

Abstract
Dendritic cells (DCs) are professional antigen-presenting cells that act as sentinels in the cell-mediated response against invading pathogens associated with septic challenge. The purpose of the present study was to determine whether there is a loss of dendritic cells and/or changes in function of these cells in septic mice. Here we report that the number of DCs, in both spleen and peritoneum, decreased over 24 h postsepsis [cecal ligation and puncture (CLP)] when compared with sham. The most dramatic change was seen in the peritoneal cavity. This decrease appeared to be caused mainly by the depletion of immature DCs rather than mature DCs. This change was LPS independent and minimally affected by FasL; however, overexpression of human Bcl-2 gene provides protection of the septic peritoneal DCs. Moreover, although the level of IL-12 release decreased significantly in splenic DCs obtained from CLP mice, IL-12 secretion was markedly elevated by peritoneal DCs as well as in both plasma and peritoneal fluid at 24 h post-CLP. In peritoneal cells, the expression of CD40, CD80, and CD86 was unchanged, but their respective ligands CD40L, CD28, and CD152 all increased in mice 24 h after CLP, although no such change was observed in splenocytes. Regardless of the presence or absence of antigen, peritoneal DCs from CLP mice showed higher capacity to stimulate T-cell proliferation than those cells from the sham control. However, splenic DCs from CLP mice only showed augmented capacity to induce antigen-dependent stimulation of T-cell proliferation. Together, these data indicate that sepsis produces divergent functional changes in splenic and peritoneal DC populations.
Keywords: Sepsis, dendritic cells, costimulatory molecules, antigen presentation
INTRODUCTION
Polymicrobial sepsis with concomitant multiple organ failure is one of the leading causes of death in the surgical intensive care unit despite advances in operative intervention, the use of specific antibiotics, and other therapeutic and supportive agents (1-6). Studies indicate that sepsis causes marked immune suppression, which is associated with enhanced plasma anti-inflammatory mediator release, increased lymphocyte Th2 cytokine release, and changes at the intracellular level (7-9).
Dendritic cells (DC) are potent antigen-presenting cells that play an important role in orchestrating the body’s cell-mediated immune response by activating T lymphocytes (10-12). Although a considerably smaller component of the immune cell population (13), DCs retain a much higher capacity to present antigen to T lymphocytes than macrophages. Originating from hematopoietic precursors in the bone marrow, DCs migrate to the periphery as immature cells and reside there. They subsequently become mature in the presence of microbial stimuli and proinflammatory cytokines (i.e., IL-12) or by interacting with CD40L-expressing T cells (10, 14) during an infectious and/or inflammatory event. Immature DCs capture and process foreign antigens and then differentiate into mature DCs. The mature DC is characterized by its expression of high levels of MHC II costimulatory molecules such as CD40, CD80, and CD86 as well as the production of great amounts of Th1 cytokine IL-12 (15, 16). Meanwhile, the expression of the corresponding ligands (CD40L, CD28, and CD152/CTLA-4 on T cells) for the costimulatory molecules expressed on DCs is necessary for the completion of the antigen presentation process and cognate T-cell activation (16). Any defects in this cascade will result in the failure of competent antigen presentation, which will impair the body’s ability to clear a microbial challenge such as that seen with sepsis.
Despite the implication of the above findings concerning the essential role DCs play in cell-mediated immunity, little is understood about the effects of polymicrobial sepsis on DCs. In this respect, Hotchkiss et al. reported recently that DCs were depleted in the spleens of patients with sepsis (17). However, little insight was provided concerning the mechanism for this process of DC loss in the spleen and less is known about the mechanisms that underpin these changes or the consistency at divergent DC sites. Therefore, the objectives of the present study were to determine (a) whether there was a depletion of DCs in a septic mouse model; (b) if so, was it comparable in a site of inflammation (the peritoneum versus the spleen) and what population of DCs (immature versus mature) were affected; (c) whether DCs isolated from septic mice had a reduced capacity to present antigens; (d) if so, was it associated with a loss of the MHC II molecules, the expression of membrane costimulatory molecules, and/or changes in the capacity to release proinflammatory cytokines such as IL-12.
MATERIALS AND METHODS
Animals
Male mice 7-10 weeks old and weighing 20-25 g were used in all experiments. Inbred C3H/HeN mice were purchased from Charles River Laboratories (Wilmington, MA), C3H/HeJ (endotoxin tolerant TLR-4 deficient), and C3H/HeJ-FasLgld (FasL-gld, TLR4, and Fas Ligand deficient, FasL -/-) mice were from Jackson Laboratories (Bar Harbor, ME). The myeloid restricted human Bcl-2 transgenic mice (hBcl2myl) (18) were kindly provided by Dr. I. L. Weissman at Stanford University School of Medicine (Stanford, CA), and their control mice, C57BL/6, were obtained from Jackson Laboratories. The studies performed here were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Rhode Island Hospital Committee on Animal Use and Care.
Cecal ligation and puncture
Polymicrobial sepsis was induced in mice by the method described by Baker et al. (19) and Ayala et al. (20). Briefly, mice were lightly anesthetized with isoflurane, shaved at the abdomen, and scrubbed with betadine. A midline incision (approximately 1 cm) was made below the diaphragm to expose the cecum. The cecum was ligated and punctured twice with a 22-gauge needle and gently compressed to extrude a small amount of cecal contents through the punctured holes. The cecum was returned to the abdomen, and the incision was closed in layers with 6-0 Ethilon suture (Ethicon, Inc., Somerville, NJ). The animals were then resuscitated with 0.6 mL of lactated Ringer’s solution by subcutaneous injection. For the sham controls (sham), the animals were subjected to the same surgical procedure, i.e., laparotomy and cecal isolation, but the cecum was neither ligated nor punctured.
Isolation of splenocytes and peritoneal lavage cells
Mice were sacrificed by inhalation of CO2 at the designated time points following either cecal ligation and puncture (CLP) or sham operation. The peritoneal cells were collected by lavage, and the spleens were dissected. The splenocytes were isolated according to the methods described previously (20). Following hypotonic lysis of the associated erythrocytes, splenocytes were washed twice and counted. Cell viability was assessed by Trypan blue exclusion.
Flow cytometric analysis
Cell suspensions were first blocked with 1 μg/mL Fc block (clone: 2.4G2) antibody (Ab) for 15 min on ice and then stained with various combinations of Abs against surface markers including CD11c (clone: HL3) with MHC II (clone: 10-3.6), CD40 (clone HM40-3), CD80 (clone: 1G10), or CD86 (clone: GL1) for DCs and CD3 (clone 17A1) with CD28 (clone: 37.51), CD40L/CD154 (clone: MRI), or CD152/CTLA-4 (clone: UC10-4F10-11) for T cells (BD Pharmingen, San Diego, CA). Cells were acquired using a FACSCalibur™ (Becton Dickinson), and 50,000 to 300,000 events were collected for analysis.
Isolation of dendritic cells
Spleens were digested by collagenase D (Roche, Indianapolis, IN) and teased apart by repeated pipetting in PBS containing 5% FCS and 5 mM EDTA (21). The splenocytes and peritoneal leukocytes were osmotically lysed and blocked with 50 μg/mL of Fc block for 15 min on ice. Cell suspensions were enriched with anti-CD11c magnetic beads and positive selection columns MS+ according to manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). The flowthrough was also collected to retrieve CD11c- cells.
Antigen presentation
The capacity of either purified peritoneal or splenic CD11c+ cells to present antigen to D10.G4.1 cloned helper T-cells (ATCC, Manassas, VA) was carried out according to the method of Kaye et al. (22) and Ayala et al. (23). In brief, the CD11c+ cells were incubated for 30 min (37°C, 5% CO2, in the dark) with 50 μg/mL mitomycin C (Sigma Chemical Co., St. Louis, MO). The cells were then washed three or four times in PBS, and a series of dilutions was made. The DCs were then cocultured with 2 × 104 cells of D10.G4.1 in the presence or absence of 300 μg/mL conalbumin (Sigma Chemical Co.) for about 2½ days (37°C, 5% CO2). After incubation, the cells were collected by centrifugation and kept in -80°C for at least 30 min. The cell pellets were then allowed to thaw completely at room temperature, and the T-cell proliferation was determined using CyQUANT® cell proliferation assay kit (Molecular Probes, Eugene, OR).
Determination of IL-12 level in plasma, peritoneal lavage fluid, and CD11c+ cell culture supernatants
Purified CD11c+ cells from spleen and peritoneal lavage were cultured in 96-well tissue culture plates in RPMI-1640 medium containing 10% FCS and 50 ng/mL gentamycin. After 24 h incubation at 37°C, 5% CO2, the plate was centrifuged at 1500 rpm for 10 min. Supernatants were collected and frozen at -80°C until use. Plasma and lavage fluid samples were collected and clarified by centrifugation. IL-12 levels in cell supernatants, plasma, or lavage fluid were measured by ELISA (BD Pharmingen) as described previously in our laboratory (24).
Presentation of data and statistical analysis
The data are presented as mean ± SD for each group. Differences in percentile (nonparametric) data were considered to be significant if P < 0.05, as determined using either a Mann-Whitney U test (for pairwise comparison) or the Student-Newman-Keuls test (multiple group pairwise comparison). Alternatively, cytokine values data were considered to be significant if P < 0.05, as determined by two-way ANOVA.
RESULTS
Phenotype of DCs in C3H/HeN mice after sham or CLP
Four, 24, and 48 h after sham or CLP, splenocytes were isolated and stained with anti-CD11c and anti-MHCII Abs (Fig. 1). At the 4-h time point, no change was observed between sham and CLP in the percentages of total CD11c+ cells, CD11c+/MHCII- (immature) and CD11c+/MHCII+ (mature) DCs (Fig. 1, C-E). Compared with sham, 24, and 48 h after surgery, CLP mice showed decreased percentages of total CD11c+ cells and immature, but not mature, DCs (Fig. 1, F-K). Peritoneal cells were also collected 24 h after surgery and stained with CD11c and MHCII Abs (Fig. 2). CLP mice showed a significant decrease in percentages of total CD11c+ cells and immature DCs (Fig. 2, C-D). Percentage of mature DCs also trended toward reduced levels; however, it did not reach statistical significance (Fig. 2E).
Fig. 1. Phenotype of splenic DCs following sepsis.

Splenocytes were isolated 4, 24, and 48 h after CLP or sham and stained for the expression of CD11c/MHCII. The results of a typical flow cytogram of splenocytes harvested 24 h after surgery on C3H/HeN mice are shown in A and B. Cumulative results from different time points are presented (4 h, C-E; 24 h, F-H; 48 h, I-K) as percentage of total DCs (% of CD11c+), percentage of immature DCs (% of CD11c+/MHC II-), and percentage of mature DCs (% of CD11c+/MHCII+). n = 5-6 mice/group. *P < 0.05 versus sham, Mann-Whitney U test.
Fig. 2. Phenotype of peritoneal DCs following sepsis.

Peritoneal lavage cells were isolated 24 h after CLP or sham and stained for CD11c/MHCII expression. Flow cytometric analysis was used to determine the percentage of peritoneal DCs among the peritoneal leukocytes from C3H/HeN mice. The results of a typical flow cytogram of peritoneal leukocytes harvested 24 h after surgery are shown in A and B. Cumulative results from repeated experiments are presented as percentage of total DCs (% of CD11c+, C), percentage of immature DCs (% of CD11c+/MHCII-, D), and percentage of mature DCs (% of CD11c+/MHCII+, E). n = 5-6 mice/group. *P < 0.05 versus the equivalent sham group, Mann-Whitney U test.
The loss of DCs is differentially effected by the lack of TLR4
Studies indicate that endotoxin, a common component of gram-negative bacteria present in CLP mice, can have a marked effect on DC function/maturation and possibly DC numbers (25-27). To address this, we initially examined the effect of functional TLR4 receptor deficiency, using C3H/HeJ mice, on CD11c+ cell numbers following CLP. Although the number of total CD11c+ cells from spleen was consistently lower initially in sham C3H/HeJ as compared with sham C3H/HeN mice (Fig. 3, A-C), there was a further marked decline in the percentage of total CD11c+ cells in septic C3H/HeJ mice. Interestingly, this was caused primarily by a loss of mature DCs and less so by the loss of immature DCs (Fig. 3, B-C). Alternatively, the number of total CD11c+ cells were comparable in the peritoneum (Fig. 3, D-F), and an equivalent decline was seen in immature DCs from both septic C3H/HeJ and C3H/HeN mice.
Fig. 3. Effects of TLR4 and TLR4-FasL deficiency on the changes of DC percentages following sepsis.

Splenocytes (A-C) and peritoneal lavage cells (D-F) from C3H/HeN, C3H/HeJ (TLR4-/-), or C3H/HeJ-FasL-gld (TLR4-/-, FasL-/-) mice were isolated 24 h after surgery. Cells were stained for CD11c/MHCII expression and analyzed by flow cytometry. The results are presented as percentage of total DCs (% of CD11c+), percentage of immature DCs (% of CD11c+/MHC II-), and percentage of mature DCs (% of CD11c+/MHCII+). n = 5-6 animals/group. *P < 0.05 versus the equivalent sham group. #P < 0.05 versus C3H/HeN sham group using the Student-Newman-Keuls test.
The decrease of DCs was minimally affected by FasL
To address whether the decrease of CD11c+ cells was FasL dependent, a subsequent phenotypic assessment of cells from FasL-/- or their background control, the C3H/HeJ mice, was also conducted. The results showed that the overall percentage of total CD11c+ cells in the spleen from sham or CLP FasL -/- mice was typically lower than that seen in either C3H/HeN or C3H/HeJ mice (Fig. 3, A-B). Interestingly, there was a small decline in total CD11c+ cells following CLP, and most of the cell loss was immature DCs (Fig. 3, A-B). Again, with similar changes in peritoneal DCs compared with either C3H/HeN or C3H/HeJ mice, FasL was not involved in the loss of this DC population seen after sepsis (Fig. 3, D-F).
Overexpression of human Bcl-2 protects from depletion of DCs in peritoneum after CLP
To examine whether the depletion of CD11c+ cells in CLP mice could be protected by overexpression of Bcl-2, cell phenotypes were assessed on myeloid-restricted human Bcl-2 (hBcl2myl) transgenic mice, which overexpress human Bcl-2 gene on myeloid lineage only (18) (Fig. 4). The percentage of total CD11c+ cells in the peritoneum of CLP Bcl-2 transgenic mice did not decline when compared with the C57BL/6 background control animal or the equivalent hBcl2myl transgenic sham mouse but was typically three to fourfold higher then the background mouse CLP group. Unlike other mouse strains (C57BL/6 here or C3H mouse strain assessed earlier), Bcl-2 overexpression showed protection from the loss of DCs after sepsis. There was a slight (though not significant) increase in total peritoneal CD11c+ cells in septic Bcl-2 transgenic mice (Fig. 4D). This increase was mainly in immature DCs and was about six- to sevenfold higher than that of the CLP background control mice (Fig. 4E). Interestingly, although no change was observed in mature DCs from Bcl-2 transgenic mice after sepsis compared with their equivalent shams, the percentage of this DC population from both sham and septic mice was twice as high as that seen in other C57BL/6 groups (Fig. 4F). Conversely, Bcl-2 overexpression had minimal protective effects on splenic DC decline following sepsis (Fig. 4, A-C).
Fig. 4. Effects of myeloid-restricted overexpression of human Bcl2 on the changes of DC percentages following sepsis.

Splenocytes (A-C) and peritoneal lavage cells (D-F) from C57BL/6 (background control), myeloid-restricted hBcl2 transgenic (hBcl2myl, ++) overexpressor mice were isolated 24 h after surgery. Cells were stained for CD11c/MHCII expression and analyzed by flow cytometry. The results are presented as percentage of total DCs (% of CD11c+), percentage of immature DCs (% of CD11c+/MHC II-), and percentage of mature DCs (% of CD11c+/MHCII+). n = 4 animals/group. *P < 0.05 versus the equivalent sham group. #P < 0.05 versus C57BL/6 sham group using the Student-Newman-Keuls test.
Antigen-presenting capacity of DCs from the spleen and peritoneum are differentially effected by sepsis
To determine if sepsis induced a change in the function of CD11c+ cells, antigen-presenting capacity was assessed using D10.G4.1 cells. The results showed that CD11c+ cells isolated from the spleen of both C3H/HeN sham and CLP mice stimulated more D10.G4.1 T-cell line proliferation in the presence of conalbumin (the antigen to which the D10.G4.1 cell responds following appropriate antigen processing and presentation by the DC) than in its absence (Fig. 5A). Further, the CD11c+ cells isolated from CLP mice showed a small but consistently higher capacity to stimulate T-cell proliferation than those from sham controls (Fig. 5A). Alternatively, the capacity of peritoneal DCs to present antigen was minimal, irrespective of whether they were derived from Sham or CLP mice. There was no real difference seen in D10.G4.1 T-cell proliferation in the presence or absence of conalbumin (Fig. 5B). Interestingly, there was a substantial difference in extent of basal/conalbumin (antigen)-independent proliferation evident in the D10.G4.1 T cells in the presence of septic mouse peritoneal DCs as opposed to sham mouse DCs.
Fig. 5. Antigen-presenting capacity of DCs.

Antigen-presenting capacity was determined using antigen conalbumin specifically to stimulate D10.G4.1 clonal T-cell proliferation. Splenic and peritoneal DCs were isolated and purified from C3H/HeN mice 24 h after surgery. The result of a typical experiment (representative of three repeat independent experiments) depicts the extent of D10.G4.1 T-cell proliferation [as determined by fluorescent dye intercalation into DNA (Arbitrary Fluorescent U)] in response to increasing numbers of splenic (A) or peritoneal (B) DCs with or without the conalbumin.
Expression of DC costimulatory molecules and their T-cell ligands
To the degree that these changes in CD11c+ cell number and function seen in CLP were associated with alterations in costimulatory molecules CD40, CD80, and CD86 expression, we evaluated their cell surface expression by flow cytometric analysis. Similarly, because alterations in the cognate T-cell ligands (CD3+ cell), CD40L, CD28, and CD152, respectively, of these costimulatory molecules, might also contribute to these functional changes in CD11c+ cells, those markers were evaluated concurrently. Both peritoneal exudate cells and splenocytes were isolated from C3H/HeN mice 24 h after surgery. In the peritoneal cell population, the percentages of CD11c+/CD40+, CD11c+/CD80+, and CD11c+/CD86+ cells either did not change (CD40, CD80) or showed a trend (statistically not significantly different) toward increased CD86 expression in CLP mice compared with sham animals (Fig. 6, A-C). Concomitantly, the expression of their ligands CD40L, CD28, and CD152 on CD3+ T cells increased as well (Fig. 6, D-F). In CD11c+ splenocytes, on the other hand, after CLP, no change was detected in the expressions of either costimulatory molecules or any of their T-cell ligands when compared with the sham controls 24 h after CLP (data not shown).
Fig. 6. Expression of costimulatory molecules and their receptors on peritoneal leukocytes.
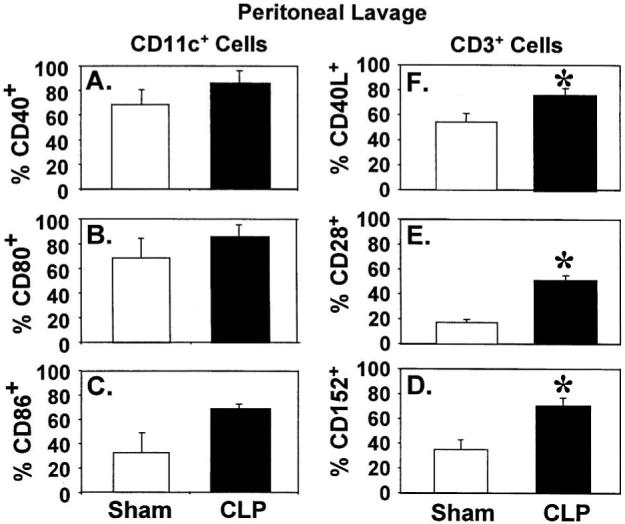
Peritoneal cells were isolated from C3H/HeN mice 24 h after surgery. For costimulatory molecule expression, enriched DCs (by anti-CD11c-coated magnetic beads) were stained with CD40, CD80, or CD86 Abs (A-C). For the expression of receptors on CD3+ cells, peritoneal cells were stained with CD3 in combination of CD40L, CD28, or CD152 Abs (D-F). The results are presented as percentage positively stained with Abs. *P < 0.05 versus sham, Mann-Whittney U test, mean ± SEM; n = 4-5 animals/group.
Levels of IL-12 in plasma, peritoneal lavage fluid, and conditioned medium of cultured CD11c+ cells
Because IL-12 is a critical cytokine in DC maturation and lymphoid as well as macrophage activation, we attempted to comparatively assess the change in IL-12 levels in plasma, lavage fluid, and the endogenous capacity to release IL-12 in CD11c+ cells from spleen and peritoneum of C3H/HeN mice. Twenty-four hours after CLP, the levels of IL-12 in both plasma and peritoneal lavage fluid increased significantly compared with shams (Fig. 7). The level of IL-12 released by cultured mixed peritoneal cell population was markedly increased following sepsis (Fig. 8). This increase in the overall release of IL-12 appears to be attributable to the CD11c+ cell population and not to the CD11c- cells because, following cell separation, the CD11c+ cells retained their capacity to produce markedly higher levels of IL-12 than sham animals 24 h after surgery (Fig. 8). Conversely, the level of IL-12 produced by cultured splenic CD11c+ cells from septic mice was markedly lower than that from control mice which was consistent with the loss of IL-12 production seen in the mixed splenocyte septic mouse cultures (Fig. 8).
Fig. 7. IL-12 level in plasma and peritoneal lavage fluid.

Plasma and peritoneal lavage fluid were collected from each animal 24 h after surgery. IL-12 levels were measured by ELISA. n = 6 animals/group. *P < 0.05 versus the equivalent sham, two-way ANOVA.
Fig. 8. Levels of IL-12 in conditioned medium of cultured cells from the spleen or peritoneum.

Total splenocytes (total), CD11c- splenocytes, CD11c+ splenocytes, total peritoneal leukocytes (total), CD11c- peritoneal cells, and CD11c+ peritoneal cells were isolated 24 h after surgeries. Triplets of equal number (106) of cells (total, CD11c-, CD11c+) were cultured for 24 h (at 37°C, 5% CO2). The supernatants were collected and pooled for IL-12 measurement by ELISA. n = 4-5 independent repeat experiments. *P < 0.05 versus the equivalent sham by two-way ANOVA.
DISCUSSION
Despite the importance of DCs in the maintenance and development of cell-mediated immunity, studies assessing the behavior of both splenic and peritoneal DCs in septic animals are virtually unknown. This in large part is probably linked to the difficulty in working with the low numbers of DCs residing in the body and to the technical difficulties associated with isolating them. To our knowledge, the function of DCs and the expression of MHC II and costimulatory molecules and their ligands on both splenic (a distal-tertiary lymphoid site) and peritoneal (a proximal-inflammatory site) cells isolated from mice suffering from polymicrobial sepsis have not been studied before.
For the first time, we report that the number of DCs in peritoneum and the percentage of immature versus mature DCs change in septic mice. Moreover, we report that there was a depletion of both splenic and peritoneal DCs 24 to 48 h after CLP. Importantly, we found that this depletion of DCs was caused primarily by the loss of immature DCs rather than mature DCs. The present study does provide some insight into the nature of this DC depletion. What we found was that this depletion of immature DCs seemed to be neither LPS nor FasL dependent. The observation that the depletion of DCs was not FasL sensitive is in keeping with the observation of several groups (28-30). That said, the myeloid restricted overexpression of the human Bcl-2 gene did provide some degree of protection against the loss of peritoneal DCs during sepsis. Although no decline in peritoneal DCs was observed, the fraction of immature DCs actually increased. Thus, non-death-receptor-mediated apoptotic process may contribute to the loss of peritoneal DCs seen here but not in the splenic compartment.
As shown in studies carried out both by other groups and by our own group, sepsis causes marked immune suppression (7, 31-36). We therefore would have expected that there would be a dysfunction of antigen-presenting capacity of DCs derived from septic mice, similar to what has been reported for macrophages at these sites. To our surprise, no change (if anything a slight increase) was found for splenic DCs’ antigen-presenting capacity in septic mice. This was supported by our data showing that (a) no decrease in numbers of mature splenic DCs was found, (b) no change for the expression of costimulatory molecules on splenic DCs from septic mice was evident, and (c) no change in the expression of the ligands for the costimulatory molecules on T cells was found. Interestingly, the peritoneal DCs showed a more active response to septic challenge because the capacity of peritoneal DCs isolated from septic mice to trigger T-cell proliferation was enhanced. These changes were associated with (a) an increase of the expression of DC costimulatory molecules, (b) an increase of the expression of the T-cell ligands for these costimulatory molecules, and (c) an increased level of IL-12 in both peritoneal lavage fluid and DC-conditioned medium. However, this was contradicted by the observation that the percentage of mature peritoneal DCs was also decreased. Note that in the proinflammatory stage of sepsis (the first 0-12 h in this model) (31, 37, 38), there is a large influx of neutrophils and macrophages into the peritoneum, increasing the peritoneal exudate cell yield by two- to threefold. In this respect, we have previously observed that the peritoneal exudate cell yield rises from 6.1 ± 0.3 × 106 cells in sham mice at 24 h to 17.3 ± 7.0 × 106 cells in CLP mice at 24 h (31). No such change occurs in the spleen (sham 4.2 ± 0.7 × 107 splenocytes vs. 4.8 ± 0.9 × 107 cells in CLP mice) (39). Therefore, the decreased percentage of peritoneal DCs may not actually reflect a true decrease in DC number but a relative decrease in proportions. In any case, the elevated capacity of peritoneal DCs to stimulate T-cell proliferation was antigen independent. We speculate that other inflammatory mediators in the peritoneal fluid may have contributed to the enhanced capacity of DCs to stimulate T-cell proliferation (10). Thus, polymicrobial sepsis seemed to cause divergent effects on splenic DCs and peritoneal DCs. That said, the impact of these changes on the induction of lymphoid (cell-mediated) immune dysfunction previously reported in septic animals (40) and/or septic patients (41, 42) can only be speculated on here because neither mice genetically deficient in DCs nor good selective DC ablative in vivo protocols are presently available.
IL-12 is a proinflammatory cytokine that plays an important role in modulating the body’s immune response. DCs are the major source of IL-12 production (43, 44). It is noteworthy that in the septic mice, the peritoneal DCs produced significantly higher amounts of IL-12 than control mice, indicating DCs’ contribution to the elevated level of IL-12 in peritoneal lavage fluid. The splenic DCs from septic mice, however, produced a lower amount of IL-12 in vitro. It remains to be determined whether this is because IL-12 was already released into circulation before these cells were sampled or if the cells’ capacity to produce IL-12 had been directly or indirectly suppressed. We might also speculate that the potentiation of the antigen-independent proliferative response seen with isolated peritoneal DCs from septic mice was a result of enhanced release of IL-12 by these cells; however, this remains to be tested.
One limitation of the current study is that only CD11c was used as the marker to identify DCs, although MHCII and costimulatory molecules were used to determine the developmental stages of DCs. Subsets of DCs can be derived from different origins and may behave functionally differently (45, 46). Therefore, in future studies, it will be important to use different combinations of markers to study each DC subset in more detail.
It is also worth noting that all the studies done here examining the effect of sepsis on DC function and cell loss were carried out in the absence of antibiotics, which are common components of the septic patient’s treatment. In this regard, although the presence of antibiotics might have created conditions that more closely approximate septic patient treatment, it is clear from the work from a number of groups that using antibiotics would not have ameliorated much of the morbidity and mortality that is evident in this mouse model of polymicrobial sepsis (47, 48). Depending on the nature of the antibiotic or antibiotic combination used, they may have potentially significant confounding effects on DC/immune cell function (49). That said, although the mechanism underpinning the loss of splenic DCs was not examined, their loss has been reported to occur both in patients succumbing from sepsis and in mice subjected to CLP despite the provision of antibiotics (17, 50). Thus, in an effort to reduce variables that might further confound our interpretation of the results, antibiotics were not included in these initial studies.
In summary, the above data indicate that sepsis produces divergent phenotypic and functional changes in the splenic and peritoneal DC population. The loss of immature DCs and the inability of mature splenic DCs to produce Th1 cytokines such as IL-12, versus the potentiation of the mature peritoneal DCs’ capacity to activate T cells, produces immune-suppressive effects. From a clinical perspective, this illustrates that DCs are also affected by sepsis, but the response differs in a tissue-specific fashion. Thus, one could speculate that in some instances replacement of DCs lost during sepsis (51), and/or DC-targeted gene therapy (52), might have some potentially beneficial contributions to the host’s response to infectious challenge. However, because these differences in the septic DC response appear to relate to divergent processes, inflammatory and/or apoptotic as seen here, they support a need for further study to understand the pathological significance of these changes and/or their potential as therapeutic targets.
ACKNOWLEDGMENTS
The authors would like to thank Ms. Sara Spangenberger at Rhode Island Hospital Core Research Laboratories for her help in acquiring flow cytometry data.
This study was supported by the National Institutes of Health (grant R01-GM46354 to A. A.).
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Baue AE. Multiple organ failure. In: Baue AE, editor. Multiple Organ Failure: Patient Care and Prevention. Mosby Year Book; St. Louis: 1990. pp. 421–470. [Google Scholar]
- 3.Goris RJA. Sepsis and multiple organ failure: The result of whole body inflammation. In: Faist E, Meakins J, Schildberg FW, editors. Host Defense Dysfunction in Trauma, Shock and Sepsis. Springer-Verlag; Berlin-Heidelberg: 1993. pp. 161–170. [Google Scholar]
- 4.Suffredini AF. Current prospects for the treatment of clinical sepsis. Crit Care Med. 1994;22:S12–S18. [PubMed] [Google Scholar]
- 5.Barriere SL, Lowry SF. An overview of mortality risk prediction in sepsis. Crit Care Med. 1995;23:376–393. doi: 10.1097/00003246-199502000-00026. [DOI] [PubMed] [Google Scholar]
- 6.Abraham E. Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med. 1999;25:556–566. doi: 10.1007/s001340050903. [DOI] [PubMed] [Google Scholar]
- 7.Ayala A, Deol ZK, Lehman DL, Herdon CD, Chaudry IH. Polymicrobial sepsis but not low dose endotoxin infusion causes decreased splenocyte IL-2/IFN-gamma release while increasing IL-4/IL-10 production. J Surg Res. 1994;56:579–585. doi: 10.1006/jsre.1994.1092. [DOI] [PubMed] [Google Scholar]
- 8.Ertel W, Keel M, Steckholzer U, Ungethüm U, Trentz O. Interleukin-10 attenuates the release of proinflammatory cytokines but depresses splenocyte functions in murine endotoxemia. Arch Surg. 1996;131:51–56. doi: 10.1001/archsurg.1996.01430130053009. [DOI] [PubMed] [Google Scholar]
- 9.Song GY, Chung CS, Schwacha MG, Jarrar D, Chaudry IH, Ayala A. Splenic immune suppression in sepsis: a role for IL-10 induced changes in P38 MAPK signaling. J Surg Res. 1999;83:36–43. doi: 10.1006/jsre.1998.5556. [DOI] [PubMed] [Google Scholar]
- 10.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 11.Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med. 2001;68:106–166. [PubMed] [Google Scholar]
- 12.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 13.Crowley MT, Inaba K, Witmer-Pack MD, Gezelter S, Steinman RM. Use of the fluorescence activated cell sorter to enrich dendritic cells from mouse spleen. J Immunol Methods. 1990;133:55–66. doi: 10.1016/0022-1759(90)90318-p. [DOI] [PubMed] [Google Scholar]
- 14.Reis e Sousa C. Dendritic cells as sensors of infection. Immunity. 2001;14:495–498. doi: 10.1016/s1074-7613(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 15.Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83–243. doi: 10.1016/s0065-2776(08)60387-9. [DOI] [PubMed] [Google Scholar]
- 16.Lee KP, Harlan DM, June CH. Role of co-stimulation in the host response to infection. In: Gallin JI, Snyderman R, editors. Inflammation; Basic Principles and Clinical Correlates. Lippincott, Williams & Wilkins; Philadelphia: 1999. pp. 191–206. [Google Scholar]
- 17.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, Karl IE. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. 2002;168:2493–2500. doi: 10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 18.Lagasse E, Weissman IL. Bcl-2 inhibits apoptosis of neutrophils but not the engulfment by macrophages. J Exp Med. 1994;179:1047–1052. doi: 10.1084/jem.179.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in murine cecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- 20.Ayala A, Kisala JM, Felt JA, Perrin MM, Chaudry IH. Does endotoxin tolerance prevent the release of inflammatory monokines (IL-1, IL-6, or TNF) during sepsis? Arch Surg. 1992;127:191–197. doi: 10.1001/archsurg.1992.01420020077011. [DOI] [PubMed] [Google Scholar]
- 21.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaye J, Porcelli S, Tite J, Jones B, Janeway CA., Jr Both a monoclonal antibody and antisera specific for determinants unique to individual cloned helper T-cell line can substitute for antigen and antigen-presenting cells in the activation of T-cells. J Exp Med. 1983;158:836–856. doi: 10.1084/jem.158.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayala A, Perrin MM, Chaudry IH. Defective macrophage antigen presentation following haemorrhage is associated with the loss of MHC class II (Ia) antigens. Immunology. 1990;70:33–39. [PMC free article] [PubMed] [Google Scholar]
- 24.Song GY, Chung CS, Jarrar D, Chaudry IH, Ayala A. Evolution of an immune suppressive macrophage phenotype as a product of p38 MAPK activation in polymicrobial sepsis. Shock. 2001;15:42–48. doi: 10.1097/00024382-200115010-00007. [DOI] [PubMed] [Google Scholar]
- 25.Zitvogel L. Dendritic and natural killer cells cooperate in the control/switch of innate immunity. J Exp Med. 2002;195:F9–F14. doi: 10.1084/jem.20012040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 27.Medzhitov R. Toll-like receptors and innate immunity. Nature Rev. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 28.Rescigno M, Piguet V, Valzasina B, Lens S, Zubler R, French L, Kindler V, Tschopp J, Ricciardi-Castagnoli P. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J Exp Med. 2000;192:1661–1668. doi: 10.1084/jem.192.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoves S, Krause SW, Halbritter D, Zhang HG, Mountz JD, Scholmerich J, Fleck M. Mature but not immature Fas ligand (CD95L)-transduced human monocyte-derived dendritic cells are protected from Fas-mediated apoptosis and can be used as killer APC. J Immunol. 2003;170:5406–5413. doi: 10.4049/jimmunol.170.11.5406. [DOI] [PubMed] [Google Scholar]
- 30.Ashany D, Savir A, Bhardwaj N, Elkon KB. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol. 1999;163:5303–5311. [PubMed] [Google Scholar]
- 31.Ayala A, Perrin MM, Kisala JM, Ertel W, Chaudry IH. Polymicrobial sepsis selectively activates peritoneal but not alveolar macrophage to release inflammatory mediators (IL-1, IL-6 and TNF) Circ Shock. 1992;36:191–199. [PubMed] [Google Scholar]
- 32.Ayala A, Knotts JB, Ertel W, Perrin MM, Morrison MH, Chaudry IH. Role of interleukin 6 and transforming growth factor-beta in the induction of depressed splenocyte responses following sepsis. Arch Surg. 1993;128:89–95. doi: 10.1001/archsurg.1993.01420130101015. [DOI] [PubMed] [Google Scholar]
- 33.Munoz C, Misset B, Fitting C, Bleriot JP, Carlet J, Cavaillon JM. Dissociation between plasma and monocyte-associated cytokines during sepsis. Eur J Immunol. 1991;21:2177–2184. doi: 10.1002/eji.1830210928. [DOI] [PubMed] [Google Scholar]
- 34.Munoz C, Carlet J, Fitting C, Misset B, Bleriot J-P, Cavaillon J-M. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choudhry MA, Ahmad S, Thompson KD, Sayeed MM. Erratum: T-lymphocyte Ca2+ signalling and proliferative responses during sepsis. Shock. 1994;1:466–471. doi: 10.1097/00024382-199406000-00012. [DOI] [PubMed] [Google Scholar]
- 36.Miller-Graziano CL, Szabo G, Kodys K, Mehta B. Interactions of immunopathological mediators (tumor necrosis factor-α, TGF-β, prostaglandin E2) in traumatized individuals. In: Faist E, Meakins JL, Schildberg FW, editors. Host Defense Dysfunction in Trauma, Shock and Sepsis: Mechanisms and Therapeutic Approaches. Springer-Verlag; Berlin: 1993. pp. 637–650. [Google Scholar]
- 37.Ayala A, Karr SM, Evans TA, Chaudry IH. Factors responsible for peritoneal granulocyte apoptosis during sepsis. J Surg Res. 1997;69:67–75. doi: 10.1006/jsre.1997.5027. [DOI] [PubMed] [Google Scholar]
- 38.Ayala A, Lomas JL, Grutkoski PS, Chung CS. Pathological aspects of apoptosis in severe sepsis and shock? Int J Biochem Cell Biol. 2003;35:7–15. doi: 10.1016/s1357-2725(02)00099-7. [DOI] [PubMed] [Google Scholar]
- 39.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and nature of the mediators. Blood. 1996;87:4261–4275. [PubMed] [Google Scholar]
- 40.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6:S27–S38. [PubMed] [Google Scholar]
- 41.Faist E, Angele MK, Zedler S. Immunoregulation in shock, trauma, and sepsis. In: Marshall JC, Cohen J, Vincent J-L, editors. Update in Intensive Care and Emergency Medicine. Springer; Berlin: 2000. pp. 312–334. [Google Scholar]
- 42.Volk HD, Reinke P, Döcke WD. Immunostimulation with cytokines in patients with “immunoparalysis”. In: Marshall JC, Cohen J, Vincent J-L, editors. Update in Intensive Care and Emergency Medicine. Springer; Berlin: 2000. pp. 393–404. [Google Scholar]
- 43.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kampgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLellan AD, Kampgen E. Functions of myeloid and lymphoid dendritic cells. Immunol Lett. 2000;72:101–105. doi: 10.1016/s0165-2478(00)00167-x. [DOI] [PubMed] [Google Scholar]
- 46.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–262. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 47.Newcomb DE, Bolgos G, Green L, Remick DG. Antibiotic treatment influences outcome in murine sepsis: mediators of increased morbidity. Shock. 1998;10:110–117. doi: 10.1097/00024382-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 48.Turnbull IR, Javadi P, Buchman TG, Hotchkiss RS, Karl IE, Coopersmith CM. Antibiotics improve survival in sepsis independent of injury severity but do not chnage mortality in mice with markedly elevated interleukin 6 levels. Shock. 2004;21:121–125. doi: 10.1097/01.shk.0000108399.56565.e7. [DOI] [PubMed] [Google Scholar]
- 49.Vianna RCS, Gomes RN, Bozza FA, Amancio RT, Bozza PT, David CMN, Castro-Faria-Neto HC. Antibiotic treatment in a murine model of sepsis: impact on cytokines and endotoxin release. Shock. 2004;21:115–120. doi: 10.1097/01.shk.0000111828.07309.21. [DOI] [PubMed] [Google Scholar]
- 50.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS. Sepsis induces apoptosis and a profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. 2003;171:909–914. doi: 10.4049/jimmunol.171.2.909. [DOI] [PubMed] [Google Scholar]
- 51.Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell. 2001;106:271–274. doi: 10.1016/s0092-8674(01)00448-2. [DOI] [PubMed] [Google Scholar]
- 52.Oberholzer A, Oberholzer C, Bahjat KS, Ungaro R, Tannahill CL, Murday M, Bahjat FR, Abouhamze Z, Tsai V, LaFace D, Hutchins B, Moldawer LL, Clare-Salzler MJ. Increased survival in sepsis by in vivo adenovirus-induced expression of IL-10 in dendritic cells. J Immunol. 2002;168:3412–3418. doi: 10.4049/jimmunol.168.7.3412. [DOI] [PubMed] [Google Scholar]