

Abstract
Different strategies proposed as therapy for Alzheimer disease (AD) have aimed to reduce the level of toxic forms of Aβ peptide in the brain. Here, we directly analyze the therapeutic utility of the polyclonal anti-Aβ1–11 antibody induced in 3xTg-AD mice vaccinated with the second generation prototype epitope vaccine. Substoichiometric concentrations of purified anti-Aβ1–11 antibody prevented aggregation of Aβ42 and induced disaggregation of preformed Aβ42 fibrils down to nonfilamentous and nontoxic species. Anti-Aβ1–11 antibody delayed Aβ42 oligomer formation but ultimately appeared to stabilize nonfibrillar conformations, including oligomer-like assemblies. The reduced oligomer-mediated cytotoxicity observed upon preincubation of Aβ oligomers with the anti-Aβ1–11 antibody in the absence of oligomer disaggregation suggests a possible oligomer rearrangement in the presence of the antibody. These in vitro observations suggest that preventive vaccination may protect from AD or may delay the onset of the disease, whereas therapeutic vaccination cannot disrupt the toxic oligomers and may only minimally alleviate preexisting AD pathology.
AD4 is characterized by deposition of fibrillar forms of Aβ peptide in senile plaques, appearance of Aβ congophilic deposits and neurofibrillary tangles in the cerebrovasculature, and neuronal loss (1–4). Aβ peptide is cleaved from the amyloid precursor protein (APP) by β- and γ-secretases (5–7) and is thought to play a central role in the onset and progression of AD (8–10). In AD, the normally soluble Aβ molecule (39–43 aa) undergoes conformational changes and is deposited as insoluble fibrils, oligomers, and protofibrills. Previously, it was demonstrated that Aβ neurotoxicity requires insoluble fibril formation (11) and that these fibrils serve as inducers of neuronal apoptosis (12). Recently, emphasis has shifted to smaller soluble oligomers of Aβ42, such as the 12-mers known as Aβ-derived diffusible ligands, increased about 70-fold in AD patients' brains over controls (13). More recently, it was shown that extracellular accumulation of 56-kDa soluble Aβ assembly impairs memory in middle-aged APP/Tg 2576 mice in the absence of neuritic plaques (14). Aβ42 dimers and trimers naturally secreted from a 7PA2 cell line were also suggested to be responsible for the disruption of cognitive functions (15). Importantly, intraventricular injection of such Aβ42 small oligomers inhibited long term potentiation in rat hippocampus, and an injection of anti-Aβ monoclonal antibody 6E10 prevented this inhibition (16). It has also been demonstrated that passive immunization with monoclonal antibodies (NAB61) that specifically recognize a pathologic conformation present in Aβ oligomers resulted in a rapid improvement in spatial learning and memory (17).
The therapeutic potency of polyclonal and monoclonal anti-Aβ antibodies was documented in different mouse models of AD (18–25). Collectively, these data suggest that antibodies specific to the N-terminal region of Aβ are capable of reducing/blocking deposition of toxic forms of Aβ40/42. The protective activity of N-terminal antibodies was attributed to interference with the Aβ fibrillization pathway. In fact, it was reported that the protective ability of an antibody against 4–10 aa of Aβ42 to inhibit Aβ fibrillization and disaggregate preformed fibrils correlated with 50% reduction in plaque burden (21, 26). However, this antibody only partially inhibits fibrillization and disaggregates Aβ fibrils, at least in part, via filament breakage, which could result in an increased number of toxic structures. Another antibody, AMY-33, raised against residues 1–28 of Aβ, was shown to inhibit Aβ40 assembly into filaments. Nevertheless, large amounts of Aβ40 aggregates still form in the presence of this antibody (27). 6C6, an antibody raised against residues 1–16 of Aβ, transforms ∼80% of Aβ40 fibrillar material (28) into species that appear nontoxic, but the exact nature of these species has not been characterized. Antibody 3D6 against 1–5 aa of Aβ (23, 29) only delays fiber formation in vitro by inhibiting nucleation, and it does not appear to alter oligomer formation (30). Serum against residues 3–6 of Aβ partially (75%) disrupts preformed fibers into noncharacterized species and only partially prevents fiber toxicity (31). M266 antibody, raised against the central domain (residues 13–28) of Aβ (24), appears to completely inhibit fiber formation but has no effect on Aβ oligomerizations (30).
Here we analyze for the first time the therapeutic potency of a polyclonal anti-Aβ1–11 antibody, both in vitro and ex vivo. The antibody was raised in 3xTg-AD mice (32) immunized with the second generation of peptide epitope vaccine composed of the self B cell epitope from the immunodominant region of Aβ42 (Aβ1–11) in tandem with a universal foreign T cell epitope, PADRE. We showed that purified anti-Aβ1–11 antibody binds to different forms of Aβ42, reduces Aβ burden after intrahippocampal injection, prevents aggregation of Aβ42, and induces disaggregation of preformed Aβ42 fibrils to a heterogeneous population of nonfilamentous and nontoxic aggregates. In addition, this antibody delays Aβ42 oligomer formation significantly but ultimately appears to stabilize nonfibrillar conformations, including oligomer-like assemblies of beaded appearance. The reduced oligomer-mediated cytotoxicity observed upon preincubation of Aβ oligomers with the anti-Aβ1–11 antibody and the lack of oligomer disaggregation property suggest a possible antibody-dependent oligomer reorganization. These data along with the results demonstrating that PADRE is the universal T helper cell epitope in humans indicate that the second generation of epitope AD vaccine can induce therapeutically potent titers of anti-Aβ1–11 antibody in AD patients. However, these observations suggest that therapeutic vaccination unable to disrupt toxic oligomers may minimally inhibit preexisting AD pathology, whereas preventive vaccination could protect from AD by inhibiting/delaying and blocking formation of soluble and fibrillar species of Aβ, respectively.
Experimental Procedures
Antigen Preparation and Immunization of Mice
We synthesized two copies of the N terminus of the immunodominant B cell epitope of Aβ1–11 in tandem with a potent foreign T cell epitope, PADRE, and then conjugated them through the C terminus to MAP, (Invitrogen) as we described previously (33). 5.5–7.5-month-old hemizygous 3xTg-AD mice (32, 34) provided by Dr. LaFerla's laboratory at the University of California (Irvine, CA) were immunized four times biweekly with 2Aβ1–11-PADRE-MAP vaccine or fibrillar Aβ42 (fAβ42). Before immunization, the peptide was mixed with a Th1 type conventional adjuvant, Quil A, and mice were injected subcutaneously with 50 μg of the antigen (35). Sera were collected from individual animals 9 days after the last boost and kept at +4 °C.
Detection of Anti-Aβ Antibodies by Enzyme-linked Immunosorbent Assay
Total anti-Aβ42 antibodies were detected as described previously (35, 36) with small modifications; to develop the reaction, we used the 3,3′,5,5′-tetramethylbenzidine peroxidase substrate (Pierce), and plates were analyzed spectrophotometrically at 450 nm. The concentrations of antibody were calculated with a standard curve generated using 6E10 monoclonal antibodies. To determine the specific isotypes, pooled sera from mice were diluted 1:500 and tested in duplicate using appropriate secondary antibodies specific to IgG1, IgG2ab, IgG2b, and IgM (35, 36).
