

Abstract
The active site of thioredoxin-1 (Trx1) is oxidized in cells with increased reactive oxygen species (ROS) and is reduced by thioredoxin reductase-1 (TrxR1). The purpose of the present study was to determine the extent to which the redox state of Trx1 is sensitive to changes in these opposing reactions. Trx1 redox state and ROS generation were measured in cells exposed to the TrxR1 inhibitors aurothioglucose (ATG) and monomethylarsonous acid (MMA(III)) and in cells depleted of TrxR1 activity by siRNA knock down. The results showed that all 3 treatments inhibited TrxR1 activity to similar extents (90% inhibition), but that only MMA(III) exposure resulted in oxidation of Trx1. Similarly, ROS levels were elevated in response to MMA(III), but not in response to ATG or TrxR1 siRNA. Therefore, TrxR1 inhibition alone was not sufficient to oxidize Trx1, suggesting that Trx1-independent pathways should be considered when evaluating pharmacological and toxicological mechanisms involving TrxR1 inhibition.
Keywords: Thioredoxin, redox, reactive oxygen species, aurothioglucose, arsenic, siRNA, oxidative stress, redox signaling
INTRODUCTION
Control of the thioredoxin-1 (Trx1) redox state is fundamental to regulation of proliferation, apoptosis and gene expression [1]. Trx1 reduces a number of oxidized protein substrates including ribonucleotide reductase, redox-sensitive transcription factors and proteins involved in antioxidant defense. In the process, the active site of Trx1 is oxidized to a disulfide and must be reduced by thioredoxin reductase-1 (TrxR1). Together, these two proteins constitute the nuclear/cytoplasmic thioredoxin system. There is also a mitochondrial thioredoxin system consisting of TrxR2 and Trx2, but the current report focuses on the nuclear/cytoplasmic thioredoxin system. In cells examined so far, greater than 95% of the Trx1 pool has a reduced active site [2–5]. Trx1 oxidation has been observed, but only under conditions associated with increased levels of reactive oxygen species (ROS) such as the addition of exogenous oxidants [6] or the cellular production of ROS during redox signaling in response to growth factor stimulation [2].
TrxR1 is the only enzyme thought to reduce Trx1 in cells. Although Trx1 can be reduced by TrxR2 in vitro [7], TrxR2 is typically confined to the mitochondria and does not have access to Trx1. TrxR1 contains a highly reactive selenocysteine residue in its carboxy-terminal active site [8]. This exposed selenocysteine is particularly susceptible to inhibition by electrophiles [9, 10], gold- and platinum-containing compounds [10, 11] and arsenicals [12, 13]. However, it is unknown whether inhibition of TrxR1 leads to oxidation of the Trx1 active site.
In the current studies we have examined the influence of TrxR1 inhibition and ROS generation on the redox state of Trx1 in cells. The results demonstrate that Trx1 oxidation is not an inevitable consequence of TrxR1 inhibition and that elevated intracellular ROS levels are a much better predictor of Trx1 oxidation.
MATERIALS AND METHODS
Cell culture
HeLa cells were obtained from ATCC and cultured in DMEM supplemented with 10% fetal bovine serum, penicillin and streptomycin in a humidified atmosphere containing 5% CO2.
Chemical inhibition of TrxR1 activity
Cells were treated with either aurothioglucose (ATG; US Pharmacopeia) or monomethylarsonous acid (MMA(III); a gift from J. Gandolfi) by the addition of a concentrated stock to the culture medium. Both chemical stocks were prepared in water, and control cells received an equal volume of water. Note: all arsenic compounds should be considered as potential human carcinogens. Appropriate precautions should be taken when handling and disposing of these compounds.
Transfection with siRNA
All siRNA reagents were obtained from Dharmacon. TrxR1 knock down was compared to 3 different controls: non-targeting siRNA, mock transfected and untransfected cells. HeLa cells were plated 24 h prior to transfection. TrxR1 knock down (siTR) cells were transfected by replacing medium with fresh medium containing Dharmafect1 transfection reagent and SMARTpool siRNA targeting 4 different regions specific to TrxR1. Non-targeting siRNA controls (siNT) cells were transfected with siCONTROL Non-Targeting Pool #2 (4 siRNAs with at least 4 mismatches to all known human genes). Mock transfected cells received transfection reagent without addition of RNA, while untransfected cells received media change only.
