

Abstract
2-arachidonoylglycerol (2-AG) is the most abundant endocannabinoid and it plays a critical role in cannabinoid receptor-mediated cell signaling. Although 2-AG was shown to induce ERK activation via the cannabinoid receptor 1 (CB1), only a nonspecific CB receptor agonist and antagonist was used in those studies. Whether cannabinoid receptor 2 (CB2) is involved in 2-AG-induced ERK activation is still unclear. Moreover, whether 2-AG is involved in mediation of AP-1 activity and cell transformation is also not known. In the present study, we show that 2-AG stimulates AP-1-dependent transcriptional activity and enhances EGF-induced cell transformation in mouse epidermal JB6 P+ Cl41 cells. Using JB6 P+ C141 cells, stably transfected with an AP-1 luciferase reporter, we found that 10 μM 2-AG induced up to a 3-fold stimulation of AP-1 transcriptional activity. The AP-1 stimulation appeared to be mediated by ERK, but not JNK or p38 kinase. PD98059, a specific inhibitor of MEK1, almost completely blocked 2-AG-induced ERKs phosphorylation and AP-1 activation. Using CB1/2−/− murine embryonic fibroblasts (MEFs), we present the first direct evidence that both of cannabinoid receptors 1 and 2 (CB1/2) are involved in 2-AG-induced ERK activation. 2-AG could not stimulate ERK phosphorylation or Fyn kinase activity in dominant negative Fyn. In addition, the Fyn inhibitor, PP2, blocked 2-AG-induced Fyn kinase activity and ERKs phosphorylation and activity. siRNA Fyn also suppressed 2-AG-induced ERKs phosphorylation. Interestingly, 2-AG enhanced EGF-induced AP-1 DNA binding and cell transformation. Taken together, our data provide direct evidence suggesting that 2-AG may have a novel role in cell transformation and carcinogenesis in a signaling pathway involving CB1/2 and activation of Fyn, ERKs and AP-1.
Activation of the cannabinoid receptors results in the regulation of various cellular functions. Several lines of evidence indicate that the activation of cannabinoid receptors results in the inhibition of the cAMP response element-binding (CREB) protein, nuclear factor-kappaB (NF-kappaB), nuclear factor of activated T cells (NFAT) and activator protein-1 (AP-1) DNA binding (1–8). Some of these effects may be due to the inhibition of adenyl cyclase (7, 9). However, the down regulation of transcription factor activation is not the only result of cannabinoid receptor stimulation. Cannabinoids were also shown to stimulate mitogen-activated protein (MAP) kinases, including extracellular-signal regulated kinases (ERKs) (10–12), c-Jun N-terminal kinases (JNKs) and p38 kinases (12–15) and to stimulate B-cell (16) and splenocyte (17) proliferation under certain conditions. Cannabinoids were also shown to stimulate sequence-specific AP-1 DNA-binding activity (18). Also, cannabinoid receptor ligands may have opposite effects depending on concentration (19) and other experimental conditions (11, 13). The expression of cytokine mRNAs was also shown to be differentially affected by cannabinoids depending on cell line and the cytokine tested (20). Hence, cell signaling induced by cannbinoid receptor ligands needs additional and extensive investigation.
The most thoroughly investigated cannabinoid receptor ligands are anandamide and 2-arachidonoylglycerol (2-AG). Both of these ligands occur in trace amounts in virtually all vertebrate cells and tissues (21). Although both anandamide and 2-AG are ligands for cannabinoid receptors, significant differences exist between these two agonists. Recent work has clearly shown that 2-AG is a full agonist for both cannabinoid 1 (CB1; 22–24) and cannabinoid 2 (CB2; 25, 26) receptors, whereas anandamide is only a partial agonist for CB1 and CB2. Taken together with the fact that physiological levels of 2-AG tend to exceed those of anandamide by several orders of magnitude (21), 2-AG is more likely to play a critical role in cannabinoid receptor-mediated cell signaling.
2-AG is believed to induce cell signaling through the cannabinoid receptors (10–12,39,41). However, studies utilized a relatively nonspecific cannabinoid receptor agonist and antagonist to show that 2-AG induces ERK activation via the cannabinoid receptor 1 (CB1). Thus no direct evidence exists that distinguishes between the involvement of CB1 and CB2 in 2-AG-induced ERK activation.
Recently, Carrier, et al. (11) reported that 2-AG increases cell proliferation through a cannabinoid receptor-dependent mechanism. Jorda, et al. (12) showed that 2-AG and cannabinoid receptors are related to two distinct oncogenic effects--altered migration and block of neutrophilic development. Furthermore, Porcella, et al. (18) reported that delta9-THC, a major psychoactive cannabinoid, upregulated the mRNA levels of immediate-early genes in the rat brain. Delta9-THC was shown to increase sequence-specific AP-1 DNA-binding activity by acting on the cannabinoid receptors (18). We reported that N-acyethanolamines (NAEs) and anandamide stimulate cannabinoid-receptor-independent ERKs phosphorylation and AP-1-dependent transcriptional activity up to 2-fold in mouse epidermal JB6 cells (27). Whether 2-AG directly regulates AP-1-dependent transcriptional activity and is involved in cell transformation is as yet unknown. We addressed this question by studying signal transduction initiated by 2-AG in mouse epidermal JB6 P+ Cl41 cells, which provide a model system used extensively to study signal transduction and neoplastic transformation (28–31). The signaling pathways resulting in activation of AP-1 are well characterized in these cells and JB6 cells were shown to synthesize both anandamide (27) and 2-AG (32) and also express cannabinoid receptors 1 and 2 (CB1/2) (27). The aim of the present study was to determine whether 2-AG acts directly through CB1/2 to regulate AP-1-dependent transcriptional activity and whether 2-AG is involved in cell transformation. Here we report that in JB6 P+ Cl41 cells, 2-AG stimulated AP-1-dependent transcriptional activity through the ERKs pathway and 2-AG enhances EGF-induced cell transformation. Furthermore, by using CB1/2−/− murine embryonic fibroblasts (MEFs), we present the first direct evidence indicating that 2-AG stimulates ERKs phosphorylation through both CB1 and CB2 with the subsequent activation of Fyn.
Materials and Methods
Plasmids and Reagents
The mouse CB1 plasmid (PcDNA3-CB1 plasmid) was kindly provided by Dr. Beat Lutz (Group Molecular Genetics of Behavior Max-Planck-Institute of Psychiatry, Kraepelinstr. 2-10 D-80804 Munich, Germany). The mouse CB2 plasmid (pcDNA-CB2 plasmid) was kindly provided by Dr. Ruud Delwel (Erasmus MC, Department of Hematology, Dr Molewaterplein 50, 3015GE Rotterdam, The Netherlands) Dulbecco’s modified Eagle’s medium (DMEM), minimum essential medium (MEM) and L-glutamine were purchased from Cellgro (Park Center Road, Herndon, VA) gentamicin, penicillin and streptomycin were from Biosource (Flynn Road, Camarillo California); fetal bovine serum (FBS) was from Gemini Bio-Products (Woodland, CA). The luciferase assay substrate was purchased from Promega (Madison, WI); 12-O-tetradecanoylphorbol-13-acetate (TPA), aprotinin and leupeptin were from Sigma (St. Louis, MO); LY294002, PD98059 (2’-amino-3’-methoxyflavone), and PP2 (AG 18794-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) were from Calbiochem Co. (La Jolla, CA). DNA STAT-60 was from TEL-TEST ’B’, INC. (Friendswood, TX). The antibodies against phosphorylated ERKs, JNKs, p38 kinase, Elk1 (Ser383) and non-phosphorylated ERKs, JNKs, p38 kinase and PKC-alpha were from Cell Signaling Technology (Beverly, MA). 2-AG was from Cayman Chemical Company (Ann Arbor, Michigan). AM251 was from Biomol Research Laboratories Inc. (Plymouth Meeting, PA). The anti-Fyn antibody was from Santa Cruz Biotechnology (Santa Cruz, CA).
Preparation of CB1/2+/+ and CB1/2−/− murine embryonic fibroblasts (MEFs)
CB1/2 double knockout mice were provided by Dr. Andreas Zimmer from the Laboratory of Molecular Biology Clinic of Psychiatry, University Hospital (Bonn, Germany). CB1/2+/+ and CB1/2−/− MEFs were isolated from the embryos of c57/BL wild type mice and CB1/CB2 double knockout mice, respectively.
Verification of genotype of CB1/2−/− knockout mice and MEFs
DNA were obtained from mouse tails by digesting the tail with proteinase K (0.5 mg/ml in 50 mM, pH 7.5 Tris buffer). DNA was extracted with phenol/chloroform (91:1) and chlorophorm/iosoamyl alcohol (24:1) solutions. DNA was further purified using the DNA STAT-60 reagent. The primers used for PCR verification of CB1 from tail DNA and cellular DNA, were: primer 1 (5’CTCCTGGCACCTCTTTCTCAGTCACG3’); primer 2 (5’TCTCTCGTGGGATCATTGTTTTTCTCTTG A3’); and primer 3 (5’TGTGTCTCCTGCTGGAACCAACGG3’).
The PCR amplification was done at 95°C for 45 seconds, 65°C for 45 seconds, 72°C for 1 min, and was carried out for 35 cycles using Taq Polymerase. For CB1/2+/+ cells and mice, we got a 284bp band, and for CB1/2−/− cells and mice, we got a 334bp band (data not shown).
The primers used for PCR verification of CB2 from tail DNA and cellular DNA were: primer 1 (5’AAATGCTTGATTGGTGTCAGCCTCTC3’); primer 2 (5’TAAAGCGCATGCTCCAGACTGCCTT3’); and primer 3 (5’GGCTCCTAGGTGGTTTTCACATCAGCCT CT3’).
The PCR amplification was done at 95°C for 1 min, 60°C for 1 min, 72°C for 2 min, and was carried out for 30 cycles using Taq Polymerase. For CB1/2+/+ cells and mice, we got an 1100bp band, and for CB1/2−/− cells and mice, we got a 850bp band (data not shown). Therefore the genotype for the CB1/2−/− cells and mice is correct.
Cell transfection
For stable transfections, according to the protocol from Invitrogen (Carlsbad, CA), we transfected CB1/2−/− cells with CB1 or CB2 expression constructs using the Lipofectamine method (Invitrogen) as specified by the supplier. All the transfected CB1/2−/− cells were selected for 2 weeks in media containing 400 μg/ml of G418 after which time the G418 concentration was decreased to 200 μg/ml and maintained.
Establishing dominant negative (DN) Fyn and small interfering (si)-RNA Fyn expressing JB6 Cl41 cells
The K299M Fyn pLJ plasmid was provided by Dr. Moses V. Chao from the New York University Medical Center (First Avenue, New York). Using these plasmids, we established stable transfections according to the protocol from Invitrogen (Carlsbad, CA). All the transfected JB6 cells were selected for 2 weeks in media containing 400 μg/ml of G418 after which time the G418 concentration was decreased to 200 μg/ml and maintained. si-RNA Fyn JB6 cells were established as previously reported (33). Briefly, we “knocked down” Fyn expression by the siRNA method. The sense oligonucleotide of Fyn used for siRNA was 5’-TTTGCAGCTCGGAAGGAGATTGGTTCAAGA GACAATCTCCTTCCGAGCTGTTT TT-3’ and the antisense was 5’-CTAGAAAAACAGCTCGGAAGGAGATTGGT CTCTTGAA CCAATCTCCTTCCGAGCTG-3’. The ligated pair of oligonucleotides was inserted into the mU6pro vector. The oligonucleotide synthesis and sequencing of the inserted sequences in the mU6pro vector were performed by Sigma (St. Louis, MO). The plasmid siRNA-Fyn-mU6pro was stably transfected into JB6 Cl41 cells using the LipofectAMINETM 2000 reagent.
Cell Culture
JB6 P+ mouse epidermal cells (C141) and AP-1 luciferase reporter (Cl41 AP-1 mass1) stable transfectants were cultured in monolayers at 37 °C in MEM containing 5% heat-inactivated FBS, 2mM L-glutamine and 25 μg/ml gentamicin in a humidified atmosphere containing 5% CO2.
AP-1 Activity Assay
Confluent monolayers of JB6 P+ C141 cells (5x103), stably transfected with the luciferase reporter driven by AP-1, were suspended in 200 μl of 5% FBS/MEM and added into each well of a 96-well plate. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. Twenty four to forty eight hours later, cells were starved by culturing in 100 μl of 0.1% FBS/MEM for 24 h before treatment. After treatment in a total volume of 200 μl for 8-24 h, cells were extracted with lysis buffer and luciferase activity was measured using the Promega Luciferase Assay System (Promega, Madison, WI) and the Luminoskan Ascent (ThermoElectron Corp. Helsinki, Finland). The results are expressed as relative AP-1 activity compared to an untreated control value of 1.
ERKs Activity Assay
The assay for ERKs activity was carried out as described in the protocol provided by Cell Signal Technology (Beverly, MA). In brief, JB6 P+ cells (wild type Fyn) and dominant negative (DN)-Fyn cells were starved for 48 h in 0.1% FBS/MEM and then treated for 45 min with 1.0 μM PP2 or its vehicle, DMSO (<0.1%), which was used as a negative control. This was followed by treatment with 10 μM 2-AG for 30 min. The cells were then washed once with ice-cold phosphate-buffered saline (PBS) and disrupted in 300 μl of cell lysis buffer. The lysates were sonicated and centrifuged. Endogenenous ERKs were immunoprecipitated from the supernatant fraction containing 300 □g of protein by incubating with the specific phospho-specific ERKs antibody (Thr202/Tyr204) overnight at 4 °C, followed by incubation with protein A/G plus-agarose beads for another 4 h. The beads were washed twice with 500 μl of lysis buffer and twice with 500 μl of kinase buffer (25 mM Tris, pH7.5, 5 mM βeta-glycerolphosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, and 10 mM MgCl2). For determination of ERKs-induced phosphorylation of Elk-1, the kinase reactions were performed at 30 °C for 30 min in 50 μl of the kinase buffer containing the immunoprecipitates and 200 μM ATP with 2 μg Elk-1 fusion protein as the substrate. Phosphorylation of Elk-1 was then analyzed by western blotting using a chemiluminescent detection system and specific antibodies against phosphorylation of Elk-1 at serine 383.
Fyn Kinase Assay
Analysis of Fyn kinase activity was carried out as described in the protocol provided by Upstate (Forest St. Charlottesville, VA). In brief, the cells were treated for the indicated time with 10 μM 2-AG and different doses of PP2 or its vehicle, DMSO (<0.1%), which was used as a negative control. The procedure was essentially the same as that for assay of ERKs activity except that the immunoprecipitates were combined with 10 μl of [32P]ATP and 2.5 μl of the Src substrate peptide. Fyn kinase activity was then determined by scintillation counter.
Western Blotting
ERKs, JNKs, and p38 kinase phosphorylation were determined by immunoblotting with phospho-specific antibodies against ERK1/2 (Thr202/Tyr204), JNK1/2 (Thr183/Tyr185) and p38 kinases (Thr180/Tyr182), respectively. Antibodies bound to proteins were detected by chemofluorescence (ECF substrate, Amersham Pharmacia Biotech Inc., Piscataway, NJ) using the Storm 840 Imaging System (Molecular Dynamics, East Arques Avenue Sunnyvale, CA). Some membranes were stripped (7 M guanidine hydrochloride, 50 mM glycine (pH10.8), 0.05 mM EDTA, 0.1 M KCl, 20 mM 2-mercaptoethanol) and re-probed.
Anchorage-independent Transformation Assay
The role of 2-AG in EGF-promoted cell transformation was investigated in JB6 C141 cells (33). In brief, 8x103/ml cells were exposed to EGF (0–0.1 ng/ml) with or without 2-AG (1–10μM) in 1 ml of 0.3% basal medium Eagle (BME) agar containing 10% FBS. The cultures were maintained at 37 °C in a 5% CO2 incubator for 10 days, and the cell colonies were scored as described by Colburn et al. (19). The effect of 2-AG and EGF on JB6 Cl41 cell transformation is presented as colony number per 8,000 seeded JB6 C141 cells in soft agar.
AP-1 DNA Binding Study
Nuclear protein extracts were prepared from cells by a modification of the method of Ye et al. (33). Briefly, JB6 P+ C141 cells were cultured in 10-cm dishes and starved in 0.1% FBS/MEM at 37 °C in a 5% CO2 incubator. After 24 h of starvation, the cells were exposed to different concentrations of EGF or 2-AG or EGF and 2-AG for 12 h. The cells were then harvested and disrupted in 500 μl of lysis buffer A (50 mM KCl, 0.5% Nonidet P-40, 100 μM dithiothreitol, 25 mM HEPES, pH 7.8, 10 μg/ml leupeptin, 25 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride). After a 1-min centrifugation (16,000 xg at 4 °C), the pellets containing the nuclei were washed once with 500 μl of Buffer B (Buffer A without Nonidet P-40). The pellets were then resuspended in 100 μl of extraction buffer (Buffer B, but with 500 mM KCl and 10% glycerol) and strongly shaken at 4 °C for 30 min. After a 10-min centrifugation (16,000 x g at 4 °C), the supernatant solutions were moved into fresh tubes and stored at -70 °C until analysis. The DNA binding reaction was incubated at room temperature for 30 min in a mixture containing 5 mu;g of nuclear protein, 1 mu;g of poly(dI•dC), and 15,000 cpm of a α-32P-labeled double-stranded AP-1 oligonucleotide (5’ CGCTTGATGAGTCAGCCGGAA-3’). The samples were separated on a 5% polyacrylamide gel, and the gels were analyzed using the Storm 840 phosphorimaging system (Amersham Biosciences).
RESULTS
2-AG Stimulated ERKs Phosphorylation and AP-1 Transcriptional Activity
MAP kinases are important elements of cell signaling cascades that regulate cell growth, differentiation and tumorigenic transformation. When cells were treated with 2-AG, this endocannabonoid was found to stimulate ERKs phosphorylation in a dose dependent manner in JB6 P+ cells (Fig. 1A) but had no effect on phosphorylation of JNKs or p38 kinase (Fig. 1A). Because ERKs are upstream of and known to activate the AP-1 transcription factor (36), 2-AG-induced ERKs activation may also result in enhanced AP-1-dependent transcriptional activity. Indeed, we found that 10 μM 2-AG significantly stimulated AP-1 dependent transcription activity (Fig. 1B, * p < 0.05).
Fig. 1.
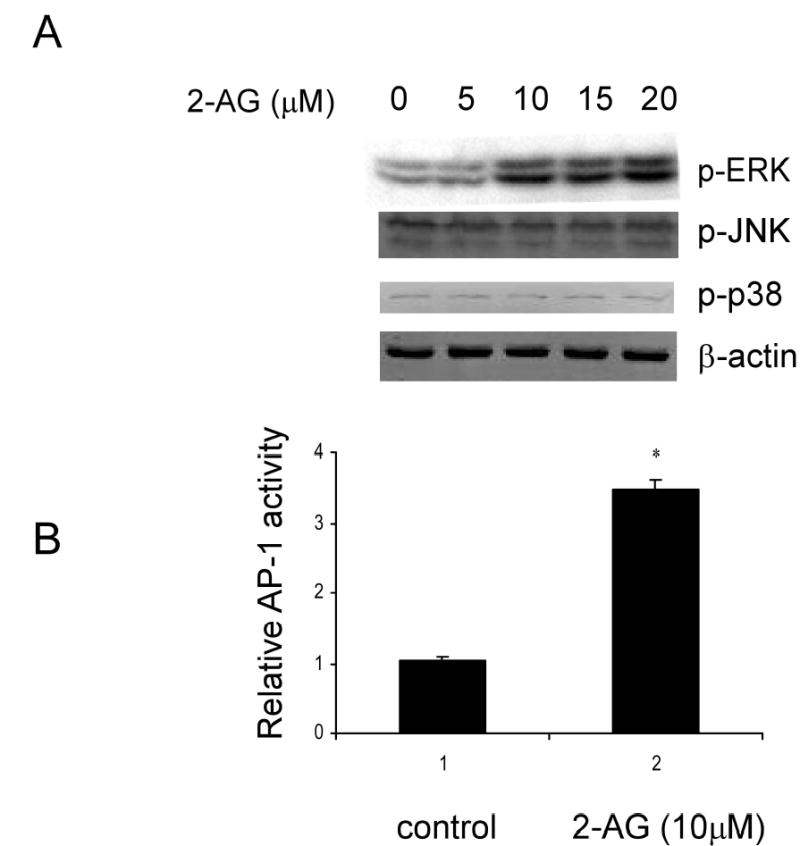
The effect of 2-AG on MAP kinase phosphorylation and AP-1 activation in JB6 P+ cells. (A) JB6 P+ C141 cells (5x104/well) were cultured in monolayers in 6-well plates until they reached 90% confluence and then were starved for 48 h in 0.1% FBS/MEM. The cells were then treated with various concentrations of 2-AG for 30 min and harvested with SDS sample buffer. Samples were analyzed by western blotting with antibodies against phosphorylation of ERKs, JNKs, and p38 as described under “Experimental Procedures.” Beta-actin was used as a loading control. (B) JB6 P+ C141 AP-1 luciferase reporter transfected cells (8x103/well) were seeded into 96-well plates. After reaching 80% confluence, cells were starved for 24 h by replacing the medium with 0.1% FBS/MEM. The cells were then treated with 10 μM 2-AG for another 24 h and AP-1 activity was measured using a luminometer as described under “Experimental Procedures.” The results are presented as relative AP-1 activity versus a control (no 2-AG) value of 1. Data from three independent experiments were averaged and are presented as mean ± SD, * p < 0.05. p-ERK (phosphorylated ERKs); p-JNK (phosphorylated JNKs); p-p38 (phosphorylated p38).
2-AG Stimulated ERKs Phosphorylation and AP-1 Transcriptional Activity Was Blocked by PD98059
To determine if 2-AG stimulation of AP-1 transcriptional activity is mediated directly by ERKs phosphorylation, PD98059, a specific inhibitor of MEK1, which is a kinase upstream from ERKs, was used. Results indicate that PD98059 almost completely blocked 2-AG-induced ERKs phosphorylation (Fig. 2A) and significantly inhibited AP-1-dependent transcriptional activity (Fig. 2B, *p < 0.05) in JB6 cells.
Fig. 2.

The effect of PD98059 on 2-AG-induced ERKs phosphorylation and AP-1 activation. (A) JB6 P+ Cl41 cells (5x104 /well) were starved for 48 h in 0.1% FBS/MEM. Cells were treated with PD98059 at various concentrations for 30 min and subsequently treated with 10 μM 2-AG for another 30 min. Cells were disrupted with SDS sample buffer and analyzed by western blotting as described in “Experimental Procedures”. (B) JB6 P+ C141 AP-1 luciferase reporter transfected cells (8x103/well) were seeded into 96-well plates. After reaching 80% confluence, cells were starved for 24 h by replacing the medium with 0.1% FBS/MEM. Cells were treated with PD98059 at various concentrations for 30 min and subsequently treated with 10 μM 2-AG for another 24 h. The AP-1 activity was measured using a luciferase assay as described under “Experimental Procedures.” The results are presented as relative AP-1 activity compared to control without PD98059. Data from three independent experiments were averaged and are presented as mean ± SD. *p < 0.05. p-ERK (phosphorylated ERK); np-ERK (nonphosphorylated or total ERK).
