge at E594.
Figure 4.

Results of scanning mutagenesis suggest a pore helix in GluR6. (A) Helical wheel analysis of Ala and Trp substitutions from T576 to M589. Black circles indicate positions for which substitution of tryptophan abolished polyamine block (Fig. 3 C, shaded bars). White circles indicate positions for which alanine substitution increased K d(0) to more than four times the value for wild-type GluR6 as shown in Fig. 2 C. Shaded circles indicate positions for which both Ala and Trp were effective. (B) Secondary structure for two adjacent KcsA subunits viewed along the axis of the pore helix (P) for the subunit on the left; the orientation matches that shown for the helical wheel plot; M1 and M2 indicate the outer and inner membrane spanning helices; an arrow indicates the central axis of the permeation pathway and points towards the central cavity (Fig. 1). Side chains are drawn in black for KcsA positions aligning with GluR6 residues mutation of which strongly disrupts polyamine block.
Neutralization of Charged Residues in the Cytoplasmic Linkers between M1 and M2
The results of the Ala and Trp scans revealed strong disruption of polyamine block for mutation of E594 to either alanine or tryptophan. This residue aligns with D80 in KcsA and is located immediately after the selectivity filter near the entrance to the channel (Fig. 1). Of note, in cyclic nucleotide–gated channels, the mutation E363G, which aligns with E594 and D80, also reduces block by polyamines (Guo and Lu 2000a). Together, these results suggest that it is the loss of charge resulting from mutations at E594 and not changes in side chain volume that abolishes high affinity polyamine block in GluR6. There are an additional four glutamate and aspartate residues in the linker between the first membrane spanning segment and the pore helix of GluR6, whereas, in the 68–amino acid COOH terminus of GluR6, there are an additional eight negatively charged side chains (Fig. 5). Any of these could potentially contribute to polyamine binding sites in conjunction with E594. This was tested by constructing a series of seven point mutations changing Glu to Gln or Asp to Asn, and three COOH-terminal truncation mutants at M862, K839, and M830, each of which progressively removed sets of two negative charges as shown in Fig. 5. Measurements of G+80/G−80 revealed only small changes in rectification for each of these charge neutralization mutants except for E594Q for which the ratio G+80/G−80, 198 ± 7% (n = 4), was similar to the values for values for the E594A and E594W mutants, indicating that the negative charge at this position is indeed the major determinant of polyamine block (Fig. 5). These experiments also reveal that 48 amino acids at the COOH terminus are not required for assembly, gating, or normal function of GluR6. Adjacent to E594 is a lysine at position 598; there are two other positively charged side chains at the cytoplasmic ends of the first two transmembrane segments: arginines at positions 552 near the end of M1 and 603 near the start of M2. We neutralized these residues by mutation to Gln or Asn to test whether removal of positively charged side chains would increase polyamine affinity because of decreased shielding of E594 or removal of a local positive surface charge that could repel polyamines. Although a small decrease in the ratio G+80/G−80 was recorded for K598N, 5.0 ± 1.6% vs. 9.0 ± 1.3% for wild-type GluR6, analysis of G-V plots suggested a slightly lower affinity of K598N for spermine, K d(0) 1.5 ± 0.1 μM (n = 17) vs. 1.3 ± 0.02 μM (n = 9) for wild-type GluR6, reflecting a small rightward shift observed in G-V plots for K598N, Vb −40.2 ± 0.8 mV vs. −43.6 ± 1.0 mV (P < 0.05), with no change in slope, k b 15.0 ± 0.3 vs. 15.5 ± 0.2 (mean ± SEM, P > 0.25). Clearly, despite the proximity of these two residues the positive charge of K598 does not interfere with binding of polyamines to the negative charge of E594.
Figure 5.

Neutralization of charged amino acids in the linkers between the pore helix and transmembrane helices. (A) Membrane folding model for GluR6 based on the alignments shown in Fig. 1 indicates the positions of individual residues for which glutamate and aspartate residues were neutralized one at a time by a mutation to glutamine and asparagine. M1, M2, and M3 indicate the three membrane spanning segments in GluRs; P indicates the pore helix. Scissors indicate positions at which deletion mutants were generated by introduction of a stop codon. Three basic residues were also neutralized by mutation of lysine and arginine side chains to asparagine and glutamine, respectively. (B) Plots of the ratio of conductance at +80/−80 mV for the charge neutralization and deletion mutants shown in A; the shaded box indicates the 95% confidence interval for wild-type responses. Of the 13 Glu and Asp positions tested using charge neutralization, only E594 was required for high affinity polyamine block.