TrxR activity
The standard assay described here does not discriminate between TrxR1 and TrxR2 and we will refer to the measured activity as “TrxR” activity [14]. After incubation with chemical inhibitors or transfection with siRNA, cells were lysed in TE buffer supplemented with 0.5% triton X-100, 0.5% deoxycholate, 0.1% sodium dodecylsulfate and 150 mM NaCl, as described [15]. Total protein was measured by the BioRadDC assay (BioRad) using gamma-globulin as the standard. Equal protein (11 μg) was assayed for TrxR activity using recombinant human Trx1 in the insulin reduction endpoint assay [14].
Western blotting
Cellular lysates prepared as described above for determination of TrxR activity were analyzed by western blotting using antibodies specific for human TrxR1 and beta-actin (Santa Cruz). Secondary antibodies were labeled with Alexafluor-680 (Invitrogen) and detected by the Odyssey imaging system (LiCor).
Trx1 Redox Western assay
The redox state of Trx1 was determined as described previously [5]. Briefly, cells were lysed in a denaturing buffer containing 50 mM iodoacetic acid (IAA), which carboxymethylates reduced thiols but does not react with oxidized cysteines. Proteins were separated by native polyacrylamide gel electrophoresis and analyzed for Trx1 by western blotting with a commercially available antibody (American Diagnostica). This technique is able to detect 3 distinct redox forms of Trx1: fully reduced, active site-disulfide and two-disulfide forms [5]. In the data presented here, only the fully reduced and the active site disulfide forms were observed. For simplicity, these 2 forms will be referred to as “reduced” and “oxidized.”
Measurement of ROS
Cells were pre-incubated with 20 μM 2’,7’-dichlorodihydrofluorescein diacetate (H2DCF-DA; Invitrogen) for 10 min and then the chemical thioredoxin reductase inhibitors were added for another 1 h. Cells were pelleted by centrifugation, resuspended in PBS and fluorescence of dichlorofluorescein (DCF; formed by deacetylation and oxidation of H2DCF-DA) was determined by FACS analysis (FACScan, Becton Dickinson). Results are expressed as the fold-increase in the mean fluorescence intensity in cells treated with inhibitors relative to cells exposed to vehicle and H2DCF-DA only. To measure ROS levels in TrxR1 knock down cells, cells were transfected for 47 h with TrxR1 siRNA and appropriate controls as described above, and H2DCF-DA was added to each for an additional 1 h before collection and FACS analysis.
RESULTS
The only enzyme that has been demonstrated to reduce Trx1 in cells is TrxR1. Therefore, decreased TrxR1 activity should be accompanied by increased levels of oxidized Trx1. To test this assumption we depleted cells of TrxR1 using siRNA and measured the relative levels of reduced and oxidized Trx1. As shown in Fig. 1, transfection of HeLa cells with siRNA targeted against TrxR1 effectively decreased TrxR1 protein levels and activity relative to each of the 3 different control cells. When compared to cells transfected with non-targeting siRNA sequences (siNT), there was a 95% decrease in protein and 90% decrease in activity 48 h after transfection (Fig. 1A and 1B). Surprisingly, the redox state of Trx1 was unaffected by TrxR1 depletion. Only 2% of the total Trx1 pool was in the oxidized form, regardless of the level of TrxR1 activity (Fig. 2C). In contrast, in cells exposed to 1 mM H2O2 there was a decrease in the reduced form and an increase in the oxidized form, such that 40% of the Trx1 was oxidized. These results show that although Trx1 could be oxidized in response to exogenous ROS, Trx1 remained almost completely reduced in cells depleted of 90% of their TrxR1 activity by siRNA knock down.
Fig. 1. Knock down of TrxR1 protein and activity does not oxidize Trx1.
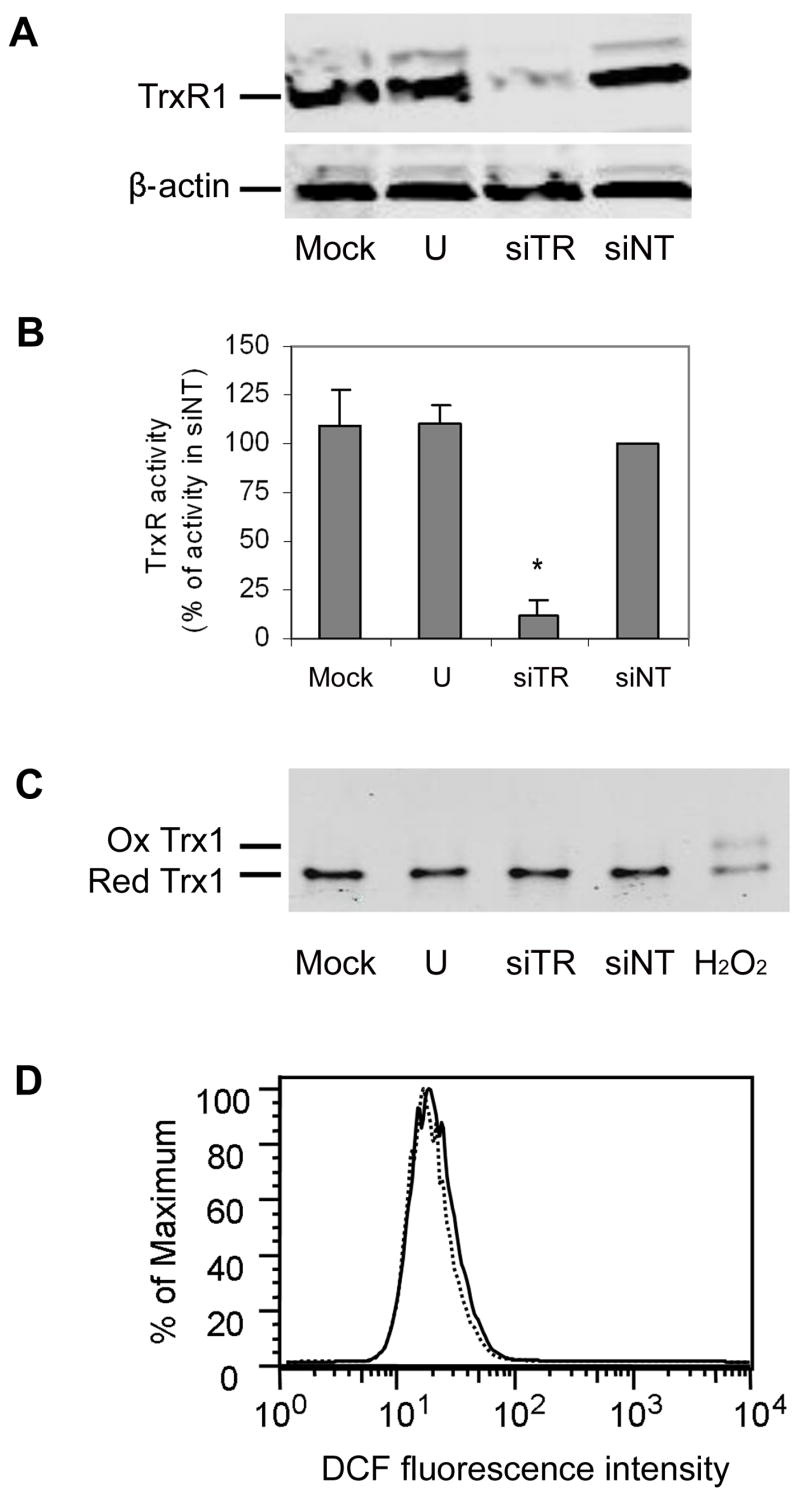
A. Cells were transfected with siRNA directed against TrxR1 (siTR), a non-targeting siRNA control (siNT), mock-transfected (mock) or untransfected (U). TrxR1 protein levels were analyzed by western blotting 48 h post-transfection. Membranes were reprobed for beta-actin to demonstrate equal loading.
B. Cells were transfected for 48 h as in A, and TrxR activity was measured as the Trx1-catalyzed reduction of insulin. Activity is expressed as a percentage of the activity from cells transfected with control siRNA (siNT). Values are the mean ± sem of 3 independent experiments performed in duplicate. An * denotes a statistically significant difference from siScr (p<0.05).