T Cell Proliferation and Production of Cytokines by Immune Splenocytes
Using [3H]thymidine, we analyzed T cell proliferation in splenocyte cultures from individual animals as we described previously (35, 36). The same splenocytes used for T cell proliferation were utilized for detection of Th1 (IFNγ) or Th2 (IL-4) lymphokines by ELISPOT technique, also described in detail in Refs. 33 and 35.
Purification of Anti-Aβ1–11 Antibody
Sera from 3xTg-AD mice immunized with 2Aβ1–11-PADRE-MAP epitope vaccine were purified by an affinity column (SulfoLink; Pierce) per the manufacturer's instructions. Briefly, 2 mg of Aβ1–11-Cys peptide (synthesized at NeoMPS, San Diego, CA) was dissolved in 2–3 ml of buffer containing 50 mm Tris, 5 mm EDTA-Na, pH 8.5, and immobilized to the iodoacetyl-activated 6% cross-linked agarose through its free sulfhydryl groups. The remaining active binding sites were blocked with a 0.05 m solution of cysteine. Sera were diluted 1:1 in PBS, added to the column, and allowed to pass through the column. The column was washed with PBS, and then bound material was eluted with glycine buffer (100 mm, pH 2.5–3.0), neutralized, and concentrated by centrifugation using a Centricon filter (cut-off 30 kDa; Millipore Corp.). Purified antibodies were analyzed via 10% Tris-SDS-PAGE, and the concentrations were determined by enzyme-linked immunosorbent assay.
Detection of Aβ Plaques in Human Brain Tissues
Anti-Aβ1–11 antibodies (1 μg/ml) were screened for the ability to bind to human Aβ plaques using 50-μm brain sections of formalin-fixed hippocampal tissue from a case with neuropathological and behavioral patterns typical of severe AD (33, 37). A digital camera (Olympus) was utilized to capture images of the plaques at ×10 magnification. The binding of anti-Aβ1–11 antibodies to the β-amyloid plaques was blocked by preabsorption of the antibodies with 2.5 μm of 2Aβ1–11 peptide as described (33).
Dot Blot Assay, Immunoprecipitation, and Western Blotting
To detect binding of Aβ1–11 antibody to different forms of Aβ42 peptide, we applied 2 μl of monomeric, oligomeric, or fibrillar forms of Aβ42 (100 μm each) to a nitrocellulose membrane as described (38). After blocking and washing, the membranes were probed with anti-Aβ1–11 (1 μg/ml). The membranes were incubated with appropriate horseradish peroxidase-conjugated anti-mouse antibody (1:1000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Blots were developed using Luminol reagent (Santa Cruz Biotechnology) and exposed to Kodak X-Omat AR film. For identification of species stabilized after disaggregation of preformed fibrils, 2 μl of resulting solutions were applied to a nitrocellulose membrane and probed with biotinylated A11 antibody (1 μg/ml) and streptavidin-conjugated secondary antibody.
To detect APP and different Aβ forms in brain tissue recognized by anti-Aβ1–11 antibody, we used a combination of immunoprecipitation with Western blotting described previously (37). Briefly, the brain tissue was processed as described previously (39), except that cortices of right hemispheres of brains were used. Frozen cortices were thawed, minced, and then homogenized in 50 mm Tris-HCl buffer containing 150 mm NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS (pH 8.0), and a mixture of protease inhibitors (MP Biomedicals). Homogenates were centrifuged (100,000 × g, 1 h, 4 °C), and the supernatants were utilized for immunoprecipitation with 20.1 monoclonal antibodies of IgG2b isotype recognizing 1–8 aa of Aβ peptide (40). Immunoprecipitated proteins were analyzed in 10% Tris-SDS-polyacrylamide gel, transferred on the nitrocellulose membrane, and visualized after incubation with anti-Aβ1–11 antibody (1 μg/ml) followed by incubation with horseradish peroxidase-anti-mouse secondary antibodies and Luminol (Santa Cruz Biotechnology).
Surface Plasmon Resonance (SPR) Analysis
Binding studies were performed on the BIAcore 2000 SPR platform (Biacore). Monomeric, oligomeric, or fibrillar forms of Aβ42 peptides were immobilized to the surface of biosensor chip (CM5) via an amine coupling of primary amino groups (N termini or Lys17 or Lys28 residues) of the appropriate peptide to carboxyl groups in the dextran matrix of the chip. Serial dilutions of anti-Aβ1–11 antibody, 6E10 antibody, or irrelevant mouse IgG (125, 250, and 500 nm) in the running buffer containing 10 mm HEPES, 150 mm NaCl, 0.05% surfactant P20, pH 7.4, were injected at 5 μl/min over each immobilized form of peptide, and the kinetics of binding/dissociation was measured as change of the SPR signal (in resonance units). Each injection was followed by a regeneration step of a 30-s pulse of 1 m NaCl, 50 mm NaOH. Fitting of experimental data was done with BIAevaluation 3.0 software.
Intrahippocampal Injection of Antibodies and Quantitative Image Analysis
Anti-Aβ1–11 antibody was administered into the hippocampus of 18-month-old homozygous 3xTg-AD mice via injections, as described previously (41). Mouse IgG to irrelevant antigen (mouse anti-osteoprotegerin antibody) was used as a negative control. Anti-Aβ1–11 antibody and control IgG were injected at a concentration of 2 μg/2 μl into the left hippocampus over a 3–5-min period. Mice were perfused and sacrificed on the 6th day after surgery, and the brains were fixed in 4% paraformaldehyde for 48 h. Serial coronal sections (50 μm) were cut using the Vibratome slicing system (Pelco) and stored in PBS. Prior to staining, sections were incubated in 90% formic acid for 4 min at room temperature. Amyloid plaques were stained using 6E10 biotinylated monoclonal antibody (Signet Laboratories) and developed using an immunodetection kit (Vector Laboratories), as we previously described (33, 37). To detect specific areas in which intrahippocampally injected anti-Aβ or irrelevant antibodies were diffused, we stained the adjacent brain sections using anti-mouse biotinylated IgG as recommended by the manufacturer (Vector Laboratories). Images were captured by an Olympus microscope. Immunostainings were observed by the means of a Sony high resolution CCD video camera (XC-77) and NIH Image version 1.59b5 software. For every animal, 10 images (525 × 410 μm each) in the hippocampus area of two adjacent sections were captured with a ×20 objective. Samples consisted of five images from the superficial layer and five from the deep layer. NIH imaging was used to analyze the area occupied by β-amyloid (Aβ load) relative to the background and expressed in the percentage units at a threshold of 125 that was established and held constant throughout the image analysis.
Inhibition of Aβ42 Fibrillization
Aβ stock solutions (2 mm) were obtained by dissolving the lyophilized peptide in 100 mm NaOH followed by water bath sonication for 30 s. The aggregation of Aβ42 into fibrils (25 μm final concentration) was initiated in 10 mm HEPES, 100 mm NaCl, 0.02% sodium azide, pH 7.4, at room temperature with stirring for up to 180 h, in the presence and absence of 0.5 μm purified anti-Aβ1–11 antibody diluted in PBS buffer. The reactions were monitored as a function of time via thioflavin T (ThT) fluorescence and at initial and final time points by transmission electron microscopy (TEM), as described below. Fluorescence spectroscopy data were fit to integral logistic equations using the Sigmaplot software (Systat Software Inc., Point Richmond, CA), and the apparent rate constant for the fibril growth, , and the lag time were determined as described before (42–47). Although is not an elementary association rate constant, it constitutes a useful parameter for comparison of fibrillization kinetics (43, 44, 48).