A Glu Scan Maps the Limits of Surface-exposed Residues in the Linker between the Pore Loop and M2
In a sequence alignment of GluR6 and KcsA, E594 corresponds to D80 (Fig. 1). In K+ channels, the sequence after D80 links the selectivity filter with the NH2 terminus of the last membrane spanning helix and forms a surface that contributes to the binding site for K+ channel toxins (MacKinnon et al. 1998). Thus, if the pore surfaces of GluR6 and KcsA have similar structures, then E594 should be located in a loop that contains multiple solvent-exposed amino side chains facing the cytoplasm (Fig. 1). Such an arrangement suggests that it might be possible to maintain high affinity polyamine block while moving the glutamate at E594 to adjacent positions. However, for side chains normally facing the protein interior, introduction of glutamate would be expected to disrupt normal function because of structural perturbations resulting from the energetic cost of burying charged residues. Implicit in this hypothesis is the assumption that E594 functions via a through space electrostatic mechanism rather than by creating a specific polyamine binding site in conjunction with surrounding residues. To test this hypothesis, we performed a glutamate scan in the E594Q mutant background. The scan ranged over 14 residues, from Q590 to R603; in KcsA, this corresponds to a region starting in the pore cavity, proceeding through the selectivity filter and entrance of the channel and extending into M2 (Fig. 1 and Fig. 8).
These experiments revealed three effects of introducing glutamate in GluR6. First, at six positions in the extracellular surface, a glutamate was able to restore high affinity polyamine block in the E594Q mutant background (Fig. 6). Second, in polyamine-free conditions, the weak biphasic rectification produced by the E594Q mutation over the range −100 to −25 mV was also attenuated, such that G-V plots for S593E and K598E recorded in outside-out patches were weakly outward rectifying like those for wild-type GluR6 (not shown). Third, at two positions (P597E and A599E) for which there was no restoration of polyamine block in the E594Q background, the introduction of glutamate in a wild-type background disrupted normal polyamine block (Fig. 7).
Figure 6.
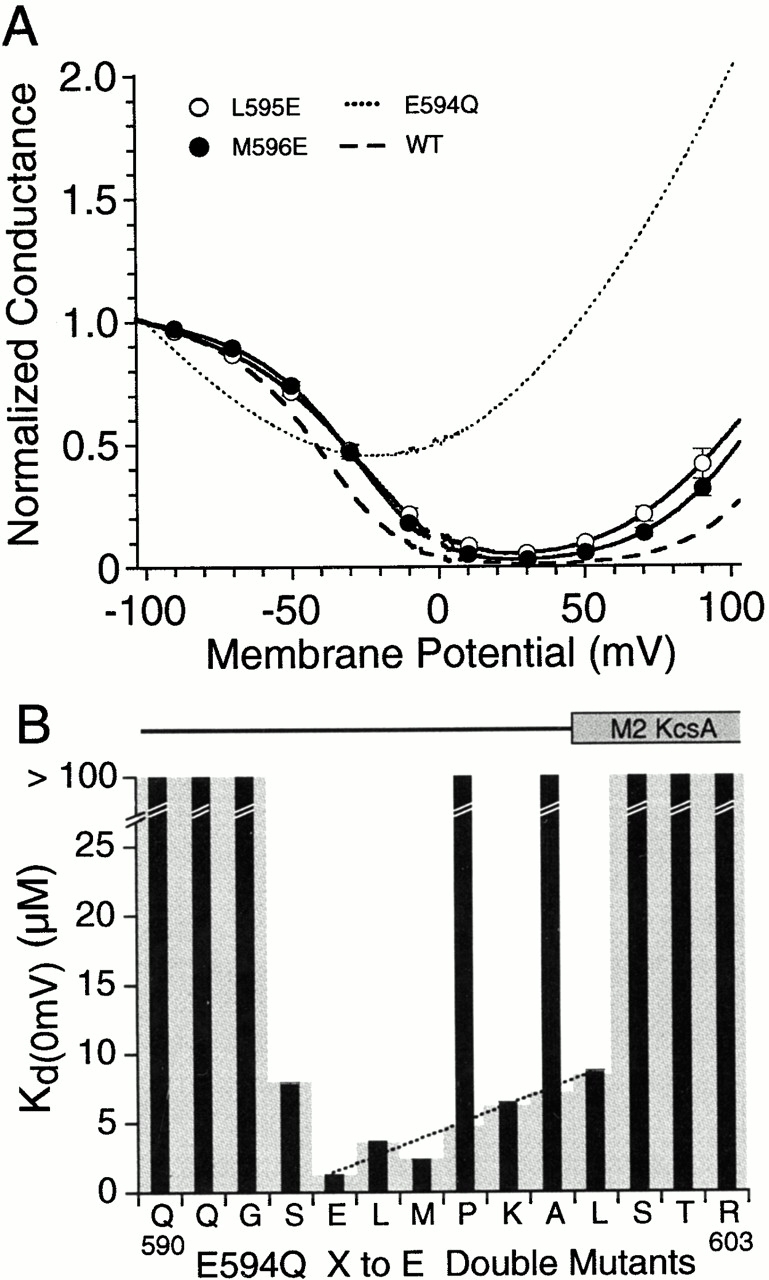
Reintroduction of glutamate into the linker between the pore loop and second transmembrane helix restores high affinity polyamine block in GluR6 E594Q mutants. (A) Mean G-V plots for L595E and M596E examples of E594Q double mutants showing restoration of polyamine block. Data points plotted every 20 mV show the mean ± SD of responses for 4–5 oocytes per mutant. The dashed line shows responses for wild-type GluR6; and a dotted line indicates responses for the charge neutralization point mutant E594Q. (B) Mean values for K d(0) measured from Boltzmann fits to responses like those for L595E and M596E. The shaded background for P597 and A599 shows a linear interpolation of K d(0) values from E594 (wild type) to L600E and emphasize that, at these positions, reintroduction of glutamate in the E594Q background fails to restore high affinity polyamine block. Note that P597 and A599 are flanked by residues for which introduction of glutamate was highly effective. α-Helical regions in the secondary structure of KcsA mapped onto the GluR6 sequence as shown in Fig. 1 are shown above the bar plot.
Figure 7.
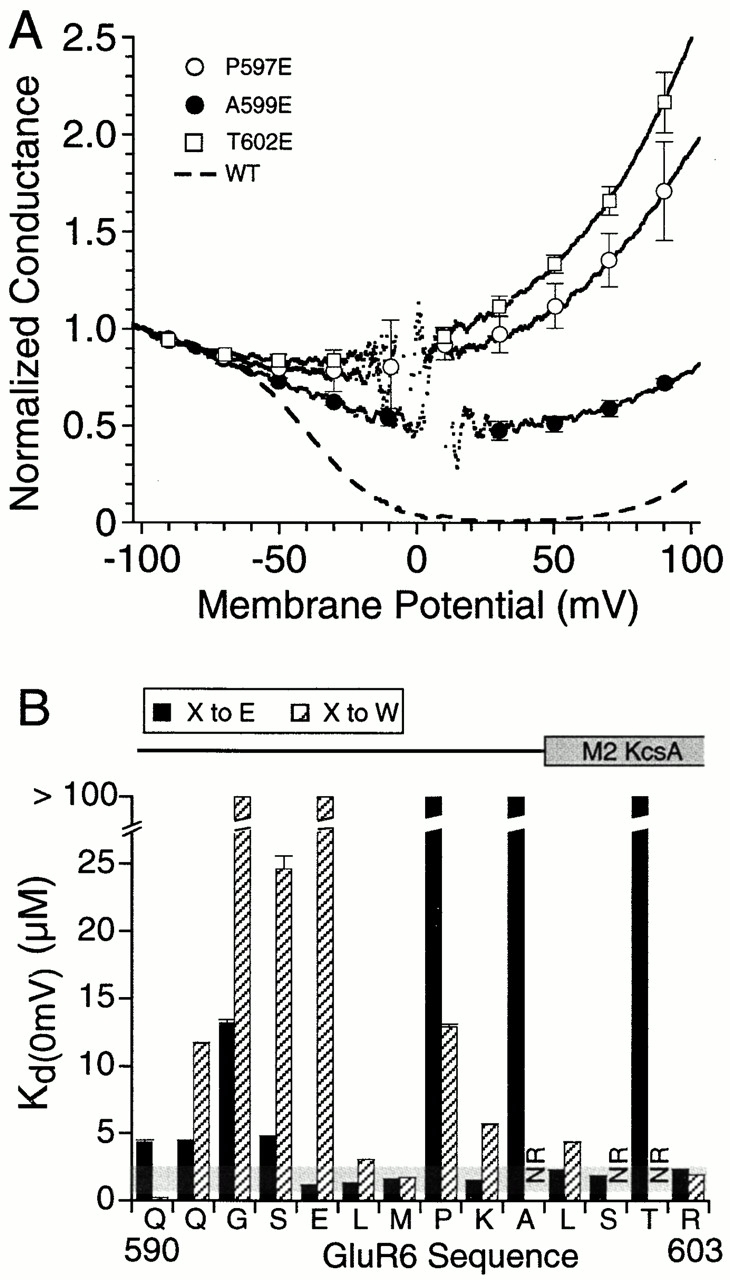
Disruption of GluR6 function by introduction of glutamate near the start of the second membrane spanning helix. (A) Mean G-V plots for P597E, A599E, and T602E in the wild-type GluR6 background. Data points plotted every 20 mV show the mean ± SD of responses for 5 oocytes per mutant. The dashed line shows the response for wild-type GluR6. (B) Changes in K d(0) for Glu and Trp point mutants; the shaded horizontal box indicates a plus/minus twofold change from the K d(0) value for wild-type GluR6. Mutants that failed to give functional responses are indicated by NR. Note that for A599 and T602, introduction of glutamate disrupted polyamine block, whereas introduction of tryptophan produced nonfunctional receptors. α-Helical regions in the secondary structure of KcsA mapped onto the GluR6 sequence (Fig. 1) are shown above the bar plot.