2-AG Stimulated ERKs Phosphorylation Occurs Through CB1 and CB2
2-AG was reported to stimulate ERKs phosphorylation in other cells (10–12, 34, 35). However, direct evidence of whether this induction occurs through CB1 and CB2 is lacking. We therefore used CB1/2+/+ and CB1/2−/− cells to address the question. CB1/2+/+ and CB1/2−/− MEFs were obtained and verified to be correct as described under “Experimental Procedures”. Treatment of cells with 10 μM 2-AG for the indicated time (Fig. 3A) or dose (Fig. 3B) resulted in a time- and dose-dependent increase in ERKs phosphorylation in CB1/2+/+ cells, but not in CB1/2−/− cells (Fig. 3, A and B). However, if CB1/2+/+ and CB1/2−/− cells were treated with 10 ng/ml of TPA, a phorbol ester known to induce ERKs phosphorylation, for the indicated time, ERKs phosphorylation increased in a time-dependent manner not only in CB1/2+/+ cells but also in CB1/2−/− cells (Fig. 3C), suggesting that ERKs response to TPA occurs independent of the CB receptor. On the other hand, 2-AG induction of ERK phosphorylation occurs through the CB1/2 receptors. To distinguish between the involvement of CB1 and CB2 in 2-AG-induced ERK phosphorylation, we transfected CB1/2−/− fibroblasts with a mammalian expression construct for CB1 or CB2 and reevaluated ERK activation after 2-AG treatment. Notably, ERK activation was observed in CB1/2−/− cells in which the wild-type CB1 or CB2 protein was re-expressed (Fig. 4). Therefore, both CB1 and CB2 are required for induction of ERK activity by 2-AG in mammalian fibroblast cells.
Fig. 3.

CB1/2-dependent and -independent 2-AG or TPA stimulated ERKs phosphorylation. (A) Time course of 2-AG stimulated ERKs phosphorylation. CB1/2+/+ or CB1/2−/− cells (5x104/well in 6-well plates) were starved for 24 h in 0.1% FBS/DMEM. Cells were treated with 10 μM 2-AG for the indicated time and then disrupted with lysis buffer. The samples were analyzed by western blotting with specific antibodies against phosphorylated ERKs. PKC-alpha was used as a loading control. (B) Dose response of 2-AG stimulated ERKs phosphorylation. CB1/2+/+or CB1/2−/− (5x104 cells/well) were cultured as described in(A). Cells were treated with different concentrations of 2-AG for 30 min and then disrupted with lysis buffer and p-ERK was detected by western blotting. (C) TPA-stimulated ERKs phosphorylation. CB1/2+/+ or CB1/2−/− (5x104 cells/well) were cultured as described in (A). Cells were treated with 10 ng/ml TPA for the indicated time and then were disrupted with lysis buffer and p-ERK was detected by western blotting.
Fig. 4.

Defect in 2-AG-induced ERK activation is rescued by reintroduction of CB1 or CB2. CB1/2−/− cells were stably transfected with the expression construct for mouse CB1 or CB2 cDNA. The cells were treated with 2-AG or left untreated. Exogenous expression of CB1 or CB2 in transfected cells was confirmed by anti-CB1 and anti-CB2 immunoblot analysis with wild-type cells as positive control; (+/+, wild-type cells; −/−, CB1/2−/− cells). ERK activation was determined by immunoblotting using an anti-pERK antibody, and CB1 and CB2 were detected with the respective CB1 or CB2 antibody. The membrane was stripped and immunoblotted to detect beta-actin as the loading control.
2-AG-induced ERKs Phosphorylation Was Not Blocked by the PI-3K Inhibitor, LY294002
Cannabinoids were reported to induce ERKs activity through recruitment of PI-3K (36, 37). Therefore, we used LY294002, a specific inhibitor of PI-3K, to study the role of PI-3K in 2-AG-induced ERKs phosphorylation in JB6 P+ cells. However, treatment of cells with 25-50 μM LY294002 for 30 min had no significant effect on 2-AG-induced ERKs phosphorylation (Fig. 5), suggesting that 2-AG-induced ERKs phosphorylation in JB6 P+ cells does not occur through the PI-3K pathway.
Fig. 5.
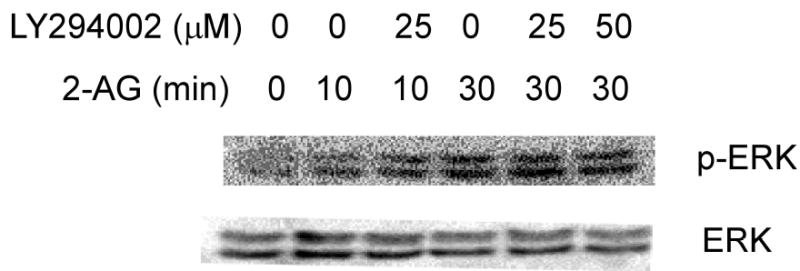
The effect of LY294002 on 2-AG–induced ERKs phosphorylation in JB6 cells. JB6 P+ Cl41 cells (5x104 cells/well) were starved for 48 h in 0.1% FBS/MEM. Cells were treated with LY294002 at the indicated concentrations for 30 min and subsequently treated with 10 μM 2-AG for the indicated times. Cells were disrupted with SDS sample buffer and analyzed by western blotting as described in “Experimental Procedures”.
2-AG-induced ERKs Phosphorylation and Activity Were Blocked by PP2 or by DN-Fyn or siRNA-Fyn
The Src-family kinase, Fyn, was reported to play a critical role in the activation of ERKs by CB1 receptors based on the finding that this activation was absent in Fyn mutant mice (34). Because PP2 is an inhibitor of Src-family kinases, including Fyn, we treated JB6 P+ cells with PP2 for 45 min, followed by treatment with 10 μM 2-AG for 30 min. Results showed that 0.5–1.0 μM PP2almost completely blocked the 2-AG-induced ERKs phosphorylation (Fig. 6A). To further determine whether 2-AG induced ERKs phosphorylation is mediated through Fyn, we employed JB6 wildtype (WT)-Fyn and dominant negative (DN)-Fyn cells and cells transfected with siRNA-Fyn. Results showed that 2-AG induced ERKs phosphorylation in JB6 WT-Fyn cells, but 2-AG-induced ERKs phosphorylation was almost completely suppressed in DN-Fyn cells (Fig. 6B) and in siRNA-Fyn cells (Fig. 6C). Additional results also indicate that 2-AG induced ERKs activity was attenuated by PP2 or DN-FYN (Fig. 7).
Fig. 6.

The effect of PP2, DN-Fyn or siRNA-Fyn on 2-AG-induced ERKs phosphorylation. (A) JB6 P+ Cl41 cells (5x104 cells/well) were cultured in monolayers in 6-well plates until they reached 90% confluence at which time they were starved for 48 h in 0.1% FBS /MEM. Cells were treated with PP2 at various concentrations for 45 min and subsequently treated with 10 μM 2-AG for another 30 min. Cells were disrupted with SDS sample buffer and analyzed by western blotting as described in “Experimental Procedures”. (B) JB6 WT-Fyn cells and DN-Fyn cells (5x104 cells/well) were starved for 48 h in 0.1% FBS/MEM. Cells were treated with 2-AG at various concentrations for 30 min and then lysed and analyzed as described in (A). (C) JB6 WT-Fyn cells and siRNA-Fyn cells (5x104 cells/well) were starved for 48 h in 0.1% FBS/MEM. Cells were treated with 2-AG at various concentrations for 30 min and then disrupted and analyzed as described in Fig. 6A.
Fig. 7.

The effect of PP2, a Fyn inhibitor, or DN-Fyn on 2-AG-induced ERKs activity. Wildtype or DN-Fyn cells (1x106) were cultured in monolayers in 10 cm-dishes until they reached 90% confluence at which time they were starved for 48 h in 0.1% FBS/MEM. Cells were then treated with 1.0 μM PP2 for 45 min and subsequently treated with 10 μM 2-AG for another 30 min. Cells were disrupted with lysis buffer and a kinase assay for ERKs activity was carried out as described in “Experimental Procedures”. The membrane was stripped and re-probed for total Elk1 protein. Data from three independent experiments were averaged and a representative blot is shown.
2-AG-induced Fyn Kinase Activity Was Blocked by PP2 or by DN-Fyn
We also studied the effect of PP2 and DN-Fyn on Fyn kinase activity. We found that 1.0 μM PP2 markedly decreased 2-AG-induced Fyn kinase activity and 2-AG-induced Fyn kinase activity was almost totally blocked in DN-Fyn cells (Fig. 8).
Fig. 8.

Effect of PP2 or DN-Fyn on 2-AG-induced Fyn kinase activity. Wildtype or DN-Fyn cells (1x106) were cultured in monolayers in 10 cm-dishes until they reached 90% confluence at which time they were starved for 48 h in 0.1% FBS/MEM. Wildtype-Fyn cells were treated with 1.0 μM PP2 for 45 min. Wildtype (lanes 1-3) or DN-Fyn (lanes 4,5) cells were subsequently treated with 10 μM 2-AG for another 30 min and then disrupted with lysis buffer. The Fyn kinase assay was carried out as described in “Experimental Procedures” and data from three independent experiments were averaged and are presented as mean ± SD. (*p < 0.05).
2-AG Stimulated Fyn Kinase Activity is Mediated Through CB1/2
To further determine whether 2-AG-stimulated Fyn kinase activity is mediated through CB1/2, we treated CB1/2+/+ or CB1/2−/− cells with 1 μM PP2 for 45 min followed by treatment with 10 μM 2-AG for 30 min and then measured Fyn kinase activity. The results indicated that 2-AG significantly increased Fyn kinase activity in CB1/2+/+ cells and that 1.0 μM PP2 almost totally blocked 2-AG stimulated Fyn kinase activity in these cells (Fig. 9, lane 4). In marked contrast, 2-AG had no effect on Fyn kinase activity in CB1/2−/− cells (Fig. 9, lane 6–9) further confirming that 2-AG-stimulated Fyn kinase activity is mediated through CB1/2.
Fig. 9.

Effect of CB1/2 knockout on 2-AG-stimulated Fyn kinase activity. CB1/2+/+ or CB1/2−/− cells (1x106) were cultured in monolayers in 10 cm-dishes until they reached 90% confluence. Then the cells were starved for 24 h in 0.1% FBS/ DMEM. CB1/2+/+ (lane 1–3) or CB1/2−/− (lane 4–6) cells were treated with 1.0 μM PP2 for 45 min and subsequently treated with 10 μM 2-AG for 30 min. The cells were disrupted with lysis buffer and a Fyn kinase activity assay was carried out according to the manufacturer’s instructions. Data from three independent experiments were averaged and are presented as mean ± SD. (*p < 0.05).
2-AG Enhancement of EGF-induced JB6 P+ cell transformation
Because our previous results and other studies demonstrated that induction of AP-1 activity is required for cell transformation, we tested whether 2-AG could induce transformation or promote EGF-induced transformation. The results showed that 2-AG could slightly induce JB6 P+ cell transformation (Fig. 10A, lane 4). Interestingly, a low concentration (0.0125ng/ml) of EGF could not alone significantly induce JB6 P+ cell transformation (Fig. 10A, lane 2). However, combining 10 μM 2-AG with 0.0125 ng/ml EGF induced significant JB6 P+ cell transformation (*p = 0.000046) (Fig.10A, lane 5). Moreover, the CB 1 and 2 inhibitor AM251 or ERK inhibitor PD98059 almost totally blocked the cell transformation induced by 2-AG and EGF (Fig. 10B, lane 4 vs lanes 5–10).
Fig. 10.
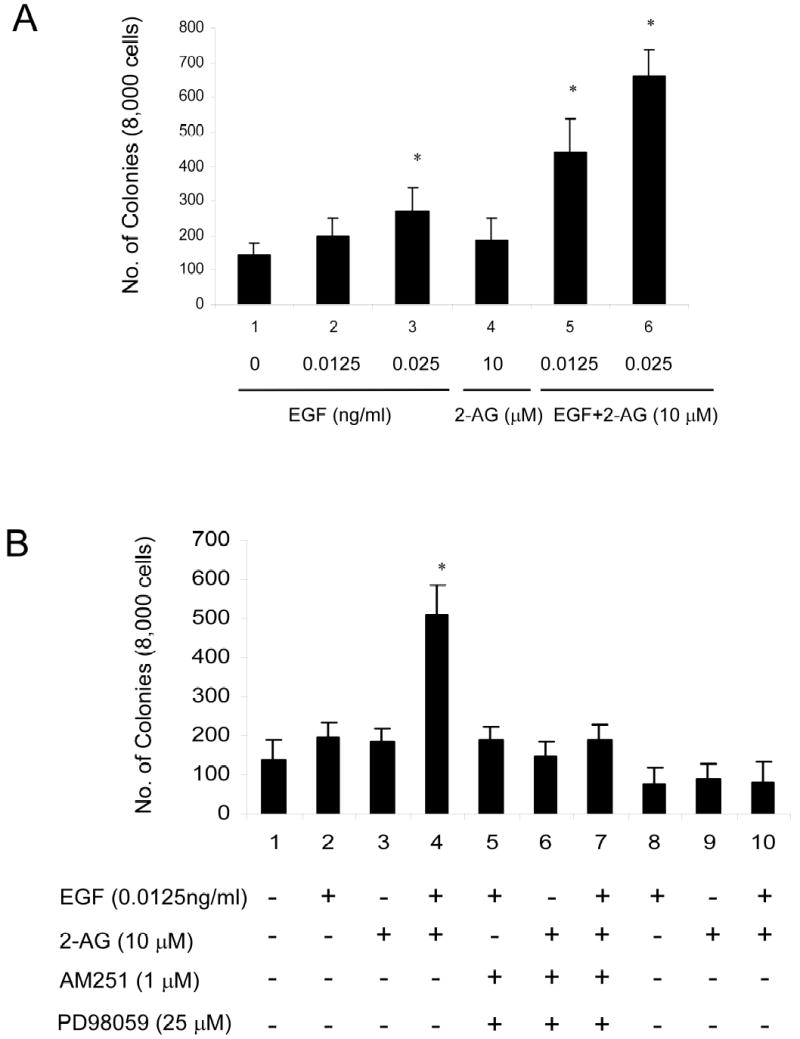
2-AG Enhancement of EGF-induced JB6 P+ Cl41 cell transformation. (A) JB6 Cl41 cells were left unexposed or were exposed simultaneously to EGF (0.0125or 0.025 ng/ml, lanes 2 and 3, respectively), or 2-AG (10 μM,lane 4), or EGF (0.0125 or 0.025 ng/ml) plus 2-AG (10 μM) (lanes 5 and 6, respectively) in 0.33% BME agar containing 10% FBS over 0.5% BME agar medium containing 10% FBS. Cell colonies were scored after 14 days of incubation at 37°C in a 5% CO2 incubator. Three dishes were made for each condition in each experiment and the average number of three independent experiments was used. Error bars indicate SD, (*p < 0.01). (B) JB6 Cl41 cells were left unexposed or were exposed simultaneously to EGF (0.0125 ng/ml, lane2), or 2-AG (10 μM, lane 3), or EGF (0.0125ng/ml) plus 2-AG (10 μM) (lane4), or with AM251 (1 μM, lanes 5–7) or with PD98059 (25 μM, lanes 8–10). Cell colonies were scored as for (A).
2-AG enhances EGF induced AP-1 DNA binding
To investigate whether 2-AG affected basal AP-1 DNA binding or EGF-induced AP-1 DNA binding in JB6 cells, we exposed JB6 P+ cells to2-AG, EGF, or EGF plus 2-AG. The results showed that 2-AG induced AP-1 DNA binding (Fig. 11, lane 7) and enhanced EGF-induced AP-1 DNA binding (Fig. 11, lanes 8 and 9).
Fig. 11.
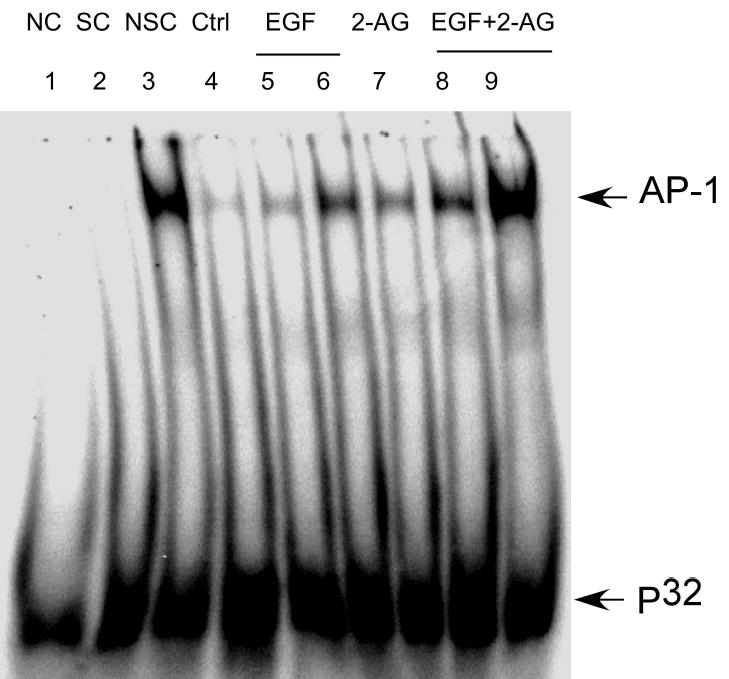
2-AG enhancement of EGF-induced AP-1 DNA binding. JB6 cells were exposed to EGF (0.0125-1.0 ng/ml, lanes5,6), or 2-AG (10 μM, lane7), EGF (0.0125 or 1.0 ng/ml) plus 2-AG (10 μM, lane8,9) for 20 h and then nuclear proteins were extracted as described in “Experimental Procedures”. Lane 1:negative control (NC); Lane 2:specific competitor (SC, non-labeled AP-1); Lane 3: nonspecific competitor (NSC; NF-κB); Lane 4: untreated control.
2-AG enhances EGF induced ERK phosphorylation
Either 2-AG or EGF can induce ERK phosphorylation but whether 2-AG can affect EGF-induced ERK phosphorylation is as yet unknown. We analyzed the effect of 2-AG on EGF induced ERK phosphorylation. As shown in Fig. 12, 2-AG not only induced ERK phosphorylation, but also enhanced EGF-induced ERK phosphorylation (Fig. 12).
Fig. 12.

2-AG enhancement of EGF-induced ERK phosphorylation. JB6 cells were exposed to EGF (1.0 ng/ml), or 2-AG (10 μM) or EGF (1.0 ng/ml) plus 2-AG (10 μM) for 15 min. ERK phosphorylation was detected by western blot. p-ERK (phosphorylated ERK); np-ERK (nonphosphorylated ERK); beta-actin was used as a loading control.
DISCUSSION
Cannabis occurs naturally in the dried flowering or fruiting tops of the Cannabis sativa plant. Cannabis is most often consumed by smoking marihuana. Recent studies suggested an association of marijuana smoking with head and neck cancers and oral lesions (38). Cannabinoids are the active compounds extracted from cannabis and endocannabinoids are defined as endogenously generated messenger molecules that bind to and activate cannabinoid receptors (21). Cannabinoid receptors are linked to multiple signaling pathways, which control DNA binding of various transcription factors and thus affect gene expression. The major signaling pathway linked to both the CB1 and CB2 receptors is the cAMP-dependent pathway, which is controlled through the inhibition of adenyl cyclase (9). Other signaling elements regulated by CB1 and CB2 receptors include MAP kinases (14, 39–41) and AP-1 (18, 27), which were shown to be activated by cannabinoids and endocannabinoids. Of the endogenous ligands for cannabinoid receptors, 2-AG is now receiving particular attention as a signaling molecule. This ligand is present in many cells at significantly higher levels than anandamide, the other known endocannabinoid (42, 43). Also, 2-AG was shown to be a full agonist for both CB1 and CB2 receptors while anandamide serves only as a partial agonist for these receptors (21). Thus 2-AG is likely to play a critical role in cannabinoid receptor-mediated cell signaling. Importantly, 2-AG but not anandamide, was shown to be generated and released within seconds upon stimulation of cultured macrophages with PAF (platelet-activating factor) (42) and can therefore be considered a new autocoid for regulating cellular function. Thus establishing the details of 2-AG-induced signaling is important in order to better understand the physiological significance of its production.
Recently, Carrier, et al. (11) reported that cultured rat microglial cells synthesize the endocannabinoid, 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-ERKs dependent mechanism. Jorda, et al. (12, 35) reported that CB2 may be involved in the induction of leukemic transformation. CB2 is aberrantly expressed in a high percentage of human acute myeloid leukemias. Aberrant expression of CB2 in hematopoietic precursor cells resulted in distinct effects depending on the ligand used. CB2-expressing myeloid precursors migrate upon stimulation by the endocannabinoid 2-arachidonoylglycerol and are blocked in neutrophilic differentiation upon exposure to another ligand, CP55940. Both effects depend on the activation of G proteins and require the mitogen-induced extracellular kinase/extracellular signal-regulated kinase (MEK/ERK) pathway.
Others and we have reported that the activation of AP-1 can induce JB6 epidermal cell transformation (27, 30, 44, 45). Berdyshev et al. (42) showed that NAEs and anandamide stimulate cannabinoid-receptor-independent ERK phosphorylation and activator protein-1 (AP-1)-dependent transcriptional activity in mouse epidermal JB6 cells. Watts et al. (44) showed that expression of dominant negative Erk2 inhibits AP-1 transactivation and neoplastic transformation. Huang, et al. (45) demonstrated that the lack of AP-1 activation and cell transformation responses to TPA or EGF in P− cells appears to be due to a low level of ERKs in these cells. Although Delta9-tetrahydrocannabinol (-THC) increases sequence-specific AP-1 DNA-binding activity (18), whether 2-AG stimulation leads to increased AP-1 activity in JB6 cells was not addressed. Our current results indicated that the activation of the ERKs pathway by 2-AG resulted in a pronounced upregulation of AP-1-dependent transcriptional activity (Fig 1B). Moreover, the MEK inhibitor, PD98059, totally blocked 2-AG-induced ERKs phosphorylation and AP-1 activation (Fig. 2, A and B), indicating 2-AG can induce AP-1 activation through ERKs and therefore may have a novel role in carcinogenesis.
Although several studies indicated that 2-AG-induced ERKs activity occurs through the cannabinoid receptors (10–12, 34, 35), these studies only used cannabinoid receptor inhibitors or MEK inhibitors to block 2-AG-induced ERKs activity and therefore direct in vivo evidence is lacking. Using stably transfected Chinese Hamster Ovary (CHO) cells expressing human CB1, Bouaboula, et al. (10) showed that cannabinoid treatment induces both ERKs phosphorylation and activation in a time- and dose-dependent manner, and also that these effects are inhibited by SR 141716A, a selective CB1 antagonist. The activation of ERKs is blocked by pertussis toxin (10). Carrier, et al. (11) reported that 2-AG induces ERKs activity in cultured rat microglial cells and increases proliferation through a CB2 receptor-ERKs dependent mechanism, the effects of which are blocked by the CB2 antagonist SR144528. Derkinderen, et al. (34) and Jorda, et al. (12, 35) showed that the MEK inhibitor PD98059 blocks 2-AG-induced ERKs activity. In the present study, we prepared and identified CB1/2+/+ and CB1/2−/− MEFs and then treated the cells with 2-AG. We found that ERKs phosphorylation increased only in the CB1/2+/+ cells, and was almost totally blocked in CB1/2−/− (Fig. 3, A and B). However, TPA induced-phosphorylation of ERKs occurred in either CB1/2+/+ or CB1/2−/− cells (Fig. 3C). These data provide strong evidence that 2-AG-induced ERKs phosphorylation is mediated almost completely through the CB1/2 receptors. To distinguish between the involvement of CB1 and CB2 in 2-AG-induced ERKs phosphorylation, we transfected a CB1 or CB2 plasmid into CB1/2−/− cells and checked the effect on 2-AG-induced ERKs phosphorylation. The result indicates that both CB1 and CB2 take part in 2-AG-induced ERK phosphorylation (Fig.4). This is the first direct evidence indicating that CB2 is involved in 2-AG-induced ERK phosphorylation.
Although 2-AG can induce ERKs phosphorylation (10–12, 34, 35), the precise mechanism of the activation of ERKs occurring through the cannabinoid receptors in JB6 cells is unclear. Our studies revealed a role for Fyn in 2-AG-induced signaling in JB6 P+ cells. 2-AG is an endocannabinoid and cannabinoids produce their effects by binding to specific plasma membrane G protein-coupled receptors (46). Src tyrosine kinase is a novel direct effector of G proteins (47) and many G protein–mediated physiologic functions are sensitive to tyrosine kinase inhibitors. Activation of many G protein–coupled receptors has been shown to increase the activity of the Src family tyrosine kinases (SFK) (47). Our data showed that 2-AG-induced ERKs phosphorylation, which was blocked by the Src inhibitor, PP2 (Figs. 6A and 7), supporting a role for SFK in 2-AG-induced ERKs activity. Li, et al. (48) reported that the activation of Fyn is coupled to the ERKs pathway. Overexpression of knockdown-Fyn (KD-Fyn) in SCC9β6 cells dramatically down regulated the activity of c-Raf and ERKs and MMP-3 promoter activity. In this study, we used DN-Fyn and siRNA-Fyn to study the role of Fyn in 2-AG-induced ERKS activity. We found that 2-AG-induced ERKs activity was almost completely blocked by DN-Fyn and siRNA-Fyn (Fig. 6, B and C, and Fig. 7). In addition, 2-AG-induced Fyn kinase activity was also blocked by PP2 or by DN-Fyn (Fig. 8), indicating an association between Fyn activation and stimulation of the ERKs-AP-1 signaling pathway in JB6 P+ Cells. These results suggested that Fyn may play a very important role in 2-AG-stimulated ERKs signaling and cancer development.
Studies in CHO cells supported a role for PI-3K in CB1-ERKs coupling (10). Activation of ERKs by G-coupled receptors can be mediated through recruitment of PI-3K, independently of the inhibition of adenyl cyclase (36). However in hippocampal slices, the cannabinoid-induced activation of ERKs was insensitive to the PI-3K inhibitor, LY294002, strongly arguing against a role for the PI-3K pathway in this effect (34). Our results indicated that LY294002 had no significant effect on 2-AG-induced ERKs phosphorylation (Fig. 5), indicating that 2-AG-induced ERKs phosphorylation in JB6 P+ cells was independent of PI-3K pathway activation.
2-AG can induce AP-1 transcriptional activity, but whether it is involved in cell transformation is unclear. Our data demonstrated that 2-AG enhances EGF-induced cell transformation (Fig.10A), and AM251 and PD98059 almost totally blocked the enhancement (Fig.10B). We further showed that this may be due to the ability of 2-AG to enhance EGF-induced AP-1 DNA binding (Fig. 11) and ERKs phosphorylation (Fig. 12).
We hypothesize that 2-AG binds to both CB1 and CB2 receptors and that this stimulation of CB1 and CB2 leads to the recruitment of Fyn and activation of ERKs by Fyn (Fig. 13). Our data support this hypothesis because Fyn kinase activity was blocked in CB1/2−/− cells. The present findings document for the first time a direct link between 2-AG stimulation and activation of the Fyn-ERK-AP-1 signaling pathway in JB6 Cl41 cells. Thus in JB6 P+ cells, 2-AG appears to have a novel role in cell transformation and carcinogenesis in a signaling pathway involving the CB1/2 receptors and activation of Fyn, ERKs and AP-1.
Fig. 13.

CB1/2, Fyn, ERKs, but not JNKs or p38 kinase, are involved in 2-AG-induced AP-1-dependent transcriptional activity in JB6 P+ cells. CB1 and CB2 are at the beginning of multiple signal transduction pathways that are activated by 2-AG. Subsequently, Fyn contributes to ERKs-mediated AP-1 signaling activation. The arrows or bars indicate activation or inhibition, respectively. The P indicates phosphorylation.
Footnotes
The work was supported in part by The Hormel Foundation and NIH grant CA27502.
The abbreviations used are: 2-AG, 2-arachidonoylglycerol; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; CBs, cannabinoids; MEF, mouse embryonic fibroblast; CB1/2−/−, CB1/CB2 double knockout; ERK, extracellular signal-regulated kinase; PI-3K, phosphoinositide-3 kinase; delta-9-THC, delta-9-tetrahydrocannabinol; TPA, 12-O-tetradecanoylphorbol-13-acetate; DN, dominant negative; PAF platelet-activating factor.
References
- 1.Condie R, Herring A, Koh WS, Lee M, Kaminski NE. J Biol Chem. 1996;271:13175–13183. doi: 10.1074/jbc.271.22.13175. [DOI] [PubMed] [Google Scholar]
- 2.Sancho R, Calzado MA, Di Marzo V, Appendino G, Munoz E. Mol Pharmacol. 2003;63:429–438. doi: 10.1124/mol.63.2.429. [DOI] [PubMed] [Google Scholar]
- 3.Gross A, Bouaboula M, Casellas P, Liautard JP, Dornand J. J Immunol. 2003;170:5607–5614. doi: 10.4049/jimmunol.170.11.5607. [DOI] [PubMed] [Google Scholar]
- 4.Herring AC, Koh WS, Kaminski NE. Biochem Pharmacol. 1998;55:1013–1023. doi: 10.1016/s0006-2952(97)00630-8. [DOI] [PubMed] [Google Scholar]
- 5.Yea SS, Yang KH, Kaminski NE. J Pharmacol Exp Ther. 2000;292:597–605. [PubMed] [Google Scholar]
- 6.Herring AC, Faubert Kaplan BL, Kaminski NE. Cell Signal. 2001;13:241–250. doi: 10.1016/s0898-6568(01)00145-0. [DOI] [PubMed] [Google Scholar]
- 7.Berdyshev EV. Chem Phys Lipids. 2000;108:169–190. doi: 10.1016/s0009-3084(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 8.Maccarrone M, Di Rienzo M, Battista N, Gasperi V, Guerrieri P, Rossi A, Finazzi-Agro A. J Biol Chem. 2003;278:33896–33903. doi: 10.1074/jbc.M303994200. [DOI] [PubMed] [Google Scholar]
- 9.Kaminski NE. J Neuroimmunol. 1998;83:124–132. doi: 10.1016/s0165-5728(97)00228-2. [DOI] [PubMed] [Google Scholar]
- 10.Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Biochem J. 1995;312 ( Pt 2):637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ. Mol Pharmacol. 2004;65:999–1007. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- 12.Alberich Jorda M, Rayman N, Tas M, Verbakel SE, Battista N, van Lom K, Lowenberg B, Maccarrone M, Delwel R. Blood. 2004;104:526–534. doi: 10.1182/blood-2003-12-4357. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Gao B, Mirshahi F, Sanyal AJ, Khanolkar AD, Makriyannis A, Kunos G. Biochem J. 2000;346(Pt 3):835–840. [PMC free article] [PubMed] [Google Scholar]
- 14.Sarker KP, Biswas KK, Yamakuchi M, Lee KY, Hahiguchi T, Kracht M, Kitajima I, Maruyama I. J Neurochem. 2003;85:50–61. doi: 10.1046/j.1471-4159.2003.01663.x. [DOI] [PubMed] [Google Scholar]
- 15.Derkinderen P, Ledent C, Parmentier M, Girault JA. J Neurochem. 2001;77:957–960. doi: 10.1046/j.1471-4159.2001.00333.x. [DOI] [PubMed] [Google Scholar]
- 16.Rayman N, Lam KH, Laman JD, Simons PJ, Lowenberg B, Sonneveld P, Delwel R. J Immunol. 2004;172:2111–2117. doi: 10.4049/jimmunol.172.4.2111. [DOI] [PubMed] [Google Scholar]
- 17.Luo YD, Patel MK, Wiederhold MD, Ou DW. Int J Immunopharmacol. 1992;14:49–56. doi: 10.1016/0192-0561(92)90104-s. [DOI] [PubMed] [Google Scholar]
- 18.Porcella A, Gessa GL, Pani L. Eur J Neurosci. 1998;10:1743–1751. doi: 10.1046/j.1460-9568.1998.00175.x. [DOI] [PubMed] [Google Scholar]
- 19.Berdyshev EV, Boichot E, Germain N, Allain N, Anger JP, Lagente V. Eur J Pharmacol. 1997;330:231–240. doi: 10.1016/s0014-2999(97)01007-8. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava MD, Srivastava BI, Brouhard B. Immunopharmacology. 1998;40:179–185. doi: 10.1016/s0162-3109(98)00041-1. [DOI] [PubMed] [Google Scholar]
- 21.Schmid HH, Schmid PC, Berdyshev EV. Chem Phys Lipids. 2002;121:111–134. doi: 10.1016/s0009-3084(02)00157-3. [DOI] [PubMed] [Google Scholar]
- 22.Sugiura T, Kodaka T, Kondo S, Nakane S, Kondo H, Waku K, Ishima Y, Watanabe K, Yamamoto I. J Biochem (Tokyo) 1997;122:890–895. doi: 10.1093/oxfordjournals.jbchem.a021838. [DOI] [PubMed] [Google Scholar]
- 23.Sugiura T, Kodaka T, Nakane S, Miyashita T, Kondo S, Suhara Y, Takayama H, Waku K, Seki C, Baba N, Ishima Y. J Biol Chem. 1999;274:2794–2801. doi: 10.1074/jbc.274.5.2794. [DOI] [PubMed] [Google Scholar]
- 24.Hillard CJ. J Pharmacol Exp Ther. 2000;294:27–32. [PubMed] [Google Scholar]
- 25.Sugiura T, Waku K. Chem Phys Lipids. 2000;108:89–106. doi: 10.1016/s0009-3084(00)00189-4. [DOI] [PubMed] [Google Scholar]
- 26.Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW. Mol Pharmacol. 2000;57:1045–1050. [PubMed] [Google Scholar]
- 27.Berdyshev EV, Schmid PC, Krebsbach RJ, Hillard CJ, Huang C, Chen N, Dong Z, Schmid HH. Biochem J. 2001;360:67–75. doi: 10.1042/0264-6021:3600067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C, Huang Y, Li J, Hu W, Aziz R, Tang MS, Sun N, Cassady J, Stoner GD. Cancer Res. 2002;62:6857–6863. [PubMed] [Google Scholar]
- 29.Dong Z, Ma W, Huang C, Yang CS. Cancer Res. 1997;57:4414–4419. [PubMed] [Google Scholar]
- 30.Dong Z. Environ Health Perspect. 2002;110(Suppl 5):757–759. doi: 10.1289/ehp.02110s5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzukawa K, Weber TJ, Colburn NH. Environ Health Perspect. 2002;110:865–870. doi: 10.1289/ehp.02110865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li JJ, Dong Z, Dawson MI, Colburn NH. Cancer Res. 1996;56:483–489. [PubMed] [Google Scholar]
- 33.He Z, Cho YY, Ma WY, Choi HS, Bode AM, Dong Z. J Biol Chem. 2004;280(4):2446–54. doi: 10.1074/jbc.M402053200. [DOI] [PubMed] [Google Scholar]
- 34.Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorda MA, Lowenberg B, Delwel R. Blood. 2003;101:1336–1343. doi: 10.1182/blood-2002-07-2034. [DOI] [PubMed] [Google Scholar]
- 36.Hawes BE, Luttrell LM, van Biesen T, Lefkowitz RJ. J Biol Chem. 1996;271:12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 37.Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 38.Hashibe M, Ford DE, Zhang ZF. J Clin Pharmacol. 2002;42:103S–107S. doi: 10.1002/j.1552-4604.2002.tb06010.x. [DOI] [PubMed] [Google Scholar]
- 39.Davis MI, Ronesi J, Lovinger DM. J Biol Chem. 2003;278:48973–48980. doi: 10.1074/jbc.M305697200. [DOI] [PubMed] [Google Scholar]
- 40.Downer EJ, Fogarty MP, Campbell VA. Br J Pharmacol. 2003;140:547–557. doi: 10.1038/sj.bjp.0705464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galve-Roperh I, Rueda D, Gomez del Pulgar T, Velasco G, Guzman M. Mol Pharmacol. 2002;62:1385–1392. doi: 10.1124/mol.62.6.1385. [DOI] [PubMed] [Google Scholar]
- 42.Berdyshev EV, Schmid PC, Krebsbach RJ, Schmid HH. FASEB J. 2001;15:2171–2178. doi: 10.1096/fj.01-0181com. [DOI] [PubMed] [Google Scholar]
- 43.Stella N, Piomelli D. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- 44.Watts RG, Huang C, Young MR, Li JJ, Dong Z, Pennie WD, Colburn NH. Oncogene. 1998;17:3493–3498. doi: 10.1038/sj.onc.1202259. [DOI] [PubMed] [Google Scholar]
- 45.Huang C, Ma WY, Young MR, Colburn N, Dong Z. Proc Natl Acad Sci USA. 1998;95:156–161. doi: 10.1073/pnas.95.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galve-Roperh I, Sanchez C, Cortes ML, del Pulgar TG, Izquierdo M, Guzman M. Nat Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- 47.Ma YC, Huang J, Ali S, Lowry W, Huang XY. Cell. 2000;102:635–646. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Yang Y, Hu Y, Dang D, Regezi J, Schmidt BL, Atakilit A, Chen B, Ellis D, Ramos DM. J Biol Chem. 2003;278:41646–41653. doi: 10.1074/jbc.M306274200. [DOI] [PubMed] [Google Scholar]