C. Cells were transfected for 48 h as in A, and the redox state of Trx1 was determined by the Redox Western blot method. As a positive control for Trx1 oxidation, 1 mM H2O2was added to an untransfected culture for 2 min. The positions of the reduced and oxidized forms are indicated. This Redox Western blot is representative of 3 independent experiments.
D. Cells were transfected with siRNA specific for TrxR1 (siTR; dashed line) or non-targeting siRNA (siNT; solid line) for 48 h. Cells were loaded with the redox-sensitive dye H2DCF-DA for the final 1 h. The histogram shows the number of cells (as a percentage of the maximum number of cells) exhibiting a given DCF fluorescence intensity as measured by FACS analysis. The data shown is representative of 2 separate experiments.
Fig. 2. Different chemical inhibitors of TrxR1 activity have different effects on cellular Trx1 redox state and ROS production.

Cells were treated with vehicle (water), ATG or MMA(III) for 1 h at the indicated concentrations. TrxR activity (A), Trx1 redox state (B) and DCF fluorescence as an indicator of ROS production (C) were measured as in Fig. 1. All values are means ± sem of 3 independent experiments. An * indicates statistical difference (p<0.05) from vehicle controls.
Because of the possibility that unknown compensatory pathways could be activated during the 48 h transfection needed to knock down TrxR1, short-term effects of TrxR1 inhibition on Trx1 redox state were explored using 2 different chemical inhibitors of TrxR1. ATG is used clinically to treat rheumatoid arthritis and has been shown to be an inhibitor of TrxR1 [16]. We found that ATG inhibited TrxR activity by greater than 90% in HeLa cells incubated with 1 mM ATG for 1 h, and there was no further increase in the degree of inhibition when the concentration of ATG was increased to 10 mM (Fig. 2A). Consistent with the results of the siRNA experiment, ATG had no effect on the redox state of Trx1, even when added at a concentration 10-fold higher than was required for maximal inhibition of TrxR1 (Fig. 2B).
We then examined the effects of MMA(III) on Trx1 oxidation. Trivalent arsenicals are known to react with protein cysteines and selenocysteines [17, 18] and to inhibit TrxR1 activity [19]. Several arsenic species have been evaluated, and the organic metabolite MMA(III) was found to be one of the most potent inhibitors of TrxR1 examined to date [12, 20]. There was a slight decrease in TrxR activity in cells exposed to 3 μM MMA(III), but this effect did not reach statistical significance (Fig. 2A). Similarly, there was a non-significant increase in the oxidized form of Trx1 in response to 3 μM MMA(III) (Fig. 2B). Incubation of HeLa cells with 10 μM MMA(III), however, resulted in 95% inhibition of TrxR activity. At this concentration MMA(III) treatment resulted in extensive oxidation of Trx1. In control cells less than 2% of the Trx1 was in the oxidized form, but following MMA(III) treatment 60% of the Trx1 was oxidized. Thus, although both ATG and MMA(III) inhibited cellular TrxR activity, only MMA(III) led to Trx1 oxidation.
The redox state of Trx1 should reflect the balance between 2 opposing forces: the oxidation of Trx1 during catalytic reduction of oxidized substrates and reduction of Trx1 by TrxR1. Because the redox state of Trx1 did not correlate with TrxR activity in the above studies, we reasoned that differences in the rates of ROS generation could account for the different responses to the 2 chemical TrxR inhibitors and TrxR1 siRNA. ROS levels were measured by incubating cells with the redox-sensitive dye H2DCF-DA. The results showed that incubation with 1 mM ATG, 10 mM ATG, or 3 μM MMA(III) had no effect on ROS levels (Fig. 2C). Similarly, knock down of TrxR1 by siRNA did not increase ROS (Fig. 1D). In contrast, 10 μM MMA(III) yielded a 5-fold increase in DCF fluorescence, indicating increased generation of ROS in these cells. These findings provide a possible explanation for the observation that Trx1 was oxidized in response to MMA(III), but not ATG or TrxR1 siRNA.
DISCUSSION
This study examined how changes in TrxR1 activity and ROS production affected the redox state of Trx1 in cells. The data showed that the redox state of cellular Trx1 was mostly reduced, consistent with previous studies [2, 4, 5, 21]. The current data further show that this reduced redox state was maintained even in cells lacking 90% of their TrxR1 activity, either through siRNA knock down or chemical inhibition by ATG. These findings demonstrate that the redox state of Trx1 is tightly regulated in favor of the reduced form, allowing Trx1 to function as a reductant under most conditions. Both the current study and our earlier studies showed that Trx1 became oxidized in cells with elevated ROS, either through the addition of exogenous oxidants (H2O2 in Fig. 1B and [6]) or the increased production of intracellular ROS (MMA(III) in Fig. 2B and [2]). Our studies did not address the source of the increased ROS observed in response to MMA(III) exposure, but other arsenic species have been reported to increase ROS production [22, 23]. It should be noted that neither ATG-treated cells nor TrxR1 knock down cells had increased ROS levels, and that these 2 treatments did not increase oxidized Trx1 levels. Therefore, TrxR1 activity was not a good predictor of Trx1 oxidation in cells without elevated ROS.
Neither of the chemical inhibitors used in this study would be expected to be specific for TrxR1 over TrxR2 [11, 13, 24]. However, Gorlatov and Stadtman reported that TrxR1 is the predominant TrxR isoform in HeLa cells [25], and the data presented here support this finding. When TrxR1 was specifically knocked down by siRNA, the cells lost 90% of their TrxR activity (a combined measure of both TrxR1 and TrxR2), suggesting that the majority of TrxR activity could be attributed to TrxR1.
The mechanism by which Trx1 redox state is maintained in TrxR1-depleted cells is unclear, but there are at least 4 possible explanations. First, the rate of Trx1 oxidation may be decreased in parallel with the decrease in the rate of Trx1 reduction. Although there was no decrease in the rate of ROS formation in ATG-treated or TrxR1 siRNA knock down cells, Trx1 may be specifically protected from oxidation. Second, HeLa cells may have an excess capacity to reduce Trx1. This seems unlikely given that there is at least some measurable oxidized Trx1 under basal conditions. Third, there may be enzymes other than TrxR1 that can reduce Trx1. Recently indirect evidence for such a protein in Arabidopsis was reported [26]. The results of the TrxR activity assay argue against the existence of another major source of Trx1 reducing activity in HeLa cells, but it is possible that other enzymes that could reduce Trx1 may not be fully active under the assay conditions used here. Finally, Trx1 may be a preferred substrate for TrxR1. TrxR1 reduces a number of small molecules and proteins [27]. We only measured the redox state of Trx1 in this study, so it remains to be determined whether other substrates are more sensitive than Trx1 to oxidation when TrxR1 activity is limiting.
The thioredoxin system makes an attractive chemotherapeutic target, and several drugs are being developed that target either Trx1 or TrxR1 [1, 13, 28–31]. This target-based drug development strategy is based on the observations that Trx1 and TrxR1 are overexpressed in a wide variety of tumors and contribute to the stimulation of proliferation and the inhibition of apoptosis in cancer cells. In the only other study examining both TrxR1 activity and Trx1 redox state, Holmgren and co-workers reported that TrxR1 was inhibited and Trx1 was oxidized 72 h after addition of cytotoxic concentrations of arsenic trioxide (another trivalent arsenic species) to MCF-7 breast cancer cells [13]. ROS were not measured as part of that study, so it remains to be determined the extent to which ROS were involved in the oxidation of Trx1 at these later time points.
One must use caution when interpreting results of experiments with TrxR1 inhibitors; substrates other than Trx1 may be mediating downstream responses, and Trx1 oxidation cannot be assumed based on loss of TrxR1 activity alone. Further research into Trx1-independent pathways will be required to gain a complete understanding of the role of TrxR1 in diseases such as cancer and rheumatoid arthritis and the mechanisms by which TrxR1 inhibitors impact these diseases.
Acknowledgments
The authors wish to thank Kylee Eblin and Jay Gandolfi for providing us with the MMA(III) used in these studies. These studies were supported by NIH grants ES012260 and ES003819.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lillig CH, Holmgren A. Thioredoxin and related molecules--from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 2.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions*. Free Radic Biol Med. 2006;40:138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1352–1359. doi: 10.1152/ajpgi.00183.2002. [DOI] [PubMed] [Google Scholar]
- 5.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 6.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003;543:144–147. doi: 10.1016/s0014-5793(03)00430-7. [DOI] [PubMed] [Google Scholar]
- 7.Turanov AA, Su D, Gladyshev VN. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. J Biol Chem. 2006;281:22953–22963. doi: 10.1074/jbc.M604326200. [DOI] [PubMed] [Google Scholar]
- 8.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc Natl Acad Sci U S A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nordberg J, Zhong L, Holmgren A, Arner ES. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J Biol Chem. 1998;273:10835–10842. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- 10.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara N, Fujii T, Fujii J, Taniguchi N. Roles of N-terminal active cysteines and C-terminal cysteine-selenocysteine in the catalytic mechanism of mammalian thioredoxin reductase. J Biochem. 2001;129:803–812. doi: 10.1093/oxfordjournals.jbchem.a002923. [DOI] [PubMed] [Google Scholar]
- 12.Lin S, Cullen WR, Thomas DJ. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem Res Toxicol. 1999;12:924–930. doi: 10.1021/tx9900775. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arner ES, Holmgren A. Current Protocols in Toxicology. John Wiley and Sons; 2005. Measurement of thioredoxin and thioredoxin reductase; pp. 7.4.1–7.4.14. [DOI] [PubMed] [Google Scholar]
- 15.Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 16.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 17.Brown SB, Turner RJ, Roche RS, Stevenson KJ. Spectroscopic characterization of thioredoxin covalently modified with monofunctional organoarsenical reagents. Biochemistry. 1987;26:863–871. doi: 10.1021/bi00377a030. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Zhang H, Li XF, Le XC. Study of interactions between arsenicals and thioredoxins (human and E. coli) using mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:3658–3666. doi: 10.1002/rcm.3263. [DOI] [PubMed] [Google Scholar]
- 19.Moore EC, Reichard P, Thelander L. Enzymatic Synthesis Of Deoxyribonucleotides.V. Purification And Properties Of Thioredoxin Reductase From Escherichia Coli B. J Biol Chem. 1964;239:3445–3452. [PubMed] [Google Scholar]
- 20.Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem Res Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- 21.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicological Sciences. 2004;82:308–317. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 22.Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic induces oxidant stress and NF-kappa B activation in cultured aortic endothelial cells. Free Radic Biol Med. 1996;21:783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- 23.Paul MK, Kumar R, Mukhopadhyay AK. Dithiothreitol abrogates the effect of arsenic trioxide on normal rat liver mitochondria and human hepatocellular carcinoma cells. Toxicol Appl Pharmacol. 2008;226:140–152. doi: 10.1016/j.taap.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 25.Gorlatov SN, Stadtman TC. Human selenium-dependent thioredoxin reductase from HeLa cells: properties of forms with differing heparin affinities. Arch Biochem Biophys. 1999;369:133–142. doi: 10.1006/abbi.1999.1356. [DOI] [PubMed] [Google Scholar]
- 26.Reichheld JP, Khafif M, Riondet C, Droux M, Bonnard G, Meyer Y. Inactivation of thioredoxin reductases reveals a complex interplay between thioredoxin and glutathione pathways in Arabidopsis development. Plant Cell. 2007;19:1851–1865. doi: 10.1105/tpc.107.050849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mustacich D, Powis G. Thioredoxin reductase. Biochem J 346 Pt. 2000;1:1–8. [PMC free article] [PubMed] [Google Scholar]
- 28.Pennington JD, Jacobs KM, Sun L, Bar-Sela G, Mishra M, Gius D. Thioredoxin and thioredoxin reductase as redox-sensitive molecular targets for cancer therapy. Curr Pharm Des. 2007;13:3368–3377. [PubMed] [Google Scholar]
- 29.Wang X, Zhang J, Xu T. Thioredoxin reductase inactivation as a pivotal mechanism of ifosfamide in cancer therapy. Eur J Pharmacol. 2008;579:66–73. doi: 10.1016/j.ejphar.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–397. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Yoo MH, Xu XM, Carlson BA, Patterson AD, Gladyshev VN, Hatfield DL. Targeting thioredoxin reductase 1 reduction in cancer cells inhibits self-sufficient growth and DNA replication. PLoS ONE. 2007;2:e1112. doi: 10.1371/journal.pone.0001112. [DOI] [PMC free article] [PubMed] [Google Scholar]