To obtain a quantitative estimate of changes in the affinity for polyamines resulting from introduction of glutamate at positions surrounding E594, G-V plots were analyzed using a two barrier one site model for permeable block. This revealed that for two positions (L595E and M596E) polyamine block occurred with an affinity only two- to threefold lower than for wild-type GluR6. The K d(0) values were 3.66 ± 0.04 μM (n = 4) for L595E and 2.39 ± 0.03 μM (n = 7) for M596E versus 1.25 ± 0.02 μM (n = 9) for wild-type GluR6. In previous work with E594Q, in which outside-out patch recording with 1 mM spermine added to the pipet solution was used to increase block and allow estimation of K d(0) for mutants with low affinity for spermine, we obtained an estimate of 1,020 ± 391 μM (n = 5; Panchenko et al. 1999), indicating a >250-fold increase in affinity produced by the L595E and M596E mutations in the E594Q background. At positions further removed from E594Q, the effectiveness of reintroducing glutamate faded out (Fig. 5). The loss of effect was steep on moving towards the NH2 terminus and into the pore loop, with S593E, K d(0) 7.80 ± 0.16 μM (n = 5), producing a 130-fold increase in affinity compared with E594Q and no effect for preceding residues. Moving away from E594 and towards M2, there was a more graded effect with a 160-fold increase in affinity compared with E594Q for K598E and a 120-fold increase for L600E, for which we measured K d(0) values of 6.38 ± 0.09 μM (n = 5) and 8.60 ± 0.14 μM (n = 4), respectively. However, the effect of reintroducing glutamate in this region was not continuous since for P597E and A599E G-V plots showed weak rectification, indicating little or no restoration of polyamine block. For the remaining positions, the G-V plots were similar in shape to responses for E594Q, suggesting no change in the K d(0) for polyamines.
To explore why in the E594Q background, the insertion of glutamate at P597 and A599 failed to restore polyamine block even though the mutation of the surrounding positions to glutamate produced at least a 100-fold increase in polyamine affinity, we repeated the Glu scan in the wild-type background. These experiments also acted as controls for Q590E, Q591E, G592E, S601E, T602E, and R603E, positions for which introduction of glutamate in the E594Q background also failed to restore polyamine block. We found that introduction of glutamate in the wild-type GluR6 background strongly disrupted polyamine block at three of these positions, with little effect at other positions. The mutants showing disruption were P597E, A599E, and T602E (Fig. 7). Analysis of G-V plots gave G+80/G−80 ratios of 206 ± 10% (n = 5) for P597E, 75 ± 2.5% (n = 5) for A599E and 206 ± 9.9% (n = 5) for T602E. A comparison of the effects of introducing glutamate in the wild-type and E594Q backgrounds with the results of the Trp scan (Fig. 3, Fig. 6, and Fig. 7) reveals that introduction of Glu and Trp were disruptive at common positions. At A599 and T602, for which the Trp mutants are nonfunctional, introduction of glutamate in the wild-type background disrupted polyamine block. As would be expected, in the E594Q background, introduction of glutamate A599 and T602 failed to restore polyamine block. At P597 the effect of introducing glutamate followed the same qualitative pattern as for A599 and T602; however, the P597W mutant was functional, albeit with a reduction in affinity for spermine (Fig. 6). For positions Q590 and Q591, the introduction of glutamate fails to restore polyamine block in the E594Q background (Fig. 6) and produces only a small increase in K d(0) in the wild-type background (Fig. 7), indicating that glutamate at these positions does not disrupt channel function.
DISCUSSION
Our experiments used scanning mutagenesis and a simple functional assay, permeable block by cytoplasmic polyamines, to examine secondary structure in the pore of glutamate receptors. The nature of the polyamine binding site(s) in GluRs and the mechanism of block are not yet well understood but were shown previously to be extremely sensitive to mutations at Q590 and E594 (Panchenko et al. 1999). We propose that similar to the effect produced by the removal of a single amino acid side chain methylene group at Q590, the RNA editing (Q/R) site, for which a Gln to Asn mutation lowers the K d(0) for spermine 16-fold (Panchenko et al. 1999), mutations of residues which alter the structural environment of the Q/R site will also disrupt the binding of polyamines via changes in the relative orientation and positions of the glutamine side chains. Similar arguments apply for E594.
Interpretation of Mutations in the Pore Helix of K+ Channels and GluRs
Our hypothesis was that the experiments reported here can be interpreted by analogy to previous work on K channels, for which mutations of side chains that are not solvent exposed and that do not directly contact K+ ions were found to disrupt ion selectivity (Doyle et al. 1998; Gross and MacKinnon 1996; Heginbotham et al. 1994; Lü and Miller 1995; Ogielska and Aldrich 1998). Thus, before the determination of the structure of KcsA numerous studies had used mutagenesis in the pore region of K+ channels in attempts to identify amino acid side chains forming the lining of the ion channel. In retrospect, although these pioneering studies clearly identified the location of the selectivity filter, it was not possible to accurately predict secondary structure or the role of individual amino acid side chains because the presence and role of the pore helix was not yet understood. Taking one example, when tryptophan residues in Shaker K+ channels equivalent to W67 and W68 in KcsA were substituted with cysteine, labeling by silver ions was interpreted as indicating that these side chains were solvent-exposed and in the lumen of the pore (Lü and Miller 1995). However, the structure of KcsA reveals that these side chains are in the pore helix and project to the interior of the protein and not to the lumen of the pore (Doyle et al. 1998). This effect suggests that mutations of amino acid side chains within the pore helix, but which are not solvent-exposed, can have major effects on channel function if changes in side chain volume and chemistry disrupt packing of the adjacent selectivity filter.
Comparable to results for Shaker K+ channels we found that for GluR6 mutation to alanine of W582 and F583 (equivalent to W67 and W68 in KcsA) strongly decreased polyamine block, whereas the mutation F581A had no effect. The aromatic side chains equivalent to W67 and W68 are highly conserved both in other K+ channels and in GluRs, which is consistent with their known structural role in KcsA. Previous work on NMDA receptors had revealed that the mutation W607L in NR2B (equivalent to W67 in KcsA) disrupts Mg2+ block. This effect was interpreted as reflecting a direct interaction of the indole ring with Mg2+ ions via cation-π bonding (Williams et al. 1998). The same mutation also reduced block by and increased the permeability of the large organic cation N1-dansyl-spermine; this led to the proposal that W607 was solvent-exposed and contributed to the narrowest constriction of the pore (Kashiwagi et al. 1997). Based on the structure of KcsA, it seems more likely that, similar to K+ channels, the tryptophan at position 607 in NR2B forms a sheet of aromatic amino acids surrounding structures that form the pore, and that mutations that decrease W607 side chain volume perturb the packing of adjacent residues which interact directly with blockers and permeant ions.
The identification of channel lining residues in the NMDA receptor NR1 and NR2 subunits (Kuner et al. 1996) and the AMPA receptor GluRD subunit (Kuner et al. 1999) has also been proposed from the results of substituted cysteine accessibility analysis. These landmark studies clearly revealed a periodic pattern of labeling consistent with an α-helical structure followed by a stretch of consecutively labeled residues. However, when mapped by amino acid sequence alignment to the KcsA structure, this pattern matches extremely well the location of the pore helix and selectivity filter. Thus, just as in the case of cysteine-substituted Shaker K+ channels for which labeling with Ag+ incorrectly assigned pore lining roles to buried aromatic residues (Lü and Miller 1995), in both NMDA and AMPA receptors side chains equivalent to W582, F583, and G586 in GluR6, which we propose play key structural roles in the pore helix, were assigned as solvent exposed residues lining the lumen of the pore (Kuner et al. 1996, Kuner et al. 1999).
Molecular Modeling Based on KcsA Coordinates
In the present experiments, particularly in the case of highly disruptive Trp mutations, we observed periodic patterns of changes in polyamine affinity for sequences that aligned with the pore helix of KcsA. When mapped to the structure of KcsA, the sensitive residues face either the selectivity filter or adjacent membrane spanning helices (Fig. 4). Using the program O (Jones and Kjeldgaard 1997) to display and manipulate KcsA coordinates, we examined the effect of introducing tryptophan (in each of the common conformers found in proteins) into sites equivalent to the 37 positions used for scanning mutagenesis in GluR6. The goal was to identify positions that had bad contacts with surrounding residues and those that tolerated introduction of Trp without collisions with adjacent residues, and to note any correlation with the experimentally observed effects on polyamine block (Table ). The results obtained must be interpreted with caution, yet, on a residue-by-residue basis, we found that with few exceptions the coordinates of KcsA predicted remarkably well the qualitative pattern of responses observed for GluR6 scanning mutagenesis in the pore helix (Table ). For example, A65W (equivalent to S580W in GluR6) produced extremely bad contacts with both M1 and M2; polyamine block was highly disrupted for S580W. The adjacent residue, L66 (equivalent to F581 in GluR6 which was insensitive to substitution with either Ala or Trp) projects into lipid in the KcsA structure. Further into the pore helix, introduction of Trp at S69 (equivalent to G584 in GluR6) also produced bad contacts with M1 within the same subunit, whereas for E71W (equivalent to G586 in GluR6), bad contacts occurred with the selectivity filter within the same subunit as well as with M2 of an adjacent subunit. In GluR6 Trp mutations at the equivalent positions abolished polyamine block consistent with major structural perturbations. In contrast, for V70 the residue flanked by these positions, the valine side chain in KcsA projects into a deep groove between subunits and probably faces lipid; there are no bad contacts when Trp is introduced at this position, and in GluR6 polyamine block for is not altered for V585W.
A more complex result aided by comparison with the KcsA structure was residue F575 for which mutation to Ala produced a sixfold increase in K d(0), with a threefold decrease in K d(0) for the Trp mutant, indicating this position was unlikely to play any major structural role. In KcsA, the equivalent residue I60 is located at the NH2 terminus of the pore helix and contributes to a solvent-exposed surface that binds K+ channel toxins. Consistent with the predicted solvent accessibility of F575 in GluR6, introduction of glutamate was also well tolerated, with a K d(0) of 0.54 ± 0.01 μM, twofold lower than wild type. In KcsA, the terminal COOH group of a Glu side chain introduced at I60 could be positioned within 12 Å of the COOH group of D80, the equivalent of which in GluR6 is a major determinant of polyamine binding. Perhaps coincidentally, a spermine molecule of length 17 Å could easily span this distance.
We continued using the KcsA structure to help interpret the results obtained for the glutamate scan. As for Trp mutations individual side chains in KcsA were changed to glutamate and a full range of conformers examined. This revealed that side chains that are solvent-exposed and on the surface of KcsA precisely corresponded to positions in GluR6 for which introduction of glutamate was effective in restoring polyamine block in the E594Q background. The Cα atoms of effective positions were separated by a distance exceeding 17 Å, indicating little positional requirement for negative surface charge (Fig. 8). At positions where glutamate was ineffective we observed either of two patterns in the KcsA structure, which corresponded to two distinct patterns for GluR6 responses. For those positions at which introduction of glutamate both failed to restore polyamine block in the E594Q background and was disruptive in the wild-type background, molecular modeling with KcsA revealed that the Glu side chain was not solvent exposed but buried in the protein (Fig. 8). This was observed for G77, P83, and T85 in KcsA, which correspond to G592E, P597E, and A599E in GluR6. For T602E, the model was not informative since this side chain (G88 in KcsA), when mutated to Glu, would project into the groove between the outer helix and pore helix, possibly disrupting the packing of M3. For positions where introduction of glutamate failed to restore polyamine block in the E594Q background but was not disruptive in the wild-type background, the mutated side chain was solvent-exposed but either projected towards the inner cavity (Q590E) or was located on the far side of the turret and shielded from the entrance of the pore by surrounding residues (S601E and R603E).
Location and Function of the RNA Editing Site in Glutamate Receptors
The high affinity for polyamines created by the Q590W mutant suggests an unusual molecular mechanism that might give insight into the normal role of the RNA editing site side chain in glutamate receptors. To address this we changed the T75 side chain, which aligns with Q590 in GluR6 to tryptophan. The W75 side chains of individual subunits in the KcsA model were rotated around the Cα–Cβ–Cγ bonds to minimize bad contacts with other residues. In the resulting structure, shown in Fig. 8, the Trp side chains point upwards and away from the selectivity filter and into the central cavity. Remarkably, in the KcsA T75W model, there are relatively few bad contacts between the Trp side chains of adjacent residues provided that they point into the central cavity. Instead, the faces of opposite pairs of indole rings are separated by 8.5 Å, and form a cage that we propose binds aliphatic cation polyamine molecules with high affinity after they pass through the sequence corresponding to the selectivity filter. A related mechanism has been proposed to create the high affinity external TEA binding site in K+ channels (Heginbotham and MacKinnon 1992). The aromatic cage shown in Fig. 8 suggests a plausible structure for creation of a high affinity spermine binding site and, in addition, gives clues as to the normal orientation and action of the Q/R site side chains in the genomically encoded and RNA-edited versions of glutamate receptors. It seems likely that in the genomically encoded and edited forms of GluR6, the Gln and Arg side chains will also point up into the central cavity. Shortening of the Gln side chains to Asn could prevent the ability of all four subunits to simultaneously bind polyamine molecules as they pass through the pore, thus, reducing block and increasing the rate of permeation and relief from block (Panchenko et al. 1999). Introduction of arginine side chains via RNA editing is not energetically prohibitive due to the presence of solvent in the pore cavity, but likely makes energetically unfavorable entry of polyamines into the pore and causes the channel to lose selectivity for cations versus anions (Burnashev et al. 1996).
The results of our study suggest conservation of the pore helix and associated cavity in K+ channels, GluRs and other members of this super family of ion channels. That these channels all have similar structures almost certainly reflects the energetic advantage that this architecture confers when moving ions across the lipid bilayer (Parsegian 1969; Roux and MacKinnon 1999) and would be consistent with evolution of an ancestral channel into a vast multigene family.
Supplemental Material
Acknowledgments
We thank Drs. P. Seeburg and E. Liman for the gift of plasmids, Ms. E. Tansey for help with immunohistochemical assays, Drs. R. Petralia and R. Wenthold for the gift of GluR6 antibody, Dr. T. Kuner for sharing results before publication, Dr. K. Swartz for numerous discussions and Drs. K. Swartz and C. Cui for comments on the manuscript.
This work was funded by the Intramural Research Program of the National Institutes of Health.
Footnotes
The online version of this article contains supplemental material.
Abbreviation used in this paper: GluR, glutamate receptor ion channel.
References
- Armstrong N., Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptorcrystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N., Sun Y., Chen G.Q., Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- Bähring R., Bowie D., Benveniste M., Mayer M.L. Permeation and block of rat GluR6 glutamate receptor channels by internal and external polyamines. J. Physiol. 1997;502:575–589. doi: 10.1111/j.1469-7793.1997.575bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D., Mayer M.L. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Bowie D., Lange G.D., Mayer M.L. Activity-dependent modulation of glutamate receptors by polyamines. J. Neurosci. 1998;18:8175–8185. doi: 10.1523/JNEUROSCI.18-20-08175.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N., Zhou Z., Neher E., Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J. Physiol. 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N., Villarroel A., Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J. Physiol. 1996;496:165–173. doi: 10.1113/jphysiol.1996.sp021674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chen G.Q., Cui C., Mayer M.L., Gouaux E. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature. 1999;402:817–821. doi: 10.1038/45568. [DOI] [PubMed] [Google Scholar]
- Cui C., Bähring R., Mayer M.L. The role of hydrophobic interactions in binding of polyamines to non NMDA receptor ion channels. Neuropharmacology. 1998;37:1381–1391. doi: 10.1016/s0028-3908(98)00112-9. [DOI] [PubMed] [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Egebjerg J., Bettler B., Hermans-Borgmeyer I., Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- Everts I., Petroski R., Kizelsztein P., Teichberg V.I., Heinemann S.F., Hollmann M. Lectin-induced inhibition of desensitization of the kainate receptor GluR6 depends on the activation state and can be mediated by a single native or ectopic N-linked carbohydrate side chain. J. Neurosci. 1999;19:916–927. doi: 10.1523/JNEUROSCI.19-03-00916.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A., MacKinnon R. Agitoxin footprinting the Shaker potassium channel pore. Neuron. 1996;16:399–406. doi: 10.1016/s0896-6273(00)80057-4. [DOI] [PubMed] [Google Scholar]
- Guo D.L., Lu Z. Mechanism of cGMP-gated channel block by intracellular polyamines J. Gen. Physiol. 115 2000. 783 797a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.L., Lu Z. Mechanism of IRK1 channel block by intracellular polyamines J. Gen. Physiol. 115 2000. 799 814b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L., MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- Heginbotham L., Lu Z., Abramson T., MacKinnon R. Mutations in the K+ channel signature sequence. Biophys. J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K.H., Miller C. The lipid–protein interface of a Shaker K+ channel. J. Gen. Physiol. 2000;115:51–58. doi: 10.1085/jgp.115.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Kjeldgaard M. Electron-density map interpretation. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K., Pahk A.J., Masuko T., Igarashi K., Williams K. Block and modulation of N-methyl-d-aspartate receptors by polyamines and protonsrole of amino acid residues in the transmembrane and pore-forming regions of NR1 and NR2 subunits. Mol. Pharmacol. 1997;52:701–713. doi: 10.1124/mol.52.4.701. [DOI] [PubMed] [Google Scholar]
- Keinänen K., Wisden W., Sommer B., Werner P., Herb A., Verdoorn T.A., Sakmann B., Seeburg P.H. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. MOLSCRIPTa program to produce both detailed and schematic plots of protein structures. J. App. Crystal. 1991;24:946–950. [Google Scholar]
- Kuner T., Wollmuth L.P., Karlin A., Seeburg P.H., Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- Kuner T., Wollmuth L.P., Sakmann B. The ion-conducting pore of glutamate receptor channels. In: Jonas P., Monyer H., editors. Ionotropic Glutamate Receptors in the CNS. Springer Verlag; Berlin, Germany: 1999. pp. 219–249. [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel Neuron. 25 2000. 411 423a [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. α-Helical structural elements within the voltage-sensing domains of a K+ channel J. Gen. Physiol. 115 2000. 33 50b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H., Wisden W., Köhler M., Keinänen K., Sommer B., Seeburg P.H. High-affinity kainate and domoate receptors in rat brain. FEBS Lett. 1992;307:139–143. doi: 10.1016/0014-5793(92)80753-4. [DOI] [PubMed] [Google Scholar]
- Lü Q., Miller C. Silver as a probe of pore-forming residues in a potassium channel. Science. 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Pore loopsan emerging theme in ion channel structure. Neuron. 1995;14:889–892. doi: 10.1016/0896-6273(95)90327-5. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Cohen S.L., Kuo A., Lee A., Chait B.T. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- Merritt E.A., Bacon D.J. Raster3Dphotorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Monks S.A., Needleman D.J., Miller C. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 1999;113:415–423. doi: 10.1085/jgp.113.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska E.M., Aldrich R.W. A mutation in S6 of Shaker potassium channels decreases the K+ affinity of an ion binding site revealing ion–ion interactions in the pore. J. Gen. Physiol. 1998;112:243–257. doi: 10.1085/jgp.112.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko V.A., Glasser C.R., Partin K.M., Mayer M.L. Amino acid substitutions in the pore of rat glutamate receptors at sites influencing block by polyamines. J. Physiol. 1999;520:337–357. doi: 10.1111/j.1469-7793.1999.t01-1-00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsegian A. Energy of an ion crossing a low dielectric membranesolutions to four relevant electrostatic problems. Nature. 1969;221:844–846. doi: 10.1038/221844a0. [DOI] [PubMed] [Google Scholar]
- Partin K.M., Patneau D.K., Winters C.A., Mayer M.L., Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Pearson W.L., Nichols C.G. Block of the Kir2.1 channel pore by alkylamine analogues of endogenous polyamines. J. Gen. Physiol. 1998;112:351–363. doi: 10.1085/jgp.112.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B., MacKinnon R. The cavity and pore helices in the KcsA K+ channelelectrostatic stabilization of monovalent cations. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- Santoro B., Tibbs G.R. The HCN gene familymolecular basis of the hyperpolarization-activated pacemaker channels. Annu. NY Acad. Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- Schreibmayer W., Lester H.A., Dascal N. Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflügers Arch. 1994;426:453–458. doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- Schrempf H., Schmidt O., Kummerlen R., Hinnah S., Muller D., Betzler M., Steinkamp T., Wagner R. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans . EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5170–5178. doi: 10.1002/j.1460-2075.1995.tb00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L., Benveniste M., Mayer M.L. Modulation of N-methyl-d-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990;428:313–331. doi: 10.1113/jphysiol.1990.sp018214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold R.J., Trumpy V.A., Zhu W.S., Petralia R.S. Biochemical and assembly properties of GluR6 and KA2, two members of the kainate receptor family, determined with subunit-specific antibodies. J. Biol. Chem. 1994;14:1332–1339. [PubMed] [Google Scholar]
- Williams K., Pahk A.J., Kashiwagi K., Masuko T., Nguyen N.D., Igarashi K. The selectivity filter of the N-methyl-d-aspartate receptora tryptophan residue controls block and permeation by Mg2+ . Mol. Pharmacol. 1998;53:933–941. [PubMed] [Google Scholar]
- Wo Z.G., Oswald R.E. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995;18:161–168. doi: 10.1016/0166-2236(95)93895-5. [DOI] [PubMed] [Google Scholar]
- Wood M.W., VanDongen H.M., VanDongen A.M. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc. Natl. Acad. Sci. USA. 1995;92:4882–4886. doi: 10.1073/pnas.92.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The bacterial K+ channel structure and its implications for neuronal channels. Curr. Opin. Neurobiol. 1999;9:267–273. doi: 10.1016/s0959-4388(99)80039-7. [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Siegelbaum S.A. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.