

Abstract
Here we describe the many applications of acid urea polyacrylamide gel electrophoresis (acid urea PAGE) followed by Northern blot analysis to studies of tRNAs. Acid urea PAGE allows the electrophoretic separation of different forms of a tRNA, discriminated by changes in bulk, charge, and/or conformation that are brought about by aminoacylation, formylation, or modification of a tRNA. Among the examples described are (i) analysis of the effect of mutations in the Escherichia coli initiator tRNA on its aminoacylation and formylation; (ii) evidence of orthogonality of suppressor tRNAs in mammalian cells and yeast; (iii) analysis of aminoacylation specificity of an archaeal prolyl-tRNA synthetase that can aminoacylate archaeal tRNAPro with cysteine, but does not aminoacylate archaeal tRNACys with cysteine; (iv) identification and characterization of the AUA-decoding minor tRNAIle in archaea; and (v) evidence that the archaeal minor tRNAIle contains a modified base in the wobble position different from lysidine found in the corresponding eubacterial tRNA.
Keywords: acid urea polyacrylamide gel electrophoresis, Northern hybridization, aminoacylation, deacylation kinetics
1. Introduction
As adapter molecules between codons in a mRNA and amino acids in proteins, transfer RNAs (tRNAs) play a critical role in protein synthesis [1]. From their biosynthesis to their function on the ribosome, tRNAs interact with various proteins that include processing enzymes (such as tRNA processing RNases), modifying enzymes, aminoacyl-tRNA synthetases, translation factors (initiation and elongation factors), and ultimately the ribosome and mRNA. Analysis of tRNAs, therefore, constitutes an integral part of studies of protein synthesis. Methods for tRNA purification and sequence analysis, including the determination of base modifications, are essential for structure-function studies and have been described elsewhere [2,3]. Methods for separation of tRNAs on polyacrylamide gels under native or denaturing conditions followed by identification of individual tRNAs by Northern hybridization have also proven to be extremely useful. Here, we describe the analysis of tRNAs by acid urea polyacrylamide gel electrophoresis (acid urea PAGE).
Acid urea PAGE provides the means for separating tRNAs at acidic pH under semi-denaturing conditions. Originally described by Ho and Kan [4] for analyzing the aminoacylation of eukaryotic suppressor tRNAs in vivo, Varshney et al. [5] adapted the approach for the separation of uncharged tRNA from aminoacylated tRNA (aa∼tRNA) and formylaminoacylated tRNA (faa∼tRNA), both of which show a clear electrophoretic mobility shift caused by a change in bulk, charge, and/or conformation of the aa∼tRNA and faa∼tRNA. In combination with the isolation of tRNAs under acidic conditions that preserve the labile ester linkage between tRNA and amino acid and thus allow the isolation of aa∼tRNAs and faa∼tRNAs, acid urea PAGE was firmly established as the method of choice to determine the in vivo state of aminoacylation or formylation of tRNAs. Initially used extensively for the in vivo analysis of mutants of the E. coli initiator tRNAfMet ([5], and reviewed in [6-8]), the technique quickly found its way into the standard repertoire of tRNA analysis from all kingdoms including organelles, such as mitochondria [9,10].
In this review, we describe the basic experimental procedures for acid urea PAGE and present examples of applications (case studies 1−5) that include the in vivo analysis of steady state levels of aminoacylation and formylation of tRNAs; characterization of aminoacylation specificity of tRNAs and aminoacyl-tRNA synthetases; and identification of base modifications.
2. Experimental Procedures
Methods described below include the (1) isolation of total RNA under acidic conditions to maintain the amino acid ester-linkage between tRNA and amino acid; (2) deacylation of aminoacylated tRNAs; (3) separation of tRNAs by acid urea polyacrylamide gel electrophoresis; and (4) Northern hybridization.
2.1. Preparation of total RNA under acidic conditions
2.1.1. Isolation of RNA from E. coli. The protocol is based on the original RNA extraction procedure described by Varshney et al. [5]
Cells (5 ml) are grown to mid-to-late log phase, chilled on ice for 5−15 minutes and pelleted at 4 °C. All subsequent steps are carried out in the cold.
The cell pellet is resuspended in 0.5 ml sodium acetate buffer (0.3 M sodium acetate pH 4.5−5.0; 10 mM Na2EDTA).
One volume of phenol equilibrated with sodium acetate buffer is added, and the sample is mixed by vortexing (10 second-pulse); the cell suspension is kept on ice for 15 min with repeated vortexing. After centrifugation at 12,000 g (10−15 minutes), the aqueous phase is transferred to a fresh tube and the phenol phase is re-extracted with 0.25 ml of sodium acetate buffer.
Aqueous phases from both centrifugation steps are combined and mixed with 2.5 volumes of ice-cold ethanol to precipitate nucleic acids (20 minutes, dry ice; or 2−3 hours at −20 °C).
RNA is recovered by centrifugation at 12,000 g (15−30 minutes); the pellet is washed twice with 70% ethanol, left to dry on ice for several minutes (with the lid open), and finally dissolved in 20−50 μl of 10 mM sodium acetate pH 4.5−5.0, quick-frozen and stored at −80 °C. Typically, 1−2 A260 units of total RNA are obtained from a 5 ml culture.
2.1.2. Isolation of total RNA from archaea, yeast, and mammalian cells
The protocol described above can also be applied to the isolation of total RNA from archaea (e.g. various Halobacterial species [11]; Methanocaldococcus jannaschii; Methanococcus maripaludis [12]), yeast (Saccharomyces cerevisiae [13,14]), and mammalian cells (e.g. COS1, HEK293, HeLa cells [15-18]). We recommend the addition of acid-washed glass beads during phenol extraction (1/4 of suspension volume; 425−600 μm; Sigma) for RNA preparations from methanogenic archaea and yeast to improve lysis efficiency.
2.1.3. Isolation of total RNA using Trizol
Alternatively, total RNA can be isolated under acidic conditions using Trizol reagent (Invitrogen) following the instructions provided by the manufacturer. This extraction procedure is adapted from a protocol described by Chomczynski and Sacchi [19] for isolation of total RNA with unbuffered phenol in the presence of guanidinium hydrochloride.
2.2. Deacylation of aminoacylated tRNA and measurement of deacylation rates
Total RNA is isolated under acidic conditions as described above and resuspended in ice-cold sterile water.
Preparation of bulk deacylated tRNA: A portion of the material is subjected to deacylation by addition of Tris-HCl pH 9.5 to a final concentration of 0.2 M. The deacylation reaction is usually performed at 37 °C for 30−120 min (most tRNAs will be deacylated after 30 min; however, certain tRNAs such as valine and isoleucine tRNAs require longer deacylation treatments [20]); deacylated tRNAs are re-precipitated with ethanol and stored in 10 mM sodium acetate pH 5.0; 10 mM Hepes-NaOH pH 7.4; or 10 mM Tris pH 7.5 at −80 °C.
Measurement of deacylation rates: Deacylation is performed in 0.2 M Tris-HCl pH 9.2 at 37 °C. Aliquots corresponding to 0.1 A260 are removed at appropriate time intervals, mixed with an equal volume of acid urea sample buffer (see section 2.3.1), and quick-frozen on dry-ice. Reaction products are subsequently analyzed by acid urea PAGE/Northern hybridization.
2.3. Acid urea polyacrylamide gel electrophoresis (acid urea PAGE)
tRNAs are separated by acid urea PAGE followed by identification of individual tRNAs using Northern hybridization [5].
2.3.1. Preparation of acid urea polyacrylamide gel
- Gel dimension:
- 0.4 mm × 20 cm × 45 cm
- Gel composition:
- 6.5% polyacrylamide (19:1 acrylamide/bisacrylamide)
- 0.1 M sodium acetate pH 5.0
- 8 M urea
- dissolve the above ingredients under stirring (without heating the solution); adjust
- volume to 50 ml and degas for 5−10 min
- add TEMED (0.15% v/v) and ammonium persulfate (0.7% w/v)
- cast gel and allow to polymerize for approximately 2 hours
- Acid urea sample buffer:
- 0.1 M sodium acetate pH 5.0
- 8 M urea
- 0.05% bromophenol blue
- 0.05% xylene cyanol FF
2.3.2. Gel electrophoresis
It is recommended that a short pre-electrophoresis (∼30 minutes) be performed prior to loading the samples and that the sample wells be cleaned carefully. The electrophoresis buffer is 0.1 M sodium acetate pH 5.0.
Typically, 0.01−0.5 OD of tRNA (< 5 μl) is mixed with one volume of sample buffer and loaded onto the gel. Electrophoresis is carried out in the coldroom at 500 V (∼12 V/cm) for 18−24 hours until the bromophenol blue dye reaches the bottom of the gel.
2.4. Northern hybridization
2.4.1. Transfer of tRNAs to nylon membranes
The portion of the gel containing the tRNAs (between xylene cyanol and bromophenol blue dyes) is transferred to Hybond-N+ membrane (Amersham GE) or Nytran Supercharge membrane (Millipore) using a Hoefer Electroblot apparatus (TE Series Tank Transphor Unit; Amersham GE). After electrotransfer, the membranes are baked for 2 hours at 70−80 °C.
- Transfer buffer:
- 40 mM Tris-HCl pH 8.0
- 2 mM Na2EDTA
- Transfer conditions:
- 10 min at 20 V
- 2 hours at 40 V
- coldroom
2.4.2. Northern hybridization
tRNAs are visualized by hybridization using [32P]-labeled DNA oligonucleotides following standard procedures [21]. Oligonucleotides are 5'-end labeled with γ-[32P]-ATP (3000 Ci/mmol; Perkin Elmer) using T4-polynucleotide kinase (New England Biolabs). Excess γ-[32P]-ATP is removed by Sephadex G-25 chromatography (e.g. MicroSpin G-25 columns; GE Healthcare).
- Prehybridization:
- 6 × SSC
- 10 × Denhardt's solution
- 0.5% SDS
- 6−12 hours at 42 °C
- Hybridization:
- 6 × SSC
- 0.1% SDS
- [32P]-labeled probe (1−2 × 106 cpm/ml)
- pass hybridization solution through 0.2 μm filter prior to addition to membrane
- 12−24 hours at 42 °C
Membranes are washed at room temperature with 6 × SSC (2 × 10 minutes; low stringency), followed by washes at 4 × and 2 × SSC (medium and high stringency). Northern blots are analyzed by autoradiography followed by PhosphorImaging.
2.4.3. Design of hybridization probes
In most cases, we have used synthetic DNA oligonucleotides with a length of 17−25 nucleotides complementary to the desired tRNA target; if necessary, longer probes (up to 45 nucleotides) can be considered. For highest specificity, we recommend the design of hybridization probes targeting the anticodon stem and loop domain. Due to base modifications present in the anticodon loop of tRNAs interfering with efficient hybridization between tRNA and DNA probe, hybridization signals might sometimes not be strong enough. In such a case, alternative hybridization targets on the tRNA (e.g. 5' or 3' terminal region, variable loop) are advised. Alternatively, hybridization signals can be enhanced using either ‘combinatorial oligonucleotide hybridization’ [22], which uses a combination of [32P]-labeled DNA oligonucleotide and a molar excess of a non-labeled DNA oligonucleotide acting as an ‘unfolder’, or [32P]-labeled RNA oligonucleotides, providing for a more stable RNA:RNA hybrid. In both cases, specificity of hybridization has to be carefully monitored on a case-by-case basis.
3. Case Studies
The following examples highlight the many applications of acid urea PAGE/Northern hybridization to studies of tRNA in E. coli, mammalian cells, yeast and archaea.
3.1. Role of unique features of E. coli initiator tRNAfMet in initiation of protein synthesis: Formylation
Initiator tRNAs are used exclusively for initiation of protein synthesis, whereas elongator tRNAs are used for the insertion of amino acids into internal peptide linkages. Translation initiation occurs universally with the amino acid methionine (Met) or its derivative formylmethionine (fMet). To fulfill their unique function in protein synthesis, initiator tRNAs possess a number of highly specific properties that direct them to the initiation pathway of translation [6-8]. These include (i) direct binding to the ribosomal P-site and (ii) exclusion from binding to elongation factor necessary for binding to the ribosomal A-site. In contrast to the protein synthesis systems of eukaryotes (cytoplasmic) and archaea, initiator tRNAs in eubacteria and eukaryotic organelles such as mitochondria and chloroplasts are unique in that the methionine attached to the initiator tRNA is formylated to formylmethionine (fMet∼tRNAfMet), by methionyl-tRNA transformylase (MTF) (Figure 1A). The formyl group acts as a positive determinant for initiation factor IF-2 to select fMet∼tRNAfMet from the pool of all cellular tRNAs and acts as a negative determinant for elongation factor EF-Tu to discriminate between fMet∼tRNAfMet and elongator tRNAs [23-25,8]. In addition, the eubacterial fMet∼tRNAfMet is insensitive to hydrolysis by peptidyl-tRNA hydrolase (PTH) [26], which cleaves the ester linkages between peptide and tRNAs dissociated from the ribosome and thereby allows tRNAs to recycle instead of accumulating as peptidyl∼tRNAs.
Figure 1.
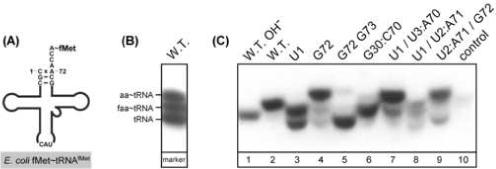
Role of unique features of E. coli initiator tRNAfMet in initiation of protein synthesis. (A) Recognition of tRNAfMet by methionyl-tRNA transformylase (MTF). (B) Separation of the three forms of E. coli initiator tRNAfMet: tRNA, aminoacyl∼tRNA (aa∼tRNA), and formylaminoacyl∼tRNA (faa∼tRNA) by polyacrylamide gel electrophoresis under acidic conditions (acid urea PAGE) followed by detection of tRNA by Northern hybridization using a [32P]-labeled DNA oligonucleotide. The markers for aa∼tRNA and faa∼tRNA were generated by in vitro aminoacylation and formylation of tRNAfMet using purified MetRS and MTF. (C) Acid urea PAGE/Northern analysis of the effect of mutations on aminoacylation and formylation of the mutant tRNAs in vivo. Analysis of tRNAfMet from E. coli transformants carrying the wild type (W.T.) gene or various mutant tRNA genes as indicated. Control, tRNA isolated from transformants carrying the plasmid vector without any tRNA gene (modified from [6]).
For in vivo studies of structure-function relationships of the E. coli initiator tRNAfMet and to establish the critical role of formylation, various mutant tRNAs were generated and examined for their activity in initiation in vivo. Acid urea PAGE followed by Northern hybridization analysis was used to determine the extent of in vivo aminoacylation and formylation of these mutant tRNAs. Control experiments using markers generated by in vitro aminoacylation and formylation of tRNAfMet show that the different forms of initiator tRNA – uncharged tRNAfMet, aminoacylated Met∼tRNAfMet, and formylaminoacylated fMet∼tRNAfMet – can be separated clearly (Figure 1B). Results in Figure 1C show that formylation is strongly affected by mutations in the acceptor stem, in particular mutations at the first, second and third base pair. Studies like these (reviewed in [6-8]) proved to be critical for the elucidation of how tRNAfMet is recognized by methionyl-tRNA synthetase (MetRS) and MTF, and how fMet∼tRNAfMet is utilized in initiation in eubacteria.
As mentioned above, a well-established difference in translation initiation between E. coli and cytoplasmic protein synthesis of eukaryotes is the use of non-formylated in the latter [27,28]. Ramesh et al. [14] showed that expression of E. coli MTF in S. cerevisiae led to formylation of the cytoplasmic initiator (up to 70%) and possibly to initiation of protein synthesis with formylmethionine. The slow growth phenotype of yeast caused by expression of E. coli MTF could be rescued by co-expressing E. coli polypeptide deformylase (DEF), an enzyme that removes formyl groups from the N-terminus of proteins. Again, the in vivo state of formylation of the cytoplasmic yeast initiator was monitored by acid urea PAGE/Northern hybridization linking the slow growth phenotype of yeast cells expressing MTF in the cytoplasm directly to formylation of the eukaryotic initiator .
A striking example of the use of acid urea PAGE of tRNAs comes from studies of a mutant derived from the E. coli elongator tRNA (Mi:2 tRNA), which is active in initiation of protein synthesis in E. coli [29]. Activity of the mutant tRNA in initiation required the overproduction of E. coli MetRS. Acid urea PAGE/Northern hybridization explained the need for overproduction of MetRS. It was found that Mi:2 tRNA is normally aminoacylated in E. coli with lysine [30] and that Mi:2 tRNA aminoacylated with lysine is not a substrate for MTF [29] (Figure 2). Overproduction of MetRS leads to aminoacylation of a portion of the Mi:2 tRNA with methionine and to its consequent formylation by MTF (Figure 2B, lane 5). Besides highlighting the importance of the amino acid attached to the tRNA for its activity as a substrate for MTF, Figure 2B also shows quite clearly the separation by acid urea PAGE of four different forms of the same tRNA; tRNA, Lys∼tRNA, Met∼tRNA, and fMet∼tRNA.
Figure 2.

Important role of the amino acid attached to tRNA in formylation and in initiation of protein synthesis in E. coli. (A) E. coli methionyl-tRNA transformylase (MTF) does not recognize Lys∼tRNAMi:2 as a substrate [29,30]. (B) Acid urea PAGE/Northern analysis of total RNA isolated from E. coli expressing the Mi:2 mutant elongator tRNA. Lanes 1 and 2, marker tRNA isolated from TG1 cells using a high-copy vector for expression of Mi:2 tRNA; tRNA isolated under acidic conditions before (lane 1) and after deacylation by base-treatment (lane 2). Lanes 3−5, tRNA isolated from CA274 transformants containing an additional plasmid that had no aminoacyl-tRNA synthetase gene (lane 3), the E. coli GlnRS gene (lane 4), or the MetRS gene (lane 5) (modified from [29]).
3.2. Orthogonal suppressor tRNAs for site-specific insertion of unnatural amino acids
An important advance in protein engineering has been the site-specific insertion of unnatural amino acids into proteins in prokaryotes or in eukaryotes. The most common approach is based on the suppression of an amber termination codon (UAG) at a predetermined site in the mRNA by an amber suppressor tRNA aminoacylated with the desired unnatural amino acid [31]. The key requirement for this approach is that the suppressor tRNA is ‘orthogonal’ (Figure 3A), i.e. not recognized by any of the aminoacyl-tRNA synthetases present in the respective expression systems. Otherwise, once the aminoacylated suppressor tRNA has inserted the unnatural amino acid at the designated site in the target protein, it will be re-aminoacylated with a natural amino acid and insert the natural amino acid instead of the analogue, thus generating a heterogeneous pool of target protein molecules. Many such orthogonal suppressor tRNAs have been established over the past years, including suppressor tRNAs derived from the eubacterial tRNATyr [31-36], archaeal tRNATyr [37], yeast tRNAPhe [38], bacterial tRNATrp [39], E. coli tRNAGln [15,18], and even human and E. coli initiator tRNAs [40,16,13]. The next requirement is the use of an orthogonal aminoacyl-tRNA synthetase that specifically recognizes the suppressor tRNA but no other tRNA in the cell (Figure 3A) (reviewed in [41-44]). The ultimate step is generation of mutants of the aminoacyl-tRNA synthetase, which use a specific unnatural amino acid instead of the natural amino acid and attach it to the orthogonal suppressor tRNA [37,35,45].
Figure 3.
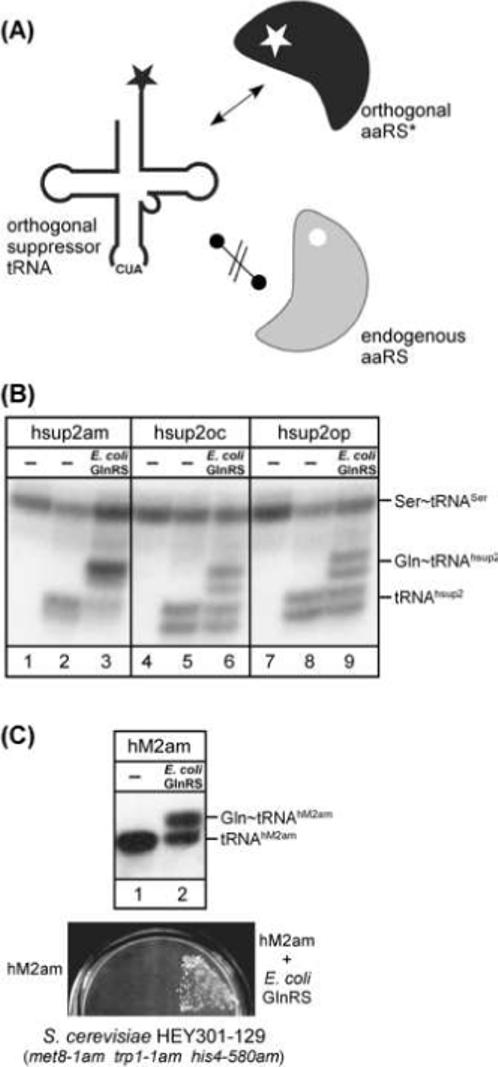
Orthogonal suppressor tRNAs for site-specific insertion of unnatural amino acids. (A) The orthogonal suppressor tRNA is not recognized by any of the endogenous aminoacyl-tRNA synthetases (grey), but is aminoacylated by the orthogonal aminoacyl-tRNA synthetase (black), which recognizes no other tRNA in the cell and has been modified to use unnatural amino acids. aaRS, aminoacyl-tRNA synthetase; the star indicates the unnatural amino acid of interest. (B) A complete set of orthogonal amber, ochre, and opal suppressor tRNAs for use in mammalian cells [18]. The suppressor tRNAs (hsup2am, hsup2oc and hsup2op) are derived from the E. coli glutamine tRNA. Acid urea PAGE/Northern analysis of total RNA isolated from HEK293T cells, transfected with plasmids carrying genes for the respective suppressor tRNA and E. coli GlnRS. The suppressor tRNAs were visualized using a [32P]-labeled oligonucleotide complementary to nucleotides 57−72; a [32P]- labeled oligonucleotide complementary to the human tRNASer was used as internal standard. (C) Orthogonal amber suppressor tRNA/aminoacyl-tRNA synthetase pair for use in yeast [13]. A suppressor tRNA (hM2am) derived from the human initiator tRNA is co-expressed alongside E. coli GlnRS in S. cerevisiae. hM2am is aminoacylated in yeast only in the presence of E. coli GlnRS (top panel; acid urea PAGE/Northern analysis) and is active in suppression (bottom panel). The test for suppression of the met8−1 amber allele was performed in S. cerevisiae HEY301−129 on a selective plate lacking methionine (modified from [13]).
The first example of orthogonal suppressor tRNA/aminoacyl-tRNA synthetase pairs for use in mammalian cells consisted of an amber suppressor tRNA derived from E. coli tRNAGln and E. coli glutaminyl-tRNA synthetase (GlnRS) [15]. The suppressor tRNA was expressed in mammalian cells, and its activity as a suppressor tRNA in readthrough of amber codons in a reporter gene was shown to be strictly dependent upon co-expression of E. coli GlnRS. Based on this orthogonal suppressor tRNA/aminoacyl-tRNA synthetase pair, a complete set of orthogonal amber, ochre, and opal suppressor tRNA/E. coli GlnRS pairs was generated [18]. Acid urea PAGE/Northern hybridization was used to confirm their orthogonality in mammalian cells (Figure 3B). Introduction of additional mutations in the anticodon loop of the amber, ochre, and opal suppressor tRNAs led to much enhanced suppression efficiencies (observed increases up to 36, 156, and 200-fold, respectively) allowing the concomitant suppression of two or three termination codons in a mRNA [18].
Another amber suppressor tRNA, orthogonal in mammalian cells and yeast, was derived from the human initiator [16,13]. Mutations in the anticodon (U35A36) and TψC-arm (U50G51:C63A64/U54/C60) of the initiator tRNA abolished its recognition by MetRS and by initiation factor eIF-2. The tRNA mutant containing a CUA anticodon (U35A36 mutation) was now a substrate for E. coli GlnRS, and acted as an amber suppressor in yeast cells expressing E. coli GlnRS [13] suggesting that the tRNA could now bind the elongation factor EF-1. In vivo aminoacylation with glutamine, estimated to be ∼50% (based on acid urea PAGE/Northern hybridization), allowed suppression of amber alleles in a yeast tester strain, S. cerevisiae HEY301−129 (met8−1am, trp1−1am, his4−580am) [32], indicated by growth on minimal media lacking methionine, tryptophan, or histidine (Figure 3C).
3.3. Formation of Cys∼tRNACys in archaea
In most organisms, the synthesis of Cys∼tRNACys is catalyzed by a canonical cysteinyl-tRNA synthetase (CysRS) that attaches cysteine to the corresponding tRNACys. Surprisingly, certain methanogenic archaea (e.g. Methanocaldococcus jannaschii, Methanothermobacter thermoautotrophicus, and Methanopyrus kandleri) lack the gene encoding a canonical CysRS, yet genes encoding tRNACys are present. This raised the intriguing question of how Cys∼tRNACys, an essential component of protein synthesis, is synthesized in these organisms. A number of studies, using incorporation of radiolabeled amino acids into total tRNA, showed that in come cases prolyl-tRNA synthetase (ProRS) from methanogenic archaea could aminoacylate total tRNA with cysteine [46,47], leading to the hypothesis that ProRS attaches proline to tRNAPro and cysteine to tRNACys. In other words, ProRS – hence called ProCysRS – could be responsible for the formation of Cys∼tRNACys.
This intriguing hypothesis was put to rest (Figure 4A) [48,12] by aminoacylation of total M. jannaschii tRNA with nonradioactive proline or cysteine, followed by acid urea PAGE and Northern hybridization, using probes specific for tRNAPro and tRNACys, to identify the tRNA species that had been aminoacylated [12]. The results showed clearly that purified recombinant ProRS – in addition to its canonical function to generate Pro∼tRNAPro – could also aminoacylate tRNAPro with cysteine, but was unable to aminoacylate tRNACys with cysteine (Figure 4B). In addition, acid urea PAGE revealed a clear difference in mobility of Pro∼tRNAPro and Cys∼tRNAPro. These data showed that ProRS cannot be responsible for generation of Cys∼tRNACys in vivo.
Figure 4.

Characterization of M. jannaschii ProRS (ProCysRS). (A) M. jannaschii ProRS (ProCysRS) catalyzes the formation of Cys∼tRNAPro but not Cys∼tRNACys [12]. (B) Acid urea PAGE/Northern analysis of the total RNA isolated from M. jannaschii cells and aminoacylated in vitro with proline and cysteine by M. jannaschii ProRS and M. maripaludis CysRS. The blots were probed with [32P]-labeled oligonucleotides complementary to M. jannaschii tRNAPro and tRNACys, as indicated. Bands indicated by 1, 2, and 3 correspond to uncharged tRNAPro, Cys∼tRNAPro, and Pro∼tRNAPro, respectively. Bands indicated by 4 and 5 correspond to uncharged tRNACys and Cys∼tRNACys, respectively. OH−, tRNA after deacylation by base-treatment (modified from [12]).
A subsequent study solved the mystery of how archaea lacking a canonical CysRS synthesize Cys∼tRNACys [49]. A two-step pathway was discovered: first, tRNACys is aminoacylated with O-phosphoserine (Sep) by O-phosphoseryl-tRNA synthetase (SepRS) forming the intermediate Sep∼tRNACys; in a second step, Sep∼tRNACys is converted to the final product Cys∼tRNACys by Sep-tRNA:Cys-tRNA synthetase (SepCysS) (Figure 5A). Acid urea PAGE/Northern hybridization analysis of tRNA samples aminoacylated with a filtered M. jannaschii S-100 total cell extract (in the absence and presence of appropriate amino acid mixtures) was used to detect the formation of both Sep∼tRNACys and Cys∼tRNACys. Sep∼tRNACys formation was further confirmed using purified recombinant M. jannaschii SepRS (Figure 5B). In requiring more that one step for the synthesis of Cys∼tRNACys, the newly identified SepRS/SepCysS pathway in archaea is similar to the biosynthesis of Asn∼tRNAAsn and Gln∼tRNAGln (in certain eubacteria and archaea), and of Sec∼tRNASec [50].
Figure 5.

Formation of Cys∼tRNACys in some methanogenic archaea. (A) Two-step conversion of tRNACys to (i) Sep∼tRNACys, catalyzed by O-phosphoseryl-tRNA synthetase (SepRS), and (ii) Cys∼tRNACys, catalyzed by Sep-tRNA:Cys-tRNA synthetase (SepCysS) [49]. (B) Acid urea PAGE/Northern analysis of total M. maripaludis RNA charged with dialyzed M. jannaschii S-100, M. maripaludis CysRS, and M. jannaschii SepRS (Mja 1660/SepRS) in the presence of 20 amino acids (20 aa), phosphoserine (Sep), or a M. jannaschii S-100 cell extract filtrate (Y3). A portion of the sample was deacylated by base-treatment (OH−). The blots were probed with [32P]-labeled oligonucleotides complementary to M. maripaludis tRNACys (modified from [49]).
3.4. Identification of the AUA-reading minor isoleucine tRNA in archaea
Annotation of the complete genome of the extreme halophilic archaeon Haloarcula marismortui does not include a tRNA for translation of AUA, the rare codon for isoleucine. This is a situation typical for most archaeal genomes sequenced to date. One hypothesis stated that the AUA-reading isoleucine tRNA is derived through alternative splicing of a transcript to produce both a tryptophan tRNA and the isoleucine tRNA [51]. However, using acid urea PAGE, we have shown that the minor AUA-decoding isoleucine tRNA in H. marismortui and other archaeal species is derived from a CAU anticodon-containing tRNA, currently annotated as methionine tRNA, in which C34 in the anticodon is post-transcriptionally modified (Figure 6A) [11]. In addition, we show that the post-transcriptional modification of C at position 34 in the anticodon of this tRNA, responsible for the switch in amino acid and decoding specificity, is different from those present at position 34 of isoleucine tRNA species in eubacteria and in eukaryotes [52,53].
Figure 6.

Identification of the minor isoleucine tRNA in archaea. (A) H. marismortui contains two different CAU anticodon-containing tRNAs, tRNA_12 and tRNA_34, currently annotated as methionine tRNAs. tRNA_12 represents the elongator methionine tRNA (); the minor isoleucine tRNA () of H. marismortui is derived from tRNA_34, and carries an unknown modified base (X) in the anticodon responsible for its aminoacylation and decoding specificity [11]. (B) Determination of deacylation rates of tRNA_12 () and tRNA_34 () from H. marismortui. Total RNA was isolated under acidic conditions and subjected to deacylation by base-treatment as described under Experimental Procedures followed by acid urea PAGE/Northern analysis using probes specific for tRNA_12 (top panel) and tRNA_34 (bottom panel), respectively. The calculated half-lives of deacylation are indicated. aa∼tRNA, aminoacyl-tRNA (modified from [11]).
To investigate whether either of the two CAU anticodon-containing elongator tRNAs, annotated as tRNA_12 and tRNA_34 (The Genomic tRNA Database at http://lowelab.ucsc.edu/GtRNAdb) in H. marismortui, is possibly aminoacylated with isoleucine in vivo, we measured the kinetics of chemical deacylation of aminoacyl-tRNAs corresponding to tRNA_12 and tRNA_34 (Figure 6A), exploiting the well-known fact that the stability of the ester link between tRNA and amino acid is determined by the nature of the amino acid attached to the tRNA [20]; with isoleucine and valine being the most stable and methionine being much less stable. Aminoacyl-tRNAs present in total RNA were isolated from H. marismortui under acidic conditions to retain the aminoacyl-ester linkage and were subjected to base-catalyzed deacylation. Samples were subsequently analyzed by acid urea PAGE and kinetics of deacylation followed by Northern hybridization using probes specific for tRNA_12 and tRNA_34, respectively (Figure 6B). Aminoacyl∼tRNA_12 is almost completely deacylated within 15 min, whereas deacylation of aminoacyl∼tRNA_34 is incomplete even after 120 min. The half-lives of deacylation of aminoacyl∼tRNA_12 is ∼5 min which is the same as that of Met∼ ; whereas that of aminoacyl∼tRNA_34 is ∼35 min, which is the same as that of [11]. These data, in combination with in vitro re-aminoacylation using purified recombinant H. marismortui MetRS and isoleucyl-tRNA synthetase (IleRS) , show that tRNA_12 is the elongator methionine , and that tRNA_34 is the minor tRNAIle (renamed , X representing an unknown modified base).
The determination of deacylation kinetics has been used earlier to characterize the nature of the amino acid attached in vivo to a mutant human initiator tRNA initiating protein synthesis from non-AUG codons [17], and to establish the importance of the anticodon sequence for recognition of tRNAs by MetRS and valyl-tRNA synthetase in archaea [54].
3.5. Base modifications
Some modified nucleosides play critical roles in the structure and function of tRNAs; in particular, base modifications in the anticodon loop are thought to be important for recognition of tRNAs by their cognate aminoacyl-tRNA synthetases and/or for codon/anticodon interactions. Post-transcriptional modifications at the wobble position 34 and the purine 37, 3' adjacent to the anticodon, are known to be involved in stabilizing the geometry of the anticodon loop and its dynamics for optimum codon/anticodon interaction [55,56]. Early work on tRNA analysis used combinations of column, paper, and thin layer chromatography of partial and complete nuclease digests of tRNAs to identify and characterize modified nucleosides [2,3]. Recent analyses include separation of tRNA fragments or nucleoside mixtures by liquid chromatography followed by mass spectrometric analysis of individual peak fractions (LC-MS), pioneered by the laboratories of Nishimura, McCloskey and Crain [57,58].
One modified base of particular interest is lysidine, a modified cytosine with a lysine side-chain attached to the O2 of cytidine. Found in the wobble position of eubacterial minor isoleucine tRNAs ( or ), lysidine has a dual role (reviewed in [53]): (i) the presence of lysidine in the anticodon allows to translate specifically the minor isoleucine codon AUA; and (ii) modification of cytidine to lysidine is necessary for recognition of by IleRS and, at the same time, for blocking its aminoacylation by MetRS. The enzyme, tRNAIle-lysidine synthetase (TilS), responsible for modifying the minor isoleucine tRNA has been identified and characterized [59] (Figure 7A), and the crystal structure of the enzyme from Aquifex aeolicus has been solved [60]. For a recent study on archaeal minor isoleucine tRNA ([11]; see section 3.4), we have devised a simple method, involving the use of acid urea PAGE, to confirm the presence of lysidine in tRNAs. Total E. coli tRNA or a T7 transcript corresponding to E. coli is incubated with sulfo-NHS-LC-biotin [N-hydroxysuccinimide (NHS) ester of biotin], which reacts with the free α-NH2 group of lysidine. Both, in vitro lysidinylation of the control T7 transcript using TilS and subsequent biotinylation of tRNAs can be monitored by acid urea PAGE followed by Northern blot analysis using a -specific probe (Figure 7B). Introduction of lysidine into the T7 transcript causes a clear mobility shift (Figure 7B, compare lanes 1 and 3). This is expected since the α-NH2 group of lysidine carries a positive charge under the acidic conditions of the gel electrophoresis. Biotinylation of the modified transcript causes an additional supershift (Figure 7B, lane 4), most likely caused by the added bulk from the biotin; as expected, a T7 transcript lacking lysidine does not show a shift (Figure 7B, lane 2). Reaction of total tRNA from E. coli with the NHS-activated biotin, also gave rise to a clear shift of confirming the presence of lysidine (Figure 7B, compare lanes 5 and 6); several minor shifts upon biotinylation were also observed, probably corresponding to partial reaction with other base modifications such as 3-(3-amino-3-carboxypropyl) uridine present in E. coli . In the course of the same study, we found that minor isoleucine tRNAs () from H. marismortui (see section 3.4) and other archaea could not be biotinylated [11]. Thus, the base modification at C34 in the archaeal tRNA is not lysidine, which is consistent with the fact that, so far, no protein with significant homology to TilS could be identified in archaea. These results are intriguing from an evolutionary point of view; the overall pathway for supplying the cell with a minor isoleucine tRNA appears to be similar in eubacteria and most archaea and requires the post-transcriptional modification of a cytidine. Nevertheless, the base modification and the enzyme(s) responsible for this switch appear to be different between eubacteria and archaea.
Figure 7.
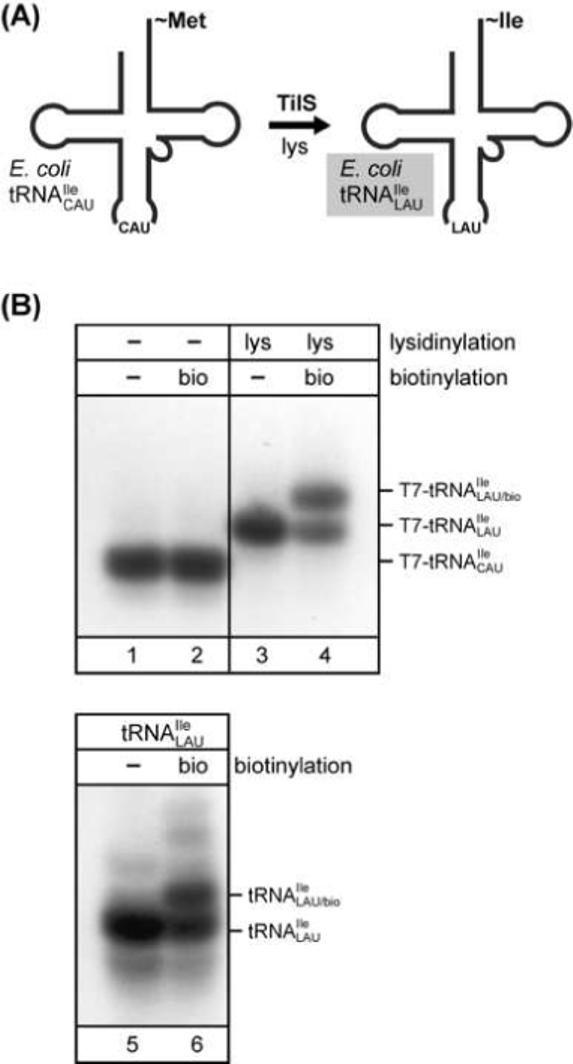
Detection of base modifications. (A) Formation of lysidine in eubacteria. A CAU anticodon-containing tRNA () is modified by tRNAIle-lysidine synthetase (TilS) generating the minor isoleucine tRNA (), which carries lysidine at position 34 in the anticodon. (B) In vitro biotinylation of the free α-NH2 group present in lysidine of from E. coli followed by acid urea PAGE/Northern analysis. A T7 transcript corresponding to was generated as a marker. Lanes 1 and 2, unmodified T7 transcript; lanes 3 and 4, T7 transcript after in vitro modification with lysidine using TilS; lanes 5 and 6, analysis of total RNA from E. coli. A comparison of tRNAs before (lanes 1, 3, 5) and after (lanes 2, 4, 6) in vitro biotinylation is shown (modified from [11]).
4. Concluding Remarks
The separation of tRNAs by acid urea PAGE followed by identification of individual tRNAs by Northern hybridization provides a unique approach to analyze the state of tRNAs in vivo and enables us to gain insight into various aspects of protein synthesis; the determination of levels of aminoacylation and formylation, the characterization of aminoacylation specificity and detection of certain base modifications. One of the most important features of acid urea PAGE/Northern hybridization is the analysis of individual tRNA species present in total tRNAs isolated from cells or in total tRNAs subjected to in vitro manipulation. The combination of acid urea PAGE/Northern hybridization with other RNA techniques (native and denaturing PAGE, in vitro aminoacylation, reverse transcription, derivatization of functional groups etc.) allows one to experimentally verify the presence of any tRNA in a cell and, thereby, make possible experimental validation of predictions of tRNA genes based on in silico studies.
Acknowledgement
We would like to thank Dr. Jae-Ho Yoo for comments and suggestions on the manuscript. This work was supported by grants from the National Institutes of Health (GM17151 and GM67741) and the U.S. Army Research Office (W911NF-04-1-0353).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crick FHC. Symp. Soc. Exp. Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- 2.RajBhandary UL, Köhrer C. J. Biosci. 2006;31:439–451. doi: 10.1007/BF02705183. [DOI] [PubMed] [Google Scholar]
- 3.Grosjean H, Droogmans L, Roovers M, Keith G. Methods Enzymol. 2007;425:57–101. doi: 10.1016/S0076-6879(07)25003-7. [DOI] [PubMed] [Google Scholar]
- 4.Ho YS, Kan YW. Proc. Natl. Acad. Sci. U S A. 1987;84:2185–2188. doi: 10.1073/pnas.84.8.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varshney U, Lee CP, RajBhandary UL. J. Biol. Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- 6.RajBhandary UL. J. Bacteriol. 1994;176:547–552. doi: 10.1128/jb.176.3.547-552.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.RajBhandary UL, Chow CM. In: tRNA: Structure, Biosynthesis, and Function, American Society for Microbiology. Söll D, RajBhandary UL, editors. Washington, DC: 1995. pp. 511–528. [Google Scholar]
- 8.Mayer C, Stortchevoi A, Köhrer C, Varshney U, RajBhandary UL. Cold Spring Harb. Symp. Quant. Biol. 2001;66:195–206. doi: 10.1101/sqb.2001.66.195. [DOI] [PubMed] [Google Scholar]
- 9.Enriquez JA, Attardi G. Proc. Natl. Acad. Sci. U S A. 1996;93:8300–8305. doi: 10.1073/pnas.93.16.8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Holmes WB, Appling DR, RajBhandary UL. J. Bacteriol. 2000;182:2886–2892. doi: 10.1128/jb.182.10.2886-2892.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Köhrer C, Srinivasan G, Mandal D, Mallick B, Ghosh Z, Chakrabarti J, RajBhandary UL. RNA. 2007 doi: 10.1261/rna.795508. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambrogelly A, Ahel I, Polycarpo C, Bunjun-Srihari S, Krett B, Jacquin-Becker C, Ruan B, Köhrer C, Stathopoulos C, RajBhandary UL, Söll D. J. Biol. Chem. 2002;277:34749–34754. doi: 10.1074/jbc.M206929200. [DOI] [PubMed] [Google Scholar]
- 13.Kowal AK, Köhrer C, RajBhandary UL. Proc. Natl. Acad. Sci. U S A. 2001;98:2268–2273. doi: 10.1073/pnas.031488298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramesh V, Köhrer C, RajBhandary UL. Mol. Cell. Biol. 2002;22:5434–5442. doi: 10.1128/MCB.22.15.5434-5442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drabkin HJ, Park HJ, RajBhandary UL. Mol. Cell. Biol. 1996;16:907–913. doi: 10.1128/mcb.16.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drabkin HJ, Estrella M, RajBhandary UL. Mol. Cell. Biol. 1998;18:1459–1466. doi: 10.1128/mcb.18.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drabkin HJ, RajBhandary UL. Mol. Cell. Biol. 1998;18:5140–5147. doi: 10.1128/mcb.18.9.5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köhrer C, Sullivan EL, RajBhandary UL. Nucleic Acids Res. 2004;32:6200–6211. doi: 10.1093/nar/gkh959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chomczynski P, Sacchi N. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 20.Matthaei JH, Voigt HP, Heller G, Neth R, Schöch G, Kübler H, Amelunxen F, Sander G, Parmeggiani A. Cold Spring Harb. Symp. Quant. Biol. 1966;31:25–38. doi: 10.1101/sqb.1966.031.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning - A Laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 22.Buvoli A, Buvoli M, Leinwand LA. RNA. 2000;6:912–918. doi: 10.1017/s1355838200000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono Y, Skoultchi A, Klein A, Lengyel P. Nature. 1968;220:1304–1307. doi: 10.1038/2201304a0. [DOI] [PubMed] [Google Scholar]
- 24.Sundari RM, Stringer EA, Schulman LH, Maitra U. J. Biol. Chem. 1976;251:3338–3345. [PubMed] [Google Scholar]
- 25.Wakao H, Romby P, Westhof E, Laalami S, Grunberg-Manago M, Ebel JP, Ehresmann C, Ehresmann B. J. Biol. Chem. 1989;264:20363–20371. [PubMed] [Google Scholar]
- 26.Kössel H, RajBhandary UL. J. Mol. Biol. 1968;35:539–560. doi: 10.1016/s0022-2836(68)80013-0. [DOI] [PubMed] [Google Scholar]
- 27.Housman D, Jacobs-Lorena M, RajBhandary UL, Lodish HF. Nature. 1970;227:913–918. doi: 10.1038/227913a0. [DOI] [PubMed] [Google Scholar]
- 28.Smith AE, Marcker KA. Nature. 1970;226:607–610. doi: 10.1038/226607a0. [DOI] [PubMed] [Google Scholar]
- 29.Varshney U, Lee CP, RajBhandary UL. Proc. Natl. Acad. Sci. U S A. 1993;90:2305–2309. doi: 10.1073/pnas.90.6.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Kumar NV, Varshney U, RajBhandary UL. J. Biol. Chem. 1996;271:1022–1028. doi: 10.1074/jbc.271.2.1022. [DOI] [PubMed] [Google Scholar]
- 31.Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 32.Edwards H, Schimmel P. Mol. Cell. Biol. 1990;10:1633–1641. doi: 10.1128/mcb.10.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Köhrer C, Xie L, Kellerer S, Varshney U, RajBhandary UL. Proc. Natl. Acad. Sci. U S A. 2001;98:14310–14315. doi: 10.1073/pnas.251438898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiga D, Sakamoto K, Kodama K, Kigawa T, Matsuda T, Yabuki T, Shirouzu M, Harada Y, Nakayama H, Takio K, Hasegawa Y, Endo Y, Hirao I, Yokoyama S. Proc. Natl. Acad. Sci. U S A. 2002;99:9715–9720. doi: 10.1073/pnas.142220099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakamoto K, Hayashi A, Sakamoto A, Kiga D, Nakayama H, Soma A, Kobayashi T, Kitabatake M, Takio K, Saito K, Shirouzu M, Hirao I, Yokoyama S. Nucleic Acids Res. 2002;30:4692–4699. doi: 10.1093/nar/gkf589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Köhrer C, Yoo J, Bennett M, Schaack J, RajBhandary UL. Chem. Biol. 2003;10:1095–1102. doi: 10.1016/j.chembiol.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 38.Mamaev SV, Laikhter AL, Arslan T, Hecht SM. J. Am. Chem. Soc. 1996;118:7243–7244. [Google Scholar]
- 39.Zhang Z, Alfonta L, Tian F, Bursulaya B, Uryu S, King DS, Schultz PG. Proc. Natl. Acad. Sci. U S A. 2004;101:8882–8887. doi: 10.1073/pnas.0307029101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CP, RajBhandary UL. Proc. Natl. Acad. Sci. U S A. 1991;88:11378–11382. doi: 10.1073/pnas.88.24.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hendrickson TL, de Crécy-Lagard V, Schimmel P. Annu. Rev. Biochem. 2004;73:147–176. doi: 10.1146/annurev.biochem.73.012803.092429. [DOI] [PubMed] [Google Scholar]
- 42.Köhrer C, RajBhandary UL. Ibba M, Francklyn C, Cusack S, editors. Aminoacyl-tRNA Synthetases, Landes Bioscience. 2005:353–363. [Google Scholar]
- 43.Wang L, Xie J, Schultz PG. Annu. Rev. Biophys. Biomol. Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 44.Xie J, Schultz PG. Nat. Rev. Mol. Cell Biol. 2006;7:775–782. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 45.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 46.Lipman RS, Sowers KR, Hou YM. Biochemistry. 2000;39:7792–7798. doi: 10.1021/bi0004955. [DOI] [PubMed] [Google Scholar]
- 47.Stathopoulos C, Li T, Longman R, Vothknecht UC, Becker HD, Ibba M, Soll D. Science. 2000;287:479–482. doi: 10.1126/science.287.5452.479. [DOI] [PubMed] [Google Scholar]
- 48.Ahel I, Stathopoulos C, Ambrogelly A, Sauerwald A, Toogood H, Hartsch T, Söll D. J. Biol. Chem. 2002;277:34743–34748. doi: 10.1074/jbc.M206928200. [DOI] [PubMed] [Google Scholar]
- 49.Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates J.r., Ibba M, Söll D. Science. 2005;307:1969–1972. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]
- 50.Feng L, Sheppard K, Namgoong S, Ambrogelly A, Polycarpo C, Randau L, Tumbula-Hansen D, Söll D. RNA Biol. 2004;1:16–20. [PubMed] [Google Scholar]
- 51.Ghosh Z, Chakrabarti J, Mallick B, Das S, Sahoo S, Sethi HS. Biochem. Biophys. Res. Commun. 2006;339:37–40. doi: 10.1016/j.bbrc.2005.10.183. [DOI] [PubMed] [Google Scholar]
- 52.Senger B, Auxilien S, Englisch U, Cramer F, Fasiolo F. Biochemistry. 1997;36:8269–8275. doi: 10.1021/bi970206l. [DOI] [PubMed] [Google Scholar]
- 53.Grosjean H, Björk GR. Trends in Biochem. Sci. 2004;29:165–168. doi: 10.1016/j.tibs.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Ramesh V, RajBhandary UL. J. Biol. Chem. 2001;276:3660–3665. doi: 10.1074/jbc.M008206200. [DOI] [PubMed] [Google Scholar]
- 55.Agris PF. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agris PF, Vendeix FAP, Graham WD. J. Mol. Biol. 2007;366:1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 57.Yokoyama S, Nishimura S. In: tRNA: Structure, Biosynthesis, and Function, American Society for Microbiology. Söll D, RajBhandary UL, editors. Washington, DC: 1995. pp. 207–223. [Google Scholar]
- 58.Crain PF, McCloskey JA. Curr. Opin. Biotechnol. 1998;9:25–34. doi: 10.1016/s0958-1669(98)80080-3. [DOI] [PubMed] [Google Scholar]
- 59.Soma A, Ikeuchi Y, Kanemasa S, Kobayashi K, Ogasawara N, Ote T, Kato J, Watanabe K, Sekine Y, Suzuki T. Mol. Cell. 2003;12:689–698. doi: 10.1016/s1097-2765(03)00346-0. [DOI] [PubMed] [Google Scholar]
- 60.Nakanishi K, Fukai S, Ikeuchi Y, Soma A, Sekine Y, Suzuki T, Nureki O. Proc. Natl. Acad. Sci. U S A. 2005;102:7487–7492. doi: 10.1073/pnas.0501003102. [DOI] [PMC free article] [PubMed] [Google Scholar]