

SUMMARY
Calcium/calmodulin (Ca2+/CaM)-dependent protein kinase II (CaMKII) couples increases in cellular Ca2+ to fundamental responses in excitable cells. CaMKII was identified over twenty years ago by activation dependence on Ca2+/CaM, but recent evidence shows CaMKII activity is also enhanced by pro-oxidant conditions. Here we show that oxidation of paired regulatory domain methionine residues sustains CaMKII activity in the absence of Ca2+/CaM. CaMKII is activated by angiotensin II (AngII) induced oxidation, leading to apoptosis in cardiomyocytes, both in vitro and in vivo. CaMKII oxidation is reversed by methionine sulfoxide reductase A (MsrA), and MsrA−/− mice show exaggerated CaMKII oxidation and myocardial apoptosis, impaired cardiac function, and increased mortality after myocardial infarction. Our data demonstrate a novel, dynamic mechanism for CaMKII activation by oxidation and highlight the critical importance of oxidation-dependent CaMKII activation to AngII and ischemic myocardial apoptosis.
INTRODUCTION
The multifunctional calcium/calmodulin (Ca2+/CaM)-dependent protein kinase II (CaMKII) couples increases in Ca2+ to activation of ion channels (Grueter et al., 2006), gene transcription (Backs et al., 2006), and apoptosis (Zhu et al., 2003; Yang et al., 2006). CaMKII is activated by enhanced intracellular Ca2+ from beta-adrenergic receptor (βAR) stimulation (Zhang et al., 2005). Excessive βAR stimulation causes apoptosis by a Ca2+, CaMKII, and caspase-3 dependent pathway (Zhu et al., 2003). The CaMKII holoenzyme is assembled from subunits containing three key domains: the association domain, which directs multimeric assembly, the regulatory domain, which controls enzyme activation and autoinhibition, and the catalytic domain, which performs the kinase function of CaMKII. Under resting conditions CaMKII is inactive, but upon binding Ca2+/CaM a conformational change relieves the autoinhibitory effect of the regulatory domain on the kinase domain, activating the enzyme (Hudmon and Schulman, 2002; Rosenberg et al., 2005). In the sustained presence of Ca2+/CaM, CaMKII undergoes intersubunit autophosphorylation at T287 (or 286; specific numbering is isoform dependent), resulting in Ca2+/CaM independent activity (Hudmon and Schulman, 2002). T287 lies within the autoinhibitory region of CaMKII, and autophosphorylation at T287 produces Ca2+ autonomous activity by preventing reassociation of the kinase domain by the autoinhibitory region (Hudmon and Schulman, 2002). Interconversion between Ca2+-dependent and Ca2+-independent forms is a critical property of CaMKII that allows transformation of a transient Ca2+ stimulus into sustained physiological or disease-causing activity.
CaMKII activity may also increase in pro-oxidant cellular environments (Howe et al., 2004; Zhu et al., 2007), suggesting CaMKII has broader functionality than originally envisioned by connecting ‘upstream’ oxidant stress and Ca2+ signals to ‘downstream’ cellular responses. Based upon the previously recognized structure-activity response of CaMKII to T287 phosphorylation, we hypothesized that oxidation directly modifies the autoinhibitory motif to confer Ca2+/CaM independent CaMKII activity by a mechanism analogous to autophosphorylation. We identified a novel direct molecular mechanism for reactive oxygen species (ROS)-dependent, Ca2+ independent CaMKII activation by modification of M281/282. Our findings show that direct activation of CaMKII by ROS engenders Ca2+ autonomous activity, a clear but previously unrecognized molecular mechanism by which CaMKII can integrate Ca2+ and ROS signals.
Elevated levels of ROS have been measured and contribute to adverse outcomes after myocardial infarction (Kinugawa et al., 2000) and in models of heart failure (Maack et al., 2003). Angiotensin II (AngII) also increases ROS in heart (Doerries et al., 2007), while AngII antagonist drugs are a mainstay for reducing mortality in patients with structural heart disease (Pfeffer et al., 1992; Pfeffer et al., 2003). We hypothesized that CaMKII is a downstream signal for ischemic and AngII stimulated apoptosis in heart and that CaMKII responses were dependent upon our newly identified M281/282 activation mechanism. Methionine sulfoxide reductase A (MsrA) specifically reverses Met oxidation, so we predicted that MsrA−/− mice would show increased CaMKII oxidation after AngII and ischemic stress. Here we show that CaMKII inhibition protects against AngII initiated apoptosis in heart and that pathological AngII responses recruit CaMKII activity by M281/282 oxidation in vitro and in vivo. MsrA−/− mice show increased CaMKII oxidation and apoptosis with AngII and ischemia and increased mortality, greater left ventricular dilation and worse in vivo mechanical function after myocardial infarction, compared to controls. Our data establish CaMKII as a downstream signal for AngII and ischemic stress and establish ROS modification of CaMKII at M281/282 as a dynamic mechanism for regulating myocardial responses to common forms of heart disease.
RESULTS
Oxidation directly activates CaMKII
CaMKII is activated by Ca2+/CaM, but autophosphorylation at T287 sustains catalytic activity after dissociation of Ca2+/CaM (Fig. 1A) because the negatively charged phosphate prevents reassociation of the catalytic domain and autoinhibitory region (Hudmon and Schulman, 2002). CaMKII activity may also be enhanced by pro-oxidant conditions (Zhu et al., 2007); we therefore hypothesized that oxidation of the regulatory domain in the vicinity of T287 could sustain CaMKII catalytic activity by an analogous mechanism. Exposure of purified CaMKII to H2O2 in the absence of any pre-treatment yielded no discernable CaMKII activity (Fig. 1B). However, exposure to H2O2 after pretreatment with Ca2+/CaM yielded persistent CaMKII activation even in the presence of EGTA. These data suggest that Ca2+/CaM binding exposed a key segment of CaMKII for oxidation, and that oxidation interfered with the interaction of the autoinhibitory and catalytic domains. Activation of wild type (WT) CaMKII by H2O2 was dose-dependent (Fig. 1C). The concentration of EGTA used was sufficient to block CaMKII activity without the addition of H2O2 (Fig. 1B), suggesting that activity observed in the pro-oxidant condition was independent of sustained Ca2+/CaM binding.