Disaggregation of Preformed Aβ42 Fibrils
Aβ42 fibrils were prepared as described above in the absence of any additive. The aggregation reaction was allowed to proceed until equilibrium was reached (∼7 days). Then PBS vehicle or 0.5 μm purified anti-Aβ1–11 antibody (25 μm final Aβ42 concentration) was added to the preformed fibrils. The reactions were monitored as a function of time via ThT fluorescence and at initial and final time points by TEM, as described below. Fluorescence spectroscopy data in the presence of the anti-Aβ1–11 antibody were fit to a complex first order-exponential decay equation,
| (Eq. 1) |
where Ft represents the ThT fluorescence at time t, Fi and Ff are the initial and plateau levels of fluorescence, and is the pseudo-first order rate constant of disaggregation. This fit was obtained using the Table Curve 2D software trial version (Systat Software Inc., Point Richmond, CA), which allows automated modeling of data to the best fit. This model describes the dissociation of fibers into two different species and assumes that the initial concentration of these species is zero.
Inhibition of Aβ42 Oligomerization
Aβ42 stock solutions (2 mm) were obtained by dissolving the lyophilized peptide in 100 mm NaOH. The aggregation reaction was initiated by diluting the stock solution in PBS, pH 7.4, 0.02% sodium azide (45 μm final Aβ42 concentration). These assembly conditions facilitate visualization of Aβ42 oligomers. The reactions were maintained at room temperature without stirring for up to 10 days in the presence of 45 μm purified anti-Aβ1–11 antibody, 6E10 monoclonal antibody, or mouse IgG to irrelevant antigen, anti-osteoprotegerin antibody (BD PharMingen) diluted in PBS buffer. The reactions were monitored via transmission electron microscopy as described below.
ThT Fluorescence Assay
To investigate the time course of Aβ42 fibril formation, 10-μl aliquots were removed at various time points during aggregation and mixed with 120 μl of ThT (3 μm, dissolved in the assembly buffer). ThT fluorescence was measured at λex = 442 nm and λex = 482 nm until equilibrium was reached in the control reactions, using a Gemini XPS plate reader (Molecular Devices, Sunnyvale CA). To monitor the time course of Aβ42 fibril disaggregation, ThT (10 μm final concentration) was added to preformed fibrils in the presence or absence of the anti-Aβ1–11 antibody. Fluorescence was measured in the plate reader in real time. Control reactions were carried out in the presence of PBS vehicle and were corrected by subtracting the buffer blank. Aggregation and disaggregation reactions in the presence of the anti-Aβ1–11 antibody were corrected using antibody-only control reactions. This assay assumes that ThT fluorescence is a measure of filament mass (49, 50).
Electron Microscopy
1-μl aliquots of the aggregation and disaggregation reactions were adsorbed onto 200-mesh Formvar/carbon-coated nickel grids (Electron Microscopy Sciences; Ft. Washington, PA) until dry. The grids were then washed with water, stained with 2% uranyl acetate, and washed again. The grids were allowed to dry between all steps and were viewed using a Phillips CM 12 microscope operated at 65 kV.
Neurotoxicity Assay
Mouse primary neurons were prepared from cortices of embryonic day 17–18 mouse embryos. Briefly, cortices were dissected and incubated in trypsin (37 °C, 7 min). The cortices were pelleted at 1000 rpm for 5 min and were resuspended in NB-N2 medium (5 μg/ml insulin, 20 nm progesterone, 100 μm putresine, 30 nm selenium, 100 μg/ml transferring) (Sigma). After 10–15 min, the cells were passed through a glass pipette four times and then through a cell strainer and washed with NB-N2. Cells were plated on polylysine-coated 96-well plates. Aβ oligomers and fibrils were prepared as described (38). Aβ42 oligomers and fibrils were incubated alone or with antibodies (anti-Aβ1–11 and irrelevant anti-osteoprotegerin) for 50 min at room temperature with occasional mixing to ensure the maximal interaction. After incubation, the peptide/antibody mixtures were diluted into culture medium to a final concentration of peptide and antibodies of 2.5 and 0.5 μm, respectively. This medium was added (100 μl) to mouse primary neurons plated at ∼104/well. The treatment time was 4 h. Untreated controls were run in parallel. Following incubation, the neurotoxicity was assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay per the manufacturer's instructions.
Statistical Analysis
All statistical parameters (average values, S.D., and significant differences) used in these studies were calculated using Prism 3.03 software (GraphPad Software, Inc.). Statistically significant differences were obtained using a t test or an analysis of variance following Tukey's or Bonferroni's multiple comparison post-test, and a p value of <0.05 was considered as significantly different.
Results
Epitope Vaccine Induced High Titers of Polyclonal Anti-Aβ Antibodies Specific to the N-terminal Region of Aβ42 without Generation of Autoreactive T Cells
Immunization of 3xTg-AD mice of H-2b immune haplotype with the second generation peptide epitope vaccine induced strong humoral response with an average concentration of anti-Aβ antibody of 205 μg/ml (titer ∼1:180,000) (Fig. 1a) of IgG1, IgG2ab, and IgG2b isotypes (Fig. 1b). Antibody isotyping has previously been used as an indirect measure of the contribution of Th1 (IgG2a) and Th2 (IgG1) cytokines to the immune response (51). In our experiments, the ratio of IgG1/IgG2ab was close to 1, implying that predominantly Th1 type immune responses were induced after immunizations with epitope vaccine formulated in a Th1-type adjuvant, Quil A. This result was confirmed by analysis of cytokine production. Vaccinated animals induced predominantly IFNγ production along with moderate levels of IL-4. Fewer splenocytes from control mice immunized with fAβ42 generated production of IFNγ or IL-4 (Fig. 1c), whereas splenocytes from naive animals did not produce Th1 and Th2 cytokines (data not shown). Immunizations with epitope vaccine also induced a robust T cell proliferation after restimulation of the immune splenocytes with PADRE but not self-Aβ40 peptide (Fig. 1d). This finding offers an indication that PADRE-specific but not autoreactive T cells provide a necessary support for production of high titers of anti-Aβ1–11 antibody in 3xTg-AD mice.
Figure 1. Vaccination with 2Aβ1–11-PADRE-MAP vaccine induced high titers of polyclonal anti-Aβ1–11 antibodies specific to the N-terminal region of Aβ42 without generation of autoreactive T cells.

Immunizations with 2Aβ1–11-PADRE-MAP or fAβ42 induced 200 and 250 μg/ml anti-Aβ antibodies respectively (a). Predominant production of IgG1, IgG2ab, and IgG2b isotypes of anti-Aβ antibody were detected in individual sera of mice vaccinated with 2Aβ1–11-PADRE-MAP or fAβ42 (b). Immunization with either 2Aβ1–11-PADRE-MAP or fAβ42 induced a significantly higher number of cells producing IFNγ than IL-4, detected by the ELISPOT assay (c). d, T cell proliferation in mice immunized with 2Aβ1–11-PADRE-MAP epitope vaccine or fAβ42 formulated in QuilA. Splenocytes from individual mice were restimulated in vitro with PADRE or Aβ40 peptides, and the average value was calculated. 2Aβ1–11-PADRE-MAP epitope vaccine induced the activation of PADRE-specific but not Aβ-specific T cells, as shown by [3H]thymidine incorporation.
Anti-Aβ1–11 Antibody Generated by the Epitope Vaccine Binds to β-Amyloid Plaques and to Aβ42 Monomeric and Aggregated Species
Therapeutic feasibility of anti-Aβ1–11 antibody was tested after isolation and purification from the sera of vaccinated 3xTg-AD mice. Purified polyclonal anti-Aβ1–11 antibody stained Aβ plaques in brain tissue from an AD clinical case (Fig. 2a). This binding was specific, since it was blocked by preabsorption with 2Aβ1–11 peptide (Fig. 2b) or Aβ42 (data not shown). Importantly, anti-Aβ1–11 antibody binds to APP, Aβ monomers, and different forms of oligomers, as shown via analysis of soluble fraction of brain extracts from aged APP/Tg 2576 mice by a combination of immunoprecipitation and Western blot (see “Experimental Procedures”). In these experiments, binding of anti-Aβ1–11 antibody to 12-mer oligomers cannot be detected due to its molecular weight similarity to the heavy chain of immunoglobulins (Fig. 2c).
Figure 2. Binding properties of anti-Aβ1–11 antibody generated after vaccination of 3xTg-AD mice.
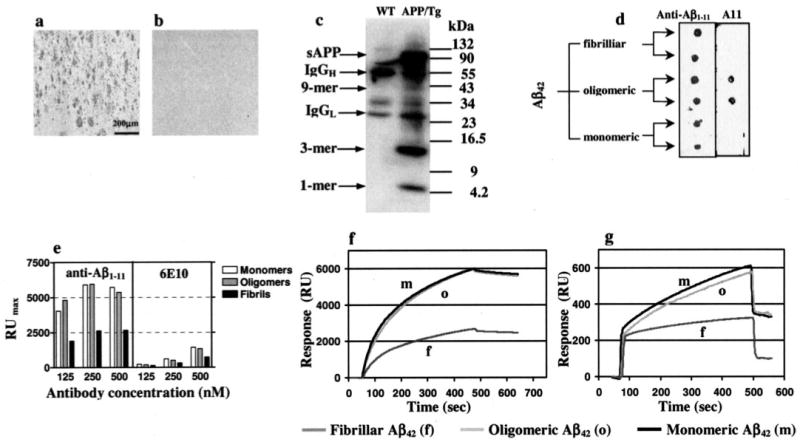
Polyclonal anti-Aβ1–11 antibody (1 μg/ml) binds amyloid plaques in the brain section of the hippocampal region from an AD case (a), and this binding was blocked after preabsorption of anti-Aβ1–11 antibody with the 2Aβ1–11 peptide (b). Original magnification was × 10; scale bar, 200 μm. c, analyses of soluble fraction of brain extract from 12-month-old APP/Tg 2576 mice using immunoprecipitation (20.1. monoclonal antibody) followed by Western blot (anti-Aβ1–11 antibody). Anti-Aβ1–11 antibody binds to all species of Aβ42 peptide, including oligomers recognized by All antibody. d, 2 μl of Aβ42 fibrils, oligomers, and monomers (100 μm each) were applied in duplicate to a nitrocellulose membrane and probed with anti-Aβ1–11. Binding of anti-Aβ1–11 (e and f) and control 6E10 antibodies (e and g) to immobilized Aβ42 monomeric, oligomeric, or fibrillar forms as measured by SPR assay.
Anti-Aβ1–11 antibody bound equally well to monomeric, oligomeric (38), and fibrillar forms of Aβ42, as demonstrated by dot blot assay (Fig. 2d). Notably, the oligomers that were used for this study included octamers, tetramers, and trimers besides higher order aggregates as detected by Western blot analysis with A11 rabbit antibodies (38 and data not shown). Next, for the first time to our knowledge, the binding capability of anti-Aβ1–11 antibody to different Aβ species has been determined using an SPR assay. This experiment revealed that anti-Aβ1–11 antibody (Fig. 2, e and f) as well as control 6E10 antibody (Fig. 2, e and g) specifically bind to immobilized monomers, oligomers, and fibrils.
Both anti-Aβ1–11 and 6E10 antibodies bound more strongly to monomeric and oligomeric forms, rather than to fibrillar Aβ42. Dissociation constants (KD) of anti-Aβ1–11-peptide complexes for monomeric, oligomeric, and fibrillar Aβ42 are 1.7 × 10−8, 1.4 × 10−8, and 5.1 × 10−8 m, respectively. The binding of anti-Aβ1–11 antibodies to all Aβ species was significantly stronger than that of 6E10, for which KD values correspond to 1.8 × 10−7, 1.2 × 10−7, and 4.3 × 10−7 m for monomeric, oligomeric, and fibrillar Aβ42, respectively (Fig. 2, e–g). Of note, irrelevant mouse IgG interacted with Aβ42 peptide nonspecifically on the base-line level (data not shown).
Intracranial Administration of Anti-Aβ1–11 Antibodies Reduces the Aβ Deposits in Brains of 3xTg-AD Mice
To further characterize in vivo therapeutic utility, anti-Aβ1–11 antibody was injected into the brains of aged 18-month-old 3xTg-AD mice (n = 4) with developed AD-like pathology. Antibodies (2 μg/2 μl) were administered via intrahippocampal injections, and mice were sacrificed on the 6th day after antibody administration. As controls, we used mice (n = 4) injected into the hippocampus with the same concentration of irrelevant mouse IgG. Quantitative image analysis shows individual variations of Aβ deposition in hippocampus of 18-month-old homozygous 3xTg-AD mice. In both experimental and control groups, there were mice with a low, medium, or high number of Aβ deposits (Table 1). A significant reduction (83%, p = 0.030) in β-amyloid load has been observed after comparison of ipsilateral (injected) hippocampus of mice treated with the anti-Aβ1–11 antibody with that of controls administrated with irrelevant antibody (Fig. 3, a–c). Comparing the Aβ ratios of immunoreactivity between the ipsilateral/contralateral (uninjected) areas also revealed a substantial (77%) and statistically significant decrease (p < 0.05) of amyloid load in mice injected with anti-Aβ1–11 antibody versus the control group (Fig. 3, d–f). This comparison has been made after quantitative image analysis of areas of the hippocampus in which antibodies/IgG diffused after the administration. These areas were determined by additional staining for mouse IgG in the adjacent brain sections of experimental and control animals (data not shown). These data suggest that anti-Aβ1–11 antibody is able to significantly reduce Aβ deposition in the brains of aged 3xTg-AD mice after intracranial administration.
Table 1. Effect of anti-Aβ1–11 antibody administration on β-amyloid deposition in homozygous 3xTg-AD mice at the age of 18 months.
| Group/Mouse number | Aβ load in hippocampus | |
|---|---|---|
| Left (injected) | Right (uninjected) | |
| Irrelevant mouse IgG | ||
| 1 | 0.88 | 0.27 |
| 2 | 2.46 | 3.09 |
| 3 | 4.09 | 3.46 |
| 4 | 1.89 | 2.77 |
|
| ||
| Average ± S.D. | 2.33 ± 1.34 | 2.40 ± 1.44 |
|
| ||
| Anti-Aβ1–11 antibodies | ||
| 1 | 0.55 | 1.53 |
| 2 | 0.70 | 1.17 |
| 3 | 0.04 | 0.07 |
| 4 | 0.29 | 0.72 |
|
| ||
| Average ± S.D. | 0.40 ± 0.29 | 0.87 ± 0.63 |
Figure 3. Anti-Aβ1–11 antibody reduces amyloid deposits in the brains of 18-month-old 3xTg-AD mice.

Quantitative image analysis of hippocampus showing significant reduction in Aβ load (p < 0.05) after administration of anti-Aβ1–11 antibody compared with that of mice injected with control IgG (a and d). Bars, mean ± S.E. of Aβ load in ipsilateral (injected) hippocampus (a) and the ratio of Aβ load in ipsilateral to contralateral (uninjected) hemispheres of hippocampus (d) from n = 4 in both groups of mice, injected with anti-Aβ1–11 antibodies or control IgG. Low and high magnification views showing a reduction in Aβ deposits in the ipsilateral hippocampus in mice receiving the anti-Aβ1–11 antibody (b and e) compared with mice administrated with control IgG (c and f). Original magnifications are as follows: ×2.5 (b and c) and ×10 (e and f). Squares show the approximate region where antibodies diffused after the injection.
Anti-Aβ1–11 Antibody Inhibits Aβ42 Assembly into Fibrils
An increasing body of evidence suggests that antibodies raised against Aβ peptide could be useful for clearance of Aβ deposits in vivo (18, 21, 23, 24, 26, 52) possibly by inhibiting or delaying Aβ aggregation (26, 27, 30). Similar data were also detected here using the anti-Aβ1–11 antibody. To understand the underlying mechanism and to further investigate the therapeutic utility of the anti-Aβ1–11 antibody, we first tested whether the anti-Aβ1–11 antibody can inhibit Aβ42 fibrillization. The aggregation of Aβ42 into fibrils initiated in the presence and absence of anti-Aβ1–11 antibody was monitored using TEM. At time 0, no aggregated species were detected either in the control (Fig. 4a) or in the antibody-treated samples (data not shown). After a 7-day incubation in the absence of antibody, Aβ42 formed fibrils with classic morphology (Fig. 4b). In contrast, in the presence of the anti-Aβ1–11 antibody, Aβ42 formed a heterogeneous population of nonfilamentous aggregates (Fig. 4c), which resembled oligomers of 3–10 nm. Occasionally, clusters of bigger oligomers up to 20 nm in length could also be detected. These clusters had a beaded appearance and did resemble mature fibrils. To confirm this observation, the time course of Aβ42 fibrillization was monitored using ThT fluorescence, in the absence or presence of the anti-Aβ1–11 antibody. The control reaction followed nucleation-dependent polymerization kinetics, with a lag time of 54.28 ± 1.67 h and an apparent rate constant of assembly, (Fig. 4d). Anti-Aβ1–11 antibody, at substoichiometric concentration, inhibited the formation of Aβ42 ThT-positive species. Although this reaction was followed for 200 h, the ThT signal maintained at levels less than 10% of the plateau signal of the control reaction. In the presence of anti-Aβ1–11 antibody, the lag time of Aβ42 aggregation increased to ∼80 h, and the decreased to 0.04 ± 0.02 h−1 (Fig. 4d). The low ThT signal detected in antibody-treated reactions could be attributed to small amounts of short fibrillar material or nonfibrillar species that have been shown before to react with ThT (30, 48). These results, generated in two independent assays, suggest that anti-Aβ1–11 antibody is a potent inhibitor of Aβ42 fibrillization at substoichiometric concentrations relative to Aβ42 protomer (1:50, antibody/peptide molar ratio). The inhibitory activity could be attributed to both inhibition of filament nucleation and extension.
Figure 4. Anti-Aβ1–11 antibody inhibits Aβ42 fibrillization.
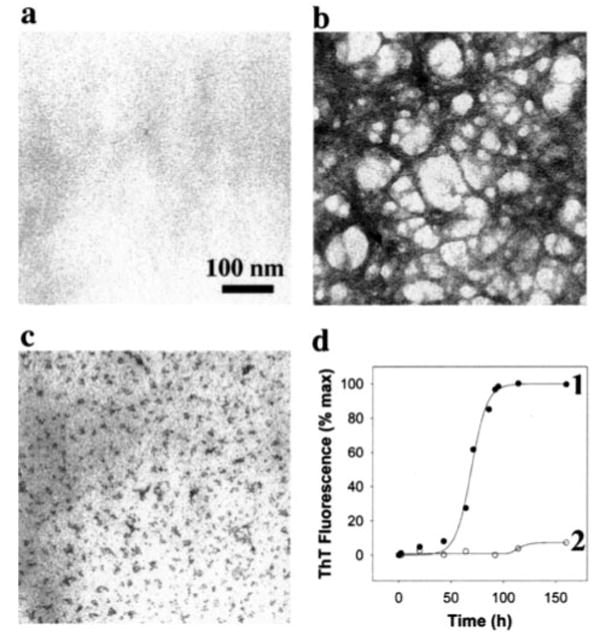
Aβ42 (25 μm) was subject to aggregation in the absence or presence of anti-Aβ11–1 antibodies (0.5 μm). Reactions were imaged by TEM (bar, 100 nm) at time 0 (a) and 160 h after the initiation of aggregation in the absence (b) or presence of anti-Aβ1–11 antibody (c). The reactions were also monitored by ThT fluorescence (d) as a function of time in the absence (●) or presence (○) of the antibody. Each data point represents the ThT fluorescence in arbitrary units, expressed as a percentage of the plateau signal.
Anti-Aβ1–11 Antibody Mediates Aβ42 Fibril Disaggregation
To determine whether plaque clearance observed in vivo could be recapitulated in in vitro models of disease, we extended our studies to investigate whether anti-Aβ1–11 antibody can disaggregate preformed Aβ42 fibrils. Aβ42 was allowed to assemble until equilibrium was reached based on ThT fluorescence (7–8 days of incubation), and preformed fibrils were treated either with the PBS vehicle or with the anti-Aβ1–11 antibody. Aliquots were removed at time 0 and 24 h after the initiation of disaggregation and imaged by TEM. At time 0, both the control (Fig. 5a) and the antibody-treated reactions (data not shown) revealed a classical fibrillar morphology. Control reactions maintained fibrillar morphology 24 h after the initiation of disaggregation (Fig. 5b). In contrast, no fibrils were detected in the antibody-treated sample at the end of the same time period (Fig. 5c). Round structures resembling oligomers ranging in size from 3 to 10 nm and oligomer clusters populated this reaction. To confirm these results and investigate the mechanism of disaggregation, the kinetics of the disaggregation reactions were monitored in real time by ThT fluorescence. In the absence of the antibody, the ThT signal continued to increase slowly over this period of time, consistent with Aβ42 fibrils being in the equilibrium phase of assembly (Fig. 5d). In contrast, in the presence of anti-Aβ1–11 antibody, ThT fluorescence decreased over the same time period down to 35 ± 0.43% of the starting level of fluorescence (Fig. 5d), consistent with Aβ42 filament disaggregation. The disaggregation reaction followed complex first-order kinetics with a first order rate constant of disaggregation, of 0.10 ± 1.4 × 10−3 h−1 (Fig. 5d). This pattern is consistent with filament dissociation into two different species, and the low ThT fluorescence and the lack of mature fibrils by TEM at equilibrium suggest that at least one of these species is ThT-reactive without showing classical fibrillar morphology. These data confirm that anti-Aβ1–11 antibody can destabilize Aβ42 fibrils at low concentrations relative to the peptide (1:50, antibody/peptide molar ratio).
Figure 5. Anti-Aβ1–11 antibody disaggregates preformed Aβ42 fibrils into nonfilamentous species.

Aβ42 (25 μm) fibers (a) were incubated in the absence (b) or presence of anti-Aβ1–11 antibody (0.5 μm) (c) for up to 24 h and imaged by TEM (bar, 100 nm). The reactions were also monitored in real time by ThT fluorescence (d) in the absence (●) or presence (○) of the antibody. Each data point represents ThT fluorescence in arbitrary units, expressed as a percentage of the signal at time 0. 2-μl aliquots of disaggregation reaction (lane 1) and control oligomers (lane 2) were applied in duplicate to a nitrocellulose membrane and were probed with biotinylated A11 oligomer-specific antibody by dot-blot assay (e).
To identify the nature of molecules stabilized by the anti-Aβ1–11 antibody, the resulting species were probed with the anti-oligomer-specific antibody A11 (38). No immunoreactivity with the A11 antibody was observed (Fig. 5e, lane 1), whereas freshly prepared oligomers used as a positive control showed strong immunoreactivity with the same antibody (Fig. 5e, lane 2). These data suggest that the anti-Aβ1–11 antibody specific to linear peptide disaggregates Aβ42 preformed fibrils to small aggregates, which are not A11-immunopositive, although their size (3–10 nm) and shape resembles the oligomers when imaged by TEM.
Anti-Aβ1–11 Antibody Delays Aβ42 Oligomerization but Does Not Disaggregate Preformed Aβ42 Oligomers
It was suggested that the most toxic forms of Aβ42 peptide in brains are soluble oligomers of various size (13–17). Thus, it was important to test the efficacy of polyclonal anti-Aβ1–11 antibody in inhibition of amyloid oligomerization or disaggregation of preformed oligomers. Aβ42 was subjected to aggregation under conditions that facilitate visualization of Aβ42 oligomers at physiological pH. Aliquots of the Aβ42 assembly reactions treated with the anti-Aβ1–11 antibody, 6E10 antibody, or irrelevant mouse IgG were removed at various time points and imaged by TEM. At time 0, few aggregated species were detected. (Fig. 6, a, d, and g). In the presence of irrelevant mouse IgG, Aβ42 aggregated into a heterogenous population of oligomers with sizes ranging from 3 to 10 nm (Fig. 6b). In the presence of anti-Aβ1–11 antibody, fewer oligomers formed, and oligomerization appeared to be delayed (Fig. 6h). In contrast, 6E10-treated Aβ42 assembled into abundant oligomers within 4 days of the initiation of the aggregation reaction (Fig. 6e). 10 days after the initiation of aggregation, the irrelevant mouse IgG (Fig. 6c) and 6E10-treated (Fig. 6f) reactions showed amyloid fibers with classical appearance. At the same time point during aggregation, and in the presence of the anti-Aβ1–11 antibody, Aβ42 showed a nonfibrillar structure, dominated by aggregates resembling oligomers and oligomeric assemblies of beaded appearance. The size of the most abundant oligomer population was ∼10 nm, and clustering of as many as seven oligomers yielded extended nonfibrillar structures of beaded appearance (Fig. 6i). Neither of the antibodies disaggregated preformed Aβ42 oligomers (data not shown).
Figure 6. Anti-Aβ1–11 antibody delays Aβ42 oligomerization.

Aβ42 (45 μm) was incubated in the presence of equimolar concentrations of irrelevant mouse IgG (a–c), 6E10 antibody (d–f) or anti-Aβ1–11 antibody (g–i). Aliquots of these reactions were removed at 0 (a, d, and g), 4 (b, e, and h), and 10 days (c, f, and i) after the initiation of aggregation and imaged by TEM (bar, 100 nm).
Together, the results described above confirm that anti-Aβ1–11 antibody is a potent Aβ42 fibrillization inhibitor and suggest that the mechanism underlying this inhibitory activity is stabilization of nonfibrillar structures resembling oligomers and oligomer assemblies. However, the formation of Aβ42 oligomers in the presence of anti-Aβ1–11 antibody is greatly delayed compared with control reactions or Aβ42 supplemented with 6E10 antibody. In addition, consistent with its activity in vivo, anti-Aβ1–11 antibody is equally potent in destabilizing preformed, mature amyloid fibers down to the structures that do not react with anti-oligomer-specific antibodies.
Anti-Aβ1–11 Antibody Protects Neuronal Cells from Aβ42 Oligomer and Fiber-mediated Toxicity
The data presented above suggested that anti-Aβ1–11 antibody is therapeutic and might exhibit a protective effect on Aβ-induced neurotoxicity. To test that possibility, we performed in vitro toxicity studies using mouse primary neurons. The viability of cells exposed to Aβ42 oligomeric and fibrillar species preincubated with or without anti-Aβ1–11 antibody was measured using the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide toxicity assay. The data showed that both Aβ42 fibrils and oligomers are cytotoxic, reducing cell viability to about 62 and 24%, respectively (Fig. 7). Preincubation of Aβ42 fibrils with anti-Aβ1–11 antibody (1:5, antibody/peptide molar ratio) resulted in the rescue of cell viability to maximum level. Similarly, preincubation of Aβ42 oligomers with anti-Aβ1–11 antibody increased cell viability to ∼46%. In contrast, preincubation of both Aβ42 species with a control mouse IgG specific to irrelevant antigen did not rescue cells from oligomer or fiber-mediated cell death. These data suggest that anti-Aβ1–11 antibody inhibits Aβ42 fiber-mediated neurotoxicity and alleviates oligomer toxicity in vitro.
Figure 7. Anti-Aβ1–11 antibody inhibits Aβ42 fibrils and oligomer-mediated toxicity.
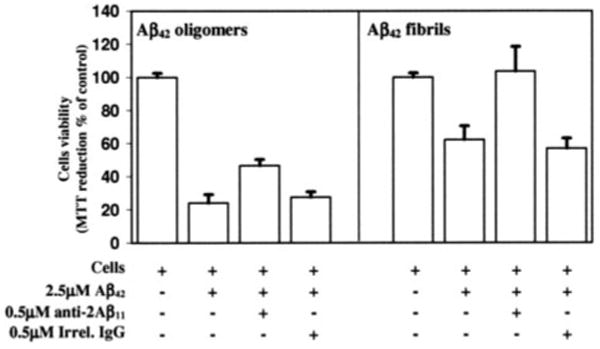
Mouse primary neurons were incubated with Aβ42 oligomers and Aβ42 fibrils, in the presence or absence of anti-Aβ1–11 antibody or mouse IgG to irrelevant antigen (1:5, antibody/peptide molar ratio). Control cells were treated with the vehicle, and cell viability was assayed in all cultures using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data were collected in triplicate and was expressed as a percentage of control ± S.D.
Discussion
Aβ immunotherapy has generated promising results in animal models of AD. Although translation to the clinic was hampered by unexpected cerebral inflammation in a subset of patients, there is still a need for further investigation of mechanisms involved in beneficial and detrimental effects of Aβ immunotherapy (53–59). It was suggested that not the anti-Aβ antibodies but rather autoreactive T cells caused the significant side effects in the vaccinated patients. Here, using a second generation prototype epitope AD vaccine, we induced a very strong anti-Aβ1–11 antibody production without generation of autoreactive T cells in 3xTg-AD mice (Fig. 1). In addition, comprehensive in vivo and in vitro analyses investigated the therapeutic utility of these molecules. For the first time, we demonstrated that autologous polyclonal anti-Aβ1–11 antibody induced after immunizations with the second generation of epitope vaccine specifically binds to Aβ plaques present in brain sections of cortical AD tissue (Fig. 2, a and b) and significantly reduces the amyloid deposits in ipsilateral hippocampus of 3xTg-AD mice (Fig. 3 and Table 1). However, it is known that in addition to populating fibrillar deposits in the brain, Aβ is also present in other conformations, such as monomers and oligomers/protofilaments. In fact, it was argued that Aβ oligomers of various size may be the most pathologic substrate responsible for disrupting neuronal functions and inducing cognitive decline in AD (13–17). Thus, we present here a comprehensive analysis aimed at investigating whether the anti-Aβ1–11 antibody generated in 3xTg-AD mice could have a therapeutic effect both on the amyloid fibers present in plaques and on other amyloid species. To clarify this, we first tested its ability to bind to different forms of human Aβ42 and then its capability to interfere with Aβ42 aggregation or to disaggregate preformed aggregates.
Dot blot analysis showed that the anti-Aβ1–11 antibody bound very well not only to monomeric but also to oligomeric and fibrillar forms of Aβ42 peptide (Fig. 2d). We also demonstrated a binding of anti-Aβ1–11 antibody to the different oligomers deposited in the brain of 12-month-old APP/Tg 2576 mice using a combination of immunoprecipitation and Western blotting. In these experiments, we could not show binding of anti-Aβ1–11 antibody to 56-kDa oligomers, probably due to its molecular weight similarity to the IgH (Fig. 2c). Anti-Aβ1–11 antibody was also bound to denatured APP that did not coincide with some published data where fAβ42 was used as an immunogen (60, 61). Antigen differences may be responsible for different specificity to the APP molecule. The binding capability of anti-Aβ1–11 antibody to different β-amyloid species was then confirmed by an SPR assay using monomeric, oligomeric, and fibrillar forms of Aβ42 as immobilized targets. Both the anti-Aβ1–11 and the 6E10 antibodies bound to all Aβ42 species, but binding to monomeric and oligomeric forms was significantly stronger than to the fibrillar form of this peptide (Fig. 2, e–g). Importantly, anti-Aβ1–11 antibody bound to all Aβ species almost 4 times more strongly than the therapeutically potent monoclonal antibody 6E10 (15, 16).
The data presented above suggest that anti-Aβ1–11 antibody could be an effective modulator of Aβ42 aggregate formation regardless of the nature of the aggregated species. Indeed, substoichiometric concentrations of anti-Aβ1–11 antibody inhibited fibril formation by 90% (Fig. 4d). TEM analysis confirmed that in the absence of anti-Aβ1–11 antibody, Aβ42 formed fibrils with classical morphology (Fig. 4b) and that the addition of the antibody completely inhibited Aβ42 fibril formation (Fig. 4c). Substoichiometric inhibition of Aβ42 fibrillization by the anti-Aβ1–11 antibody suggests that this antibody may trap multiple Aβ molecules in the early stages of aggregation and hinder filament growth. The fact that the anti-Aβ1–11 antibody binds nonfilamentous forms of amyloid peptide much more strongly than fibrils (Fig. 2, d and e) supports this hypothesis. Trapping of early assembly intermediates into assembly-incompetent conformations could explain the decrease in filament nucleation, as reflected by increased lag times and the decrease of the rate of addition of Aβ to fibers (45, 62). A similar mechanism has been proposed for M266 antibody-mediated inhibition of Aβ fibrillization (24, 30, 63), for inhibition of tau aggregation (45, 62), and for inhibition of α-synuclein fibrillization (64, 65). Alternatively, the anti-Aβ1–11 antibody could bind to exposed epitopes along the length of the fibrils, similar to Congo Red (66), or at the filament ends and cause filament destabilization and inhibition of filament growth (62). Although the anti-Aβ1–11 antibody does not completely inhibit Aβ42 aggregation, the oligomer-resembling species formed in the presence of the antibody may represent Aβ42 species or peptide-antibody complexes trapped off the fibrillization pathway. This could be therapeutically beneficial, because it would decrease the amount of assembly-competent peptide available to aggregate into toxic species that populate the fibrillization pathway. Next, we demonstrate that low concentrations of the anti-Aβ1–11 antibody can also disrupt potentially toxic Aβ42 preformed fibrils down to a heterogeneous population of nonfilamentous aggregates resembling oligomers or peptide-antibody complexes (Fig. 5). Both inhibitory mechanisms described above could account for the anti-Aβ1–11 antibody-mediated destabilization of preformed fibers. Strong antibody binding to nonfibrillar Aβ42 forms could disrupt the equilibrium between filamentous and nonfilamentous Aβ42 and therefore mediate fiber disaggregation (45, 62). Similarly, antibody binding along the length of fibrils or at the filament ends could cause filament destabilization (62, 66). Regardless of the mechanism, it is important to note that the product(s) of anti-Aβ1–11 antibody-mediated Aβ42 fiber disaggregation are A11-nonreactive (Fig. 5e). It is possible although unlikely that anti-Aβ1–11 antibody specific to linear peptide could block the binding of anti-oligomeric A11. This suggests that these oligomer-like structures may differ from the classic oligomers, which react with oligomer-specific antibodies and are toxic in cell culture (38, 67). The nontoxic nature of the end product(s) of antibody-mediated disaggregation is supported by evidence that the anti-Aβ1–11 antibody rescues cell viability to maximum level by inhibiting fibril-mediated cytotoxicity (Fig. 7).
In contrast to the results generated with Aβ42 fibrils, we demonstrate here that polyclonal anti-Aβ1–11 antibody does not disaggregate preformed oligomers and only delays Aβ42 oligomer formation when present at equimolar concentrations (Fig. 6). 6E10 antibody seemed to promote the oligomerization after 4 days of incubation with Aβ42 peptide. However, it is difficult to assess a difference in an increase of oligomers after incubation with 6E10 (IgG1) compared with that observed with irrelevant antibody (IgG1), since we cannot clearly distinguish whether these oligomers were composed of Aβ42 alone or whether they constituted Aβ42-antibody complexes. Complexes of Aβ with 6E10 and anti-Aβ1–11 but not irrelevant antibody are achievable, and the differences in 6E10 and anti-Aβ1–11 antibody affinity, specificity, and isotype composition might play a role in timing of the formation of these complexes as well as in their appearance. Consistent with this, a similar TEM image was observed after 10 days of incubation of anti-Aβ1–11 polyclonal antibody of mixed isotypes (IgG1, IgG2ab, IgG2b, and IgM) with Aβ42 peptide (Fig. 6). The longer period needed for the formation of more oligomers in the presence of anti-Aβ1–11 might be explained by the higher affinity of binding of these polyclonal antibodies to monomeric peptide than that of 6E10 (Fig. 2). Interestingly, although the anti-Aβ1–11 antibody delays Aβ42 oligomer formation, it ultimately stabilizes nonfibrillar conformations, including oligomers and oligomer assemblies of beaded appearance. The structures ultimately stabilized by the anti-Aβ1–11 antibody may differ from the classic oligomers, since the oligomer-like structures formed in the presence of the antibody are nontoxic, and preincubation of toxic oligomers with the antibody diminishes their toxicity (Fig. 7). The reduction of oligomer toxicity observed upon preincubation of Aβ oligomers with the anti-Aβ1–11 antibody in the absence of oligomer disaggregation or fiber formation suggests the possibility of oligomer trapping and rearrangement in the presence of the antibody and supports this hypothesis. Thus, oligomer trapping and reorganization into nontoxic and assembly-incompetent conformations could lead to decreasing overall oligomer-mediated cytotoxicity and inhibition of further aggregation. This seems to be possible also in the case of the 6E10 antibody, where oligomer formation is accelerated but the resulting oligomers may be less toxic than those formed by Aβ alone, and explain the protective activity of 6E10 antibody against oligomer-mediated toxicity reported in literature (15, 16).
In conclusion, here we demonstrate that the anti-Aβ1–11 antibody binds strongly to all forms of β-amyloid, prevents Aβ42 fibrillization, and induces disaggregation of preformed Aβ42 fibrils down to nonfilamentous and nontoxic species. However, this antibody does not disaggregate preformed oligomers and only delays Aβ42 oligomer formation, somewhat alleviating oligomer-mediated toxicity. These data suggest that the most promising potential for AD vaccine may be present in blocking/inhibiting formation of toxic Aβ oligomers, inhibiting memory impairment, and delaying the onset of the disease rather than in reducing the level of these molecules in the brain and improving preexisting learning and memory deficits in AD patients. Thus, our in vitro and ex vivo data support recent analyses of the AN-1792-vaccinated patients (68). It was shown that although parenchymal amyloid was focally disaggregated, vascular deposits were preserved or even increased in brains of vaccinated patients. In addition, AN-1792 vaccine increased soluble amyloid levels in the gray and white matter of immunized patients compared with AD cases. We suggest that Aβ immunotherapy would probably target oligomeric Aβ and that AD vaccines may be more effective as a prophylactic rather than therapeutic. We think this will hold true even if Aβ immunotherapy only delays the accumulation of toxic soluble forms of Aβ, postponing AD pathology by preventing oligomers to reach critical levels in the central nervous system and induce a full scale AD pathology. Further preclinical and clinical studies will demonstrate if AD therapy should be based on the prevention of accumulation of aggregates (preventive vaccination or preventive medicine) or on clearing pre-existing toxic forms of Aβ from the brains of elderly AD patients with immunosenescence (therapeutic vaccination or/and therapeutic medicine).
Acknowledgments
We thank Dr. Rakez Kayed for the A11 anti-oligomer-specific antibody and Dr. Saskia Milton for the preparation of Aβ peptides.
Footnotes
This work was supported by National Institutes of Health R01 Grants AG20241 and NS50895 (to D. H. C. and M. G. A.) and NS3I230 (to C. G. C.) and Alzheimer's Association Grant IIRG-03-6279 (to M. G. A. and D. H. C.).
The abbreviations used are: AD, Alzheimer disease; Aβ, β-amyloid peptide; All, anti-oligomer antibody; APP, amyloid precursor protein; fAβ42, fibrillar Aβ42; IFNγ, interferon γ; IL, interleukin; MAP, multiple antigenic peptide; PADRE, Pan-Dr epitope; PBS, phosphate-buffered saline; TEM, transmission electron microscopy; ThT, thioflavin T; aa, amino acid(s); SPR, surface plasmon resonance.
References
- 1.Selkoe DJ. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 2.Price DL, Sisodia SS. Annu Rev Med. 1994;45:435–446. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- 3.Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Neurobiol Aging. 2004;25:589–597. doi: 10.1016/j.neurobiolaging.2004.02.003. discussion 603-584. [DOI] [PubMed] [Google Scholar]
- 4.Zlokovic BV. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. J Neuropath Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Esler WP, Wolfe MS. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- 8.Hardy JA, Higgins GA. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 9.Sisodia SS, Price DL. FASEB J. 1995;9:366–370. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 10.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzo A, Yankner BA. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Proc Natl Acad Sci U S A. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 15.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 16.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- 17.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 18.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 19.Schenk D. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 20.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 21.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RGM. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 23.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 24.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 26.McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, George-Hyslop P. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- 27.Solomon B, Koppel R, Hanan E, Katzav T. Proc Natl Acad Sci U S A. 1996;93:452–455. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Proc Natl Acad Sci U S A. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legleiter J, Czilli DL, Gitter B, DeMattos RB, Holtzman DM, Kowalewski T. J Mol Biol. 2004;335:997–1006. doi: 10.1016/j.jmb.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 31.Frenkel D, Katz O, Solomon B. Proc Natl Acad Sci U S A. 2000;97:11455–11459. doi: 10.1073/pnas.97.21.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 33.Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- 34.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Cribbs DH, Ghochikyan A, Tran M, Vasilevko V, Petrushina I, Sadzikava N, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petrushina I, Tran M, Sadzikava N, Ghochikyan A, Vasilevko V, Agadjanyan MG, Cribbs DH. Neurosci Lett. 2003;338:5–8. doi: 10.1016/s0304-3940(02)01357-5. [DOI] [PubMed] [Google Scholar]
- 37.Ghochikyan A, Vasilevko V, Petrushina I, Tran M, Sadzikava N, Babikyan D, Movsesyan N, Tian W, Ross TM, Cribbs DH, Agadjanyan MG. Eur J Immunol. 2003;33:3232–3241. doi: 10.1002/eji.200324000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 39.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 41.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen L, Khurana R, Coats A, Frokjaer S, Brange J, Vyas S, Uversky VN, Fink AL. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 43.Uversky VN, Li J, Fink AL. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 44.Necula M, Chirita CN, Kuret J. J Biol Chem. 2003;278:46674–46680. doi: 10.1074/jbc.M308231200. [DOI] [PubMed] [Google Scholar]
- 45.Necula M, Chirita CN, Kuret J. Biochemistry. 2005;44:10227–10237. doi: 10.1021/bi050387o. [DOI] [PubMed] [Google Scholar]
- 46.Harper JD, Lansbury PT., Jr Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 47.Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT., Jr Proc Natl Acad Sci U S A. 1995;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chirita CN, Kuret J. Biochemistry. 2004;43:1704–1714. doi: 10.1021/bi036034b. [DOI] [PubMed] [Google Scholar]
- 49.Naiki H, Hasegawa K, Yamaguchi I, Nakamura H, Gejyo F, Nakakuki K. Biochemistry. 1998;37:17882–17889. doi: 10.1021/bi980550y. [DOI] [PubMed] [Google Scholar]
- 50.LeVine H., III Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finkelman FD, Holmes J, Katona IM, Urban JF, Beckmann MP, Park LS, Schooley KA, Coffman RL, Mossmann TR, Paul WE. Annu Rev Immunol. 1990;8:303–333. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- 52.Schenk D, Hagen M, Seubert P. Curr Opin Immunol. 2004;16:599–606. doi: 10.1016/j.coi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 53.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 54.Ferrer I, Rovira MB, Guerra MLS, Rey MJ, Costa-Jussa F. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 56.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 57.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 58.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 59.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 60.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, Von Rotz RC, Davey G, Moritz E, Nitsch RM. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 61.Lemere CA, Beierschmitt A, Iglesias M, Spooner ET, Bloom JK, Leverone JF, Zheng JB, Seabrook TJ, Louard D, Li D, Selkoe DJ, Palmour RM, Ervin FR. Am J Pathol. 2004;165:283–297. doi: 10.1016/s0002-9440(10)63296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chirita C, Necula M, Kuret J. Biochemistry. 2004;43:2879–2887. doi: 10.1021/bi036094h. [DOI] [PubMed] [Google Scholar]
- 63.Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 65.Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. J Biol Chem. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 66.Klunk WE, Pettegrew JW, Abraham DJ. J Histochem Cytochem. 1989;37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 67.Deshpande A, Mina E, Glabe C, Busciglio J. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]