Figure 1. CaMKII is activated by ROS.

(A) General structure of a subunit from the multimeric holoenzyme CaMKII and mechanism of CaMKII activation by autophosphorylation. The amino acid sequence of the regulatory domain is highlighted to show the autoinhibitory (AI) and calmodulin-binding (CaM-B) regions. Yellow symbols represent CaM. Pretreatment with Ca2+/CaM (1°) followed by phosphorylation at T287 (2°) yields persistent activity even after the removal of Ca2+/CaM (3°). (B) Kinase assays were performed after three distinct treatment steps: (1°) ± Ca2+/CaM, (2°) ± H2O2 or ATP, and (3°) ± EGTA. (n = 6 assays/group, * p < 0.05 vs. WT no treatment). (C) CaMKII is activated by H2O2 in a dose-dependent manner after pre-treatment with Ca2+/CaM. Oxidation-dependent CaMKII activity is ablated in M281/282V mutants (n = 6 assays/group, * p < 0.05 vs. WT no treatment). (D) M281/282V mutants have normal Ca2+/CaM-dependent and T287-autophosphorylation-dependent activation (n = 6 assays/group, * p < 0.05 vs. WT no treatment). (E) Proposed mechanism for activation of CaMKII by oxidation. After initial activation of the holoenzyme by Ca2+/CaM (1°), oxidation at M281/282 (2°) blocks reassociation of the catalytic domain, yielding persistent CaMKII activity (3°).
Pretreatment with Ca2+/CaM was also necessary for autophosphorylation-dependent CaMKII activation, indicating that autophosphorylation and oxidation of CaMKII occur by parallel mechanisms. CaMKII bearing a T287A substitution underwent normal Ca2+/CaM-dependent activation but did not maintain persistent Ca2+-independent activity in the presence of ATP (Fig. 1D). However, the T287A mutant was activated by H2O2 (Fig. 1C), and the extent of this activation was statistically indistinguishable at all but the highest concentration of H2O2 tested (1μM). We interpret these observations as evidence that activation of CaMKII by ROS and autophosphorylation occur by a similar mechanism, but by independent modifications to nearby sites. Activation of the kinase by either mechanism requires the enzyme to be initially ‘opened’ by Ca2+/CaM to allow access to the autoinhibitory domain for oxidation or autophosphorylation (Fig. 1A, E). Either of these modifications can prevent subsequent interaction of the autoinhibitory region with the catalytic domain, providing for sustained Ca2+-independent activation of CaMKII. Consistent with these ideas, direct measurements of intrinsic fluorescence revealed that autophosphorylation and oxidation of CaMKII independently induce similar conformational changes in CaMKII (Supplemental Fig. 1).
Proteomic analysis of the synthetic peptide that contains the 281/282 methionine residues was used to probe for oxidative modification upon treatment with H2O2. We observed a clear decrease in the unoxidized form coupled with an increase in the various oxidized forms of this peptide based on the chromatographic traces and on the change in the number of observed spectra (Supplemental Fig. 2). In addition to the synthetic peptide, we analyzed the peptide containing the 281/282 methionine residues after treatment of the whole protein with H2O2 followed by trypsin cleavage. We were able to determine the relative change in oxidation of this peptide upon hydrogen peroxide treatment (Supplemental Table 1,2). The MS/MS spectra of the oxidized forms of the peptide were identical to those from the synthetic peptide, verifying that the oxidized peptide was correctly identified.
Given these observations and the recognized susceptibility of methionine residues to oxidation (Hoshi and Heinemann, 2001), we made methionine to valine mutations for the paired residues (M281/282V) and for another methionine (M308V) in the CaM-binding region. These mutants were exposed to H2O2 and assayed for activity in the presence of EGTA (Fig. 1C). The H2O2-dependent activation of CaMKII was preserved in the M308V mutant. However, oxidation-dependent CaMKII activity was completely abolished in the M281/282V and M281/282/308V mutants. Our data, obtained in cell free assay conditions, point to direct oxidation of the M281/282 pair as the primary H2O2-dependent activation pathway for CaMKII. Importantly, all the methionine to valine mutants showed a normal activity response to autophosphorylation (Fig. 1D), further supporting the concept that Ca2+ autonomous CaMKII activation by ROS or T287 autophosphorylation are independent events. While the paired methionine motif is conserved in the β, γ, and δ isoforms of CaMKII, the neuronal α isoform substitutes a cysteine residue for the first methionine of the pair (position 280 in CaMKIIα). The side chain of cysteine is also susceptible to oxidation. We generated a M281C mutant of CaMKIIδ to mimic the substitution in CaMKIIα. Additionally, we generated and purified CaMKIIα. Both the M281C CaMKIIδ mutant and the purified CaMKIIα were activated by H2O2, indicating that the cysteine substitution seen in CaMKIIα also supports ROS-dependent activation (Fig. 1C). To further elucidate the role of M281 and M282 in ROS-dependent activation, these sites were individually mutated (Supplemental Fig. 3). The M282V mutation completely ablated oxidation-dependent activation, while the M281V mutation partially reduced activation by 65%, indicating that a single oxidation event within the regulatory domain is insufficient to activate CaMKII.
Autophosphorylation at T287 dramatically increases the binding affinity of CaMKII for CaM, a phenomenon known as “CaM trapping” (Meyer et al., 1992). In the absence of ATP the Ca2+/CaM/CaMKII complex was very rapidly dissociated following addition of EGTA, independent of the redox state, as measured by fluorescence anisotropy of dansylated CaM (Supplemental Fig. 4A). CaMKII exposure to H2O2 for 10 min induced Ca2+/CaM-independent activity (as in Fig 1B), but also failed to induce CaM trapping (not shown). These observations indicate that under normal experimental conditions, oxidation of CaMKII is not sufficient to induce CaM trapping. Dissociation of Ca2+/CaM from autophosporylated CaMKII and CaM was significantly slower than from nonphosphorylated enzyme, consistent with CaM trapping. However, pretreatment with H2O2 prior to EGTA had no significant effect on the dissociation kinetics. Thus, oxidation of CaM or CaMKII does not prevent or enhance CaM trapping by autophosphorylated CaMKII. CaM trapping is reduced by phosphorylation of T306/307 (Colbran 1993), so we investigated if oxidation of M308 might prevent CaM trapping by a parallel mechanism. We did observe a significant slowing of dissociation of the CaM/CaMKII complex after H2O2 treatment of the M308 mutant (Supplemental Fig. 4B,C). These data suggest that the absence of CaM trapping during oxidation is partly due to M308.
It seemed possible that conditions capable of oxidizing methionine residues would also oxidize unprotected cysteine residues. Although mutation of methionine residues at 281 and 282 was sufficient to completely ablate ROS-dependent activation of CaMKII, we created a C290V mutant to determine whether this cysteine residue within the CaMKII regulatory domain could also play a role. Both the Ca2+/CaM-dependent and ROS-dependent activity of the C290V mutant were indistinguishable from WT CaMKII (Supplemental Fig. 4D). Our finding that oxidation of paired amino acids (M281/282 in CaMKIIδ) were required for activation by H2O2 support a view that oxidation of a lone residue is insufficient to confer Ca2+/CaM autonomous CaMKII activity. In order to comprehensively test the potential role of all accessible cysteines in contributing to oxidation-dependent CaMKII activity we measured CaMKII activity responses to H2O2 in the presence of iodoacetic acid, a reagent that blocks oxidation of unprotected cysteine residues (Zangerle et al., 1992). Cysteine protected CaMKIIδ showed equivalent H2O2 activity responses compared to CaMKIIδ without iodoacetic acid (Supplemental Fig. 2D). We used an established colorometric assay to quantify the available cysteine residues and verify that cysteine protection by iodoacetic acid was effective. Our results confirmed that most or all of the 11 cysteines in CaMKIIδ were accessible to the Ellman’s reagent after Ca2+/CaM binding, while treatment with iodoacetic acid blocked the accessibility of cysteine residues to biochemical modification (data not shown). Taken together, these findings demonstrate that oxidative activation of CaMKIIδ is independent of cysteines.
Oxidation of CaMKII occurs in vivo
We developed a new immune serum against oxidized M281/282 to detect ROS effects on CaMKII in vivo. We validated the fidelity of the antiserum using purified CaMKII protein by immunoblotting against WT CaMKII and the M281/282V mutant in control conditions and after treatment with H2O2 or Ca2+/CaM/ATP. Blots were also assayed with a phospho- and site-specific antibody against T287 (p-287). WT CaMKII exposed to H2O2 after pretreatment with Ca2+/CaM showed significant reactivity to our oxidized M281/282 antiserum, but untreated and T287-phosphorylated CaMKII samples were not recognized by our antiserum (Fig. 2A). The M281/282V mutant had minimal reactivity to our antiserum among the three treatments. These findings demonstrated that phospho-T287 and oxidized M281/282 were immunologically distinct sites. We performed additional immunoblots in which oxidized CaMKII was probed with the antiserum along with increasing concentrations of the peptide antigen (Fig. 2B). Band intensity decreased with increasing peptide concentration, indicating that the immune serum was specific for oxidized CaMKII.
Figure 2. AngII induces oxidation of CaMKII in vivo.

(A) Immunoblot of WT CaMKII and M281/282V mutant after no treatment, oxidation, or autophosphorylation probed with antibodies against total, autophosphorylated (p-T287), or oxidized CaMKII. Summary data shows relative band intensity using the oxidized CaMKII antibody (n = 3 trials/group, * p < 0.05 vs. band intensity of WT CaMKII treated with H2O2). (B) Immunoblot and summary data of oxidized WT CaMKII probed with antiserum against oxidized M281/282 with increasing ratios of oxidized antigen peptide. (n = 3 trials/group, * p < 0.05 vs. band intensity with no peptide). (C) Immunofluorescent staining of heart sections from mice treated with saline, AngII, or Iso and probed for oxidized or total CaMKII. Red staining is positive for oxidized or total CaMKII and blue staining is for nuclei. Calibration bars are 100 microns. (D) Immunoblot and summary data of heart lysates from mice treated with saline (Sal), Iso, or AngII probed with antibodies against total CaMKII, oxidized CaMKII, or actin (n = 3 hearts/group, * p < 0.05 vs. band intensity of saline treatment).
To determine the role of CaMKII oxidation in apoptosis, mice were treated with saline, AngII, or isoproterenol (Iso) for one week, and transverse heart sections from these mice were probed for the production of oxidized CaMKII in vivo. WT mice treated with AngII produced more oxidized CaMKII than those treated with saline or Iso (Fig. 2C). Total CaMKII immunoreactivity remained constant regardless of treatment. Conversely, mice lacking a critical subunit of NADPH oxidase (p47−/−) did not show increased levels of oxidized CaMKII in response to AngII. The p47−/− mice do not assemble the ROS-producing complex NADPH oxidase (Munzel and Keaney, 2001), the main source of ROS due to AngII stimulation in many cell types (Lyle and Griendling, 2006). Heart sections from WT mice showed increased staining for T287-phosphorylated CaMKII after AngII treatment, while p47−/− mice were unaffected (Supplemental Fig 5). Other studies have suggested that protein phosphatase activity is decreased by pro-oxidant conditions (Howe et al., 2004), indicating the possibility of coordinate activation of CaMKII both by direct oxidation at the Met281/282 sites and by phosphatase inactivation leading to increased phosphorylation at the T287 site.
We also homogenized hearts from mice treated with saline, AngII, or Iso, and whole heart lysates were analyzed by immunoblot for oxidized CaMKII. While total CaMKII was not significantly different among the three treatment groups, heart lysates from mice treated with AngII showed significantly increased oxidized CaMKII levels (Fig. 2D). Taken together, these findings demonstrate that oxidation of CaMKII occurs in vivo, and that elevated levels of AngII increase CaMKII oxidation at M281/282 compared to saline or Iso.
AngII triggers ROS production and CaMKII-dependent apoptosis in cardiomyocytes
Given our results, we hypothesized that cells deficient in ROS production or CaMKII activity would be resistant to AngII mediated apoptosis. We treated cardiomyocytes from mice that express an inhibitory peptide against CaMKII (AC3-I, Zhang et al., 2005) with 100nM AngII for 24 hours in parallel with isolated cardiomyocytes from WT and p47−/− mice. AngII caused a significant increase in the percent of TUNEL positive nuclei in WT cells but had no significant effect in p47−/− or AC3-I cardiomyocytes (Fig. 3A). Activity assays for caspase-3, a downstream target enzyme in the CaMKII apoptotic signaling pathway in heart, recapitulated the results from the TUNEL assay (Fig. 3B). Importantly, direct addition of ROS in the form of H2O2 caused significant apoptosis in p47−/− cells, demonstrating that their resistance to AngII-induced apoptosis is a result of impaired ability to produce ROS rather than a lack of sensitivity to ROS. The apoptotic effect of H2O2 in AC3-I cells was blunted by more than half compared to WT or p47−/− cells (Fig. 3A), indicating the critical importance of CaMKII activation to ROS and Iso-dependent apoptosis.
Figure 3. AngII increases ROS production and apoptosis by a CaMKII-dependent pathway in cardiomyocytes.

(A) Percent of total isolated cardiomyocytes positive for TUNEL staining after treatment with saline, AngII, Iso, or H2O2 (n = 6 hearts/group, * p < 0.05 vs. WT with saline). (B) Caspase-3 activity induced by saline, AngII, or Iso normalized to WT cells treated with saline (n = 3 hearts/group, * p < 0.05 vs. WT with saline). (C) DHE stained cardiomyocytes after treatment with 100nM AngII or Iso. Red coloration indicates presence of ROS above control cells. Scale bars equal 50 μm. (D) Percent of total cells positive for DHE staining above control (n = 3 assays/group, * p < 0.05 vs. WT saline). (E) Example traces of intracellular calcium concentration of cultured WT cardiomyocytes treated with 100nM AngII (red symbols) or Iso (blue symbols) measured by real-time calcium imaging. The arrow indicates addition of AngII or Iso. (F) Peak intracellular Ca2+ concentration in response to either AngII or Iso for WT or p47−/− cells (n = 3 trials/group, NS = not statistically different).
In order to validate the connection between AngII and ROS in our experimental model, we treated isolated cardiomyocytes from WT and p47−/− mice with 100nM AngII and monitored production of ROS by imaging DHE, a fluorescent reporter for superoxide and hydrogen peroxide (Fig. 3C). We also incubated WT cardiomyocytes with fura-2 AM, a cell-permeant calcium indicator to observe changes in intracellular Ca2+ ([Ca2+]i). Treatment with AngII caused a significant increase in ROS production in WT but not in p47−/− cardiomyocytes (Fig. 3D). On the other hand, increases in [Ca2+]i were significantly less after AngII compared to Iso treatment (Fig. 3E, F) for cardiomyocytes from both WT and p47−/− mice. These data show AngII signaling predominantly increases ROS, while Iso predominantly increases [Ca2+]i under our experimental conditions.
CaMKII knockdown prevents AngII- and Iso-induced apoptosis
In order to further test the role of CaMKII and specifically define the effects of M281/282 on myocardial apoptosis, we used a knock down and replacement strategy in cultured neonatal cardiomyocytes. Rat cardiomyocytes were cultured and treated with shRNA encoding lentivirus against rat CaMKIIδ. After 48 hours CaMKII expression was significantly reduced, as measured by immunoblot and activity assays (Fig. 4A). Cells were then transduced with lentivirus encoding shRNA-resistant WT or M281/282V mutant CaMKII. Control cells were transduced with GFP encoding lentivirus. After 48 hours, cells transduced with CaMKII rescue constructs showed significant recovery of CaMKII expression compared to control cells. Addition of Ca2+/CaM to lysates from cells transduced with either CaMKII encoding virus had similar total activity as native cells. However, H2O2-induced activity was only rescued in cells expressing the WT CaMKII construct. These cellular studies support our earlier finding with molecular CaMKII (Fig. 1) by showing that oxidation of M281/282 is critical for ROS triggered CaMKII activity. In addition, this strategy created cardiomyocytes expressing ROS-resistant CaMKII, providing a novel system for investigating ROS and CaMKII-dependent apoptosis.
Figure 4. AngII-induced apoptosis is blocked by CaMKII silencing.
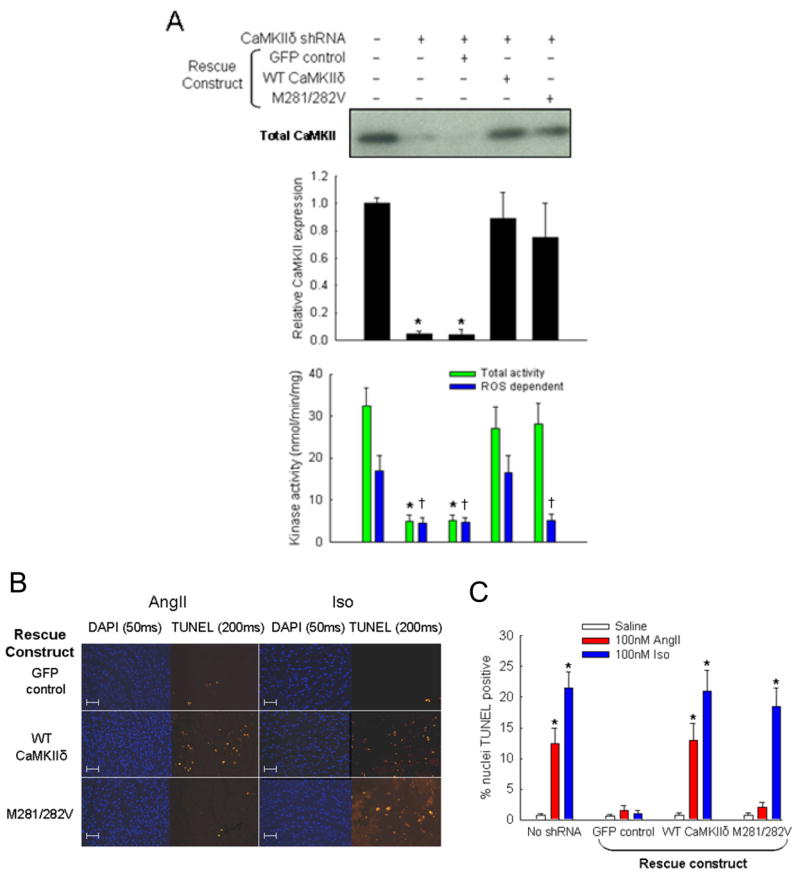
(A) Representative immunoblot with anti-CaMKII to measure protein expression after treatment with shRNA and shRNA-resistant rescue constructs. Immunoblot against actin was used as a loading control (not shown). Middle panel shows summary data of CaMKII expression relative to untreated cells (n = 3 experiments/group, * p < 0.05 vs. no treatment). Bottom panel shows summary data for CaMKII activity assays of lysates (n = 3 experiments/group, * p < 0.05 vs. total activity with no treatment, † p < 0.05 vs. ROS-dependent activity with no treatment). Only the WT CaMKII construct was able to reconstitute both Ca2+/CaM- and ROS-dependent activity observed in untreated cells. (B) Immunostaining and (C) summary data from isolated rat cardiomyocytes transduced with shRNA against CaMKII followed by rescue with WT CaMKII, M281/282V, or GFP control. Immunostaining shows total nuclei (DAPI) and DNA nicking (TUNEL) consistent with apoptosis. Scale bars equal 100 μm. Summary data show percent of total nuclei with positive TUNEL staining (n = 6 hearts/group, * p < 0.05 vs. GFP with AngII).
Cardiomyocytes treated with shRNAs/CaMKIIδ encoding lentivirus were exposed to saline, AngII or Iso as above (Fig. 4B). The apoptotic response to AngII and Iso was significantly attenuated in CaMKII knockdown cells compared to myocytes without shRNA. Moreover, expression of shRNA-resistant WT CaMKII fully rescued apoptotic responses to both agonists (Fig. 4C). In contrast, expression of the ROS-resistant M281/282V CaMKII mutant rescued the apoptotic response to Iso but, importantly, failed to rescue the apoptotic response to AngII after 24 hours (Fig. 4C). Cells expressing the M281/282V CaMKII remain susceptible to Iso-induced apoptosis (Fig. 4B, C), indicating that elimination of these residues does not affect activation of CaMKII by catecholamine stimulation. These cellular studies are performed in a complex biological environment compared to studies with isolated CaMKII, but nevertheless support a concept that direct oxidation of CaMKII by AngII is sufficient to confer enhanced CaMKII activity and trigger apoptosis.
ROS production and CaMKII activity are critical for AngII-mediated cardiac apoptosis in vivo
Intracellular ROS levels increase dramatically in models of structural heart disease (Hare, 2001), particularly those initiated by AngII (Tojo et al., 2002). Stimulation by AngII leads to activation of the NADPH oxidase complex, increasing intracellular superoxide and hydrogen peroxide levels. To establish an in vivo context for our previous findings and to test the role of CaMKII in AngII-stimulated cardiac apoptosis, p47−/−, AC3-I, and WT mice were treated with saline, AngII or Iso for one week. Transverse heart sections from these mice were stained for evidence of apoptosis. After one week WT mice treated with either AngII or Iso showed significant cardiac apoptosis, as determined by TUNEL staining of heart sections (Fig. 5). The p47−/− mice had no significant increase in cardiac apoptosis after treatment with AngII, most likely because these mice were unable to produce ROS in response to AngII stimulation (Fig. 3D). However, the p47−/− mice showed a preserved apoptotic response to Iso, suggesting that Iso-induced apoptosis occurs independently of oxidative stress generated by NADPH oxidase in vivo under these conditions. In contrast, the AC3-I mice with CaMKII inhibition were resistant to apoptosis induced by either AngII or Iso, indicating that CaMKII is a necessary signal element for apoptosis initiated by AngII or Iso in vivo.
Figure 5. AngII causes cardiac apoptosis in vivo via a ROS and CaMKII-mediated pathway.
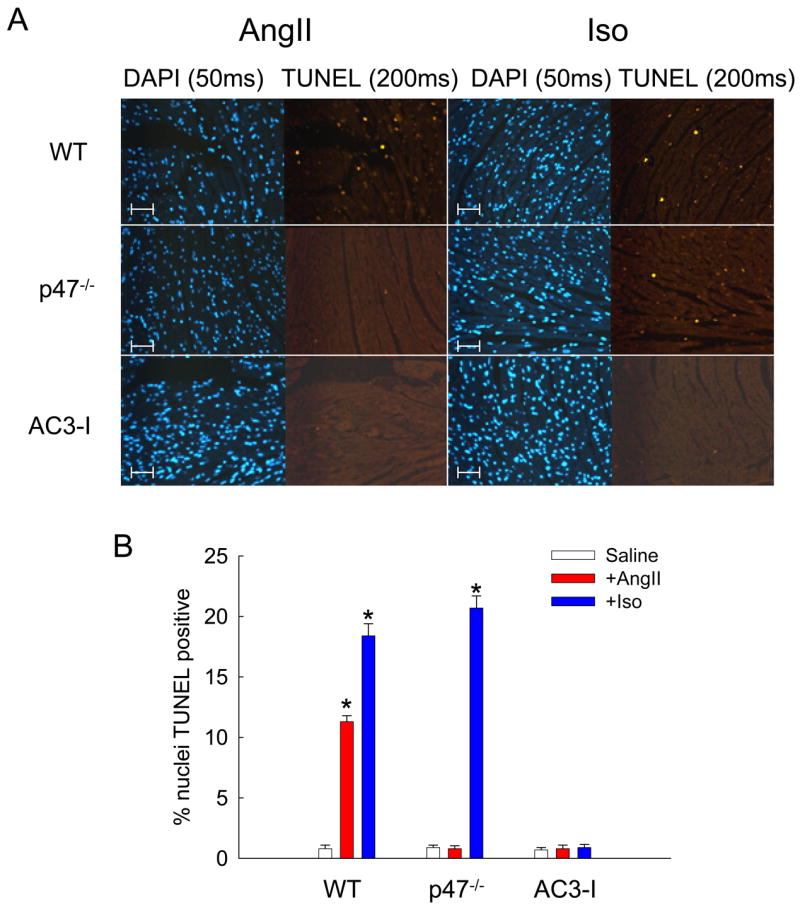
(A) Immunostaining of mouse heart sections for total nuclei (DAPI) and nuclear damage (TUNEL) consistent with apoptosis. WT, p47−/−, and AC3-I mice were treated with Ang II (3mg/kg/day) or Iso (30mg/kg/day) for seven days. Scale bars equal 100 μm. (B) Percent of total nuclei that showed positive TUNEL staining (n = 3 hearts/group, * p < 0.05 vs. WT with saline).
Increased CaMKII oxidation, apoptosis, cardiac dysfunction and death in MsrA−/− mice
Methionine oxidation is specifically reversed by MsrA (Weissbach et al., 2002), so we hypothesized that MsrA−/− mice would show enhanced vulnerability to AngII-mediated CaMKII oxidation and apoptosis. In order to test this idea we implanted MsrA−/− and WT control mice with AngII or saline eluting osmotic mini pumps. Hearts from MsrA−/− mice treated with AngII in vivo showed significantly more CaMKII oxidation (Fig. 6A,B) and increased TUNEL staining (Fig. 6C) compared to saline treated MsrA−/− mice and to saline or AngII treated control hearts. The increased CaMKII oxidation by AngII in MsrA−/− hearts showed that CaMKII oxidation is dynamically regulated by MsrA in myocardium in vivo, and suggested that MsrA−/− mice would be more vulnerable to severe myocardial stress due to increased methionine oxidation. Myocardial infarction is the most common cause of sudden cardiac death and heart failure in patients, and p47−/− (Doerries et al., 2007) and AC3-I mice (Zhang et al., 2005) are protected from left ventricular dilation and dysfunction after myocardial infarction surgery, suggesting that ROS activation of CaMKII may be important in myocardial infarction. In order to test if CaMKII oxidation and apoptosis were regulated by NADPH oxidase and MsrA in the setting of myocardial infarction, we subjected MsrA−/−, p47−/−, and WT mice to myocardial infarction surgery. MsrA−/− mice showed significantly more CaMKII oxidation (Fig. 7A, B) and myocardial apoptosis (Fig. 7C) compared to WT and p47−/− mice. These data indicate CaMKII oxidation is dynamically regulated by NADPH oxidase and MsrA in the setting of myocardial infarction. We performed myocardial infarction surgery on a larger cohort of MsrA−/− and WT control mice to determine if increased CaMKII oxidation and apoptosis in MsrA−/− mice translated into poorer functional outcomes. MsrA−/− mice were significantly more likely to die after surgery compared to WT controls (Fig. 7D, p = 0.0015) and exhibited significantly greater left ventricular dilation (Fig. 7E) and impaired systolic function (Fig. 7F) compared to controls, demonstrating that methionine oxidation increases the pathological impact of myocardial infarction in vivo.
Figure 6. MsrA−/− mice have increased susceptibility to AngII-mediated apoptosis.
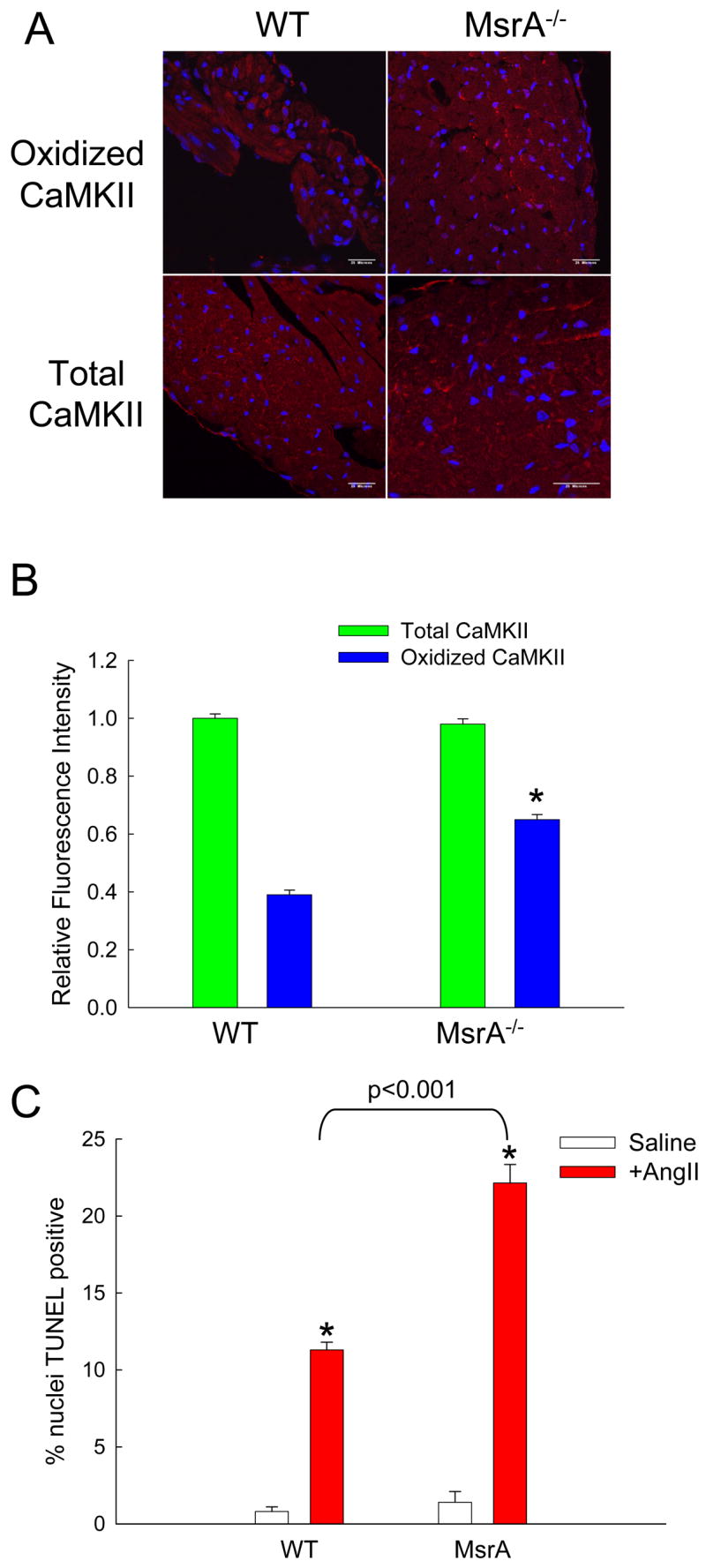
(A) Immunofluorescent staining of heart sections from WT and MsrA−/− mice treated with AngII and probed for oxidized or total CaMKII. Red staining is positive for oxidized or total CaMKII and blue staining is for nuclei. Calibration bars are 100 microns. (B) Quantification of average staining intensity for AngII treated hearts, relative to WT (n = 3 hearts/group, * p < 0.05 vs. WT with AngII). (C) Summary data for TUNEL staining of heart sections from WT and MsrA−/− mice treated with saline or AngII (n = 5 hearts/group, * p < 0.05 vs. WT with saline).
Figure 7. Mice lacking MsrA have increased CaMKII oxidation, apoptosis, reduced survival and impaired heart function after myocardial infarction.

(A) Immunostaining and (B) stain intensity quantification of oxidized CaMKII in heart sections from WT, p47−/−, and MsrA−/− mice post-MI (n = 3 hearts/group, * p < 0.05 vs. WT). (C) Summary data for TUNEL staining of heart sections from WT, p47−/−, and MsrA−/− mice post-MI (n = 3 hearts/group, * p < 0.05 vs. WT). (D) Mortality is significantly increased post-MI in MsrA−/− mice compared to WT controls. Numbers in bars represent post-MI deaths/total number of mice receiving MI. Post-MI left ventricular dilation (E) and function (F) were compromised in surviving MsrA−/− mice compared to WT controls three weeks after surgery (n = 17 hearts/group for WT, n = 9 hearts/group for MsrA−/−).
DISCUSSION
CaMKII was first identified by its dependence on Ca2+/CaM for activation (Schulman and Greengard, 1978). Later it was recognized that autophosphorylation at T287 modified the enzyme so that activity persisted even in the absence of elevated Ca2+/CaM (Saitoh and Schwartz, 1985; Lou et al., 1986; Patton et al., 1990). Our findings unveil a new dimension to CaMKII signaling by showing that oxidation of M281/282 is a distinct molecular event, but with similar consequences to Thr287 autophosphorylation for sustaining Ca2+/CaM independent activity. Oxidative activation likely is important in all known CaMKII isoforms, and relies upon paired, oxidation susceptible residues (MM in β, γ and δ). Because of the ability of CaMKII to transition between Ca2+/CaM dependent and Ca2+/CaM independent species, CaMKII is considered a ‘memory molecule’ for the history of intracellular Ca2+ elevation. ROS facilitation of Ca2+/CaM CaMKII activity suggests that the ability of CaMKII to respond to Ca2+ elevation is enhanced in pro-oxidant conditions. Because increased CaMKII activity and oxidative stress are implicated in a wide variety of physiological and disease processes, our findings have potentially broad implications for improved understanding of connections between ROS and Ca2+ in multiple cell types.
CaMKII is initially activated by Ca2+/CaM binding, which blocks the autoinhibitory association between the regulatory and catalytic domains. Phosphorylation of T287 blocks the reassociation of the regulatory and catalytic domains, conferring Ca2+/CaM independent activity on the enzyme (Hudmon and Schulman, 2002). In this study, we discovered a novel mechanism for CaMKII activation by oxidation of M281/282. As is the case for T287-autophosphorylation, activating oxidation appears to require that the regulatory domain is first exposed by Ca2+/CaM binding, whereupon oxidation at M281/282 leads to persistent Ca2+/CaM-autonomous activation of CaMKII. Oxidation of methionine residues changes both the charge and flexibility of their side chains (Hoshi and Heinemann, 2001), apparently leading to steric blockage of reassociation between the regulatory and catalytic domains. Surprisingly, cysteine residues, a common target for oxidative regulation (Barford, 2004), do not appear to play any role in oxidative activation of CaMKIIδ (or by inference CaMKIIβ or γ). The regulatory domain of all CaMKII isoforms contains a single cysteine (C290 in CaMKIIδ), but oxidation of this unpaired cysteine is insufficient to activate CaMKIIδ. Our data are aligned with the concept that Ca2+/CaM-dependent exposure of the regulatory domain sets up the CaMKII molecule for subsequent modifications that confer persistent, Ca2+ independent activity.
Our findings identified a previously unrecognized mechanism of enhancing CaMKII by direct methionine oxidation, but oxidation may affect activity of kinases by multiple mechanisms. Pro-oxidant conditions can modify the activity levels of protein kinases by direct and indirect mechanisms. For example, direct thiol oxidation within the ATP binding pocket inhibits MEK kinase 1 activity (Cross and Templeton, 2004). Oxidative stress can induce activation of ERK1/2 (Engers et al., 2006), while oxidation-dependent inactivation of protein phosphatases (Tonks, 2006) and activation of upstream kinase kinases, such as IKK-β (Reynaert et al., 2006) can indirectly lead to increased kinase activity. The present findings that AngII increases both CaMKII oxidation and autophosphorylation suggest that ROS inhibition of phosphatases further enhances CaMKII activity responses to oxidant stress in vivo.
Autophosphorylation at T287 is reversed by phosphatase activity (Zhabotinsky, 2000; Hudmon and Schulman, 2002). Because phosphorylation is a readily reversible process, activation by autophosphorylation represents a tunable regulatory mechanism for CaMKII. Oxidation of methionine residues is also a reversible biochemical modification, and the presence of methionine residues can confer functional sensitivity to oxidative stress (Santarelli et al., 2006). Methionine sulfoxide reductase (Msr) reduces the side chain of methionine to its native state (Kryukov et al., 2002), and is therefore a critical defense mechanism against cellular damage by oxidative stress. Mutant Drosophila overexpressing Msr had longer life spans, (Ruan et al., 2002) while MsrA−/− mice show increased mortality in response to oxidant induced aging (Moskovitz et al., 2001). The importance of MsrA in various biological systems suggests that reversible oxidation of methionine residues could complement a Thr287 phosphorylation/dephosphorylation cycle by serving as a ROS responsive regulatory mechanism for dynamically titering CaMKII activity. Our studies show that MsrA is essential for reversing CaMKII oxidation in myocardium in vivo and that increased methionine oxidation worsens important clinical outcomes after myocardial infarction.
Structural heart disease is one of the largest public health problems in the developed world (Jessup and Brozena, 2003). AngII and βAR receptor antagonist drugs have significantly reduced mortality in patients with structural heart disease (Lancet 1999; Pfeffer et al., 2003), and represent a remarkable success story for translating basic scientific understanding of cellular signaling into effective treatments for human disease. Increased cardiomyocyte apoptosis appears to be an important feature of advanced structural heart disease (Olivetti et al., 1997). CaMKII is activated downstream to βAR receptor stimulation (Zhang et al., 2005) by increased Ca2+i (Zhu et al., 2003). CaMKII inhibition reduces apoptosis (Zhu et al., 2003; Yang et al., 2006), and improves mortality (Khoo et al., 2006) in structural heart disease models. These findings have contributed to a growing perception that CaMKII inhibition may be a novel therapeutic strategy for treating heart failure and arrhythmias (Bers, 2005). Our data reveal the importance of M281/282 oxidation for CaMKII activation and thereby provide a new molecular mechanism for understanding the effects of AngII in cardiomyocytes and in structural heart disease. Our present findings appear to increase the potential importance of CaMKII in structural heart disease by positioning CaMKII as a critical downstream nodal signal for enhancing cardiomyocyte death in response to excessive catecholamines, AngII and ROS. CaMKII has proven to be a remarkably versatile signaling molecule and the recently recognized role of ROS in activating CaMKII provides a new way of understanding the potential for oxidant stress to engage physiological and disease pathways in excitable cells.
METHODS
Mouse Models
Mice lacking the p47 gene (p47−/−) were purchased from Jackson Labs. Mice lacking the MsrA (MsrA−/−) were supplied by NIH (Bethesda, MD). Mice with genetic CaMKII inhibition (AC3-I) were generated by us as previously described (Zhang et al., 2005).
CaMKII activity assays and protein analysis
Mutant CaMKII cDNAs were generated using a QuikChange site-directed mutagenesis kit (Stratagene). CaMKIIδ (GenBank #NP_001020609) was generated using the Bac-to-Bac baculovirus system (Invitrogen) and purified on a calmodulin-agarose column. For CaMKII activity assays, purified CaMKII was pretreated with 200μM CaCl2 and 1μM CaM on ice for 1 minute. The protein was then exposed to ATP, H2O2, or water at the described concentrations for 10 minutes. Samples exposed to ATP or H2O2 were then treated with 10mM EGTA for 10 minutes. CaMKII activity was measured as a function of 32P-ATP incorporation into a synthetic substrate (syntide-2) at 30°C, as previously described (Wu et al., 2002).
Oxidized M281/282 immune serum production and immune staining
Antiserum to oxidized CaMKII regulatory domain peptide was generated by Epitomics, Inc. Commercial antibodies were used for blots and immunostaining for total (Stressgen Biotechnologies) and phosphorylated (Santa Cruz) CaMKII.
Detection of ROS
Changes in ROS levels in cultured primary cardiac myocytes after agonist stimulation were measured using the fluorgenic probe dihydroethidium (DHE, 5μM, Molecular Probes), as previously described (Zimmerman et al., 2004). DHE fluorescent images were acquired using confocal microscopy (Zeiss LSM510).
Intracellular Calcium Concentration Measurements
Intracellular calcium concentration was assessed by Fura-2 fluorescence ratio imaging using a microscopic digital imaging system (Photon Technology International), as described previously (Sharma et al., 1995).
Fluorescence measurements
Spectra were collected at 30°C using a Fluorolog 3 (Jobin Yvon, Horiba) spectrofluorometer. For intrinsic fluorescence shift experiments, excitation wavelength was 270nm. Emission spectra were generated at 1nm increments from 280nm to 400nm. Background traces were subtracted from CaMKII spectra to eliminate the contribution from intrinsic fluorescence of CaM. For fluorescence anisotropy experiments, baseline traces of 100nM dansylated CaM in 15mM HEPES buffer, pH 7.2 were measured at baseline and after the addition of 200μM CaCl2 at 60s. At 180s, 100nM purified CaMKII was added to the CaM solution. For some trials, CaMKII became phosphorylated by the addition of 10mM ATP. 100μM H2O2 or an equivalent volume of buffer was added at 250s. Finally, addition of 10mM EGTA at 300s was used to remove free calcium from the solution, uncoupling CaM/CaMKII binding.
Cardiomyocyte TUNEL immunostaining
Myocyte isolations from neonatal mouse or rat pups were modified from previously described methods (Mohler et al., 2007). To ensure that pure populations of cardiomyocytes were obtained, cultures were immunolabeled with alpha-actinin Ig (cardiomyocyte-specific marker). Only cultures with >90% cardiomyocytes were used in experiments (see Supplemental Fig. 6). Lentiviral treatments (shRNA, rescue constructs) and apoptosis inducing agents (Iso, AngII) were used for 24 hours. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 and sodium citrate, and stained using In Situ cell death detection kits, TMR Red (Roche). TUNEL stain assays were interpreted as previously described (Yang et al., 2006). Nuclei were co-stained with DAPI. Investigators were blinded to the genetic identity and treatment of the mice in all studies.
CaMKII cDNA, shRNA, and virus generation
See supplemental text for sequences and methods.
Heart section TUNEL immunostaining
Minipumps containing saline or AngII (3mg/kg/day) were inserted or daily injections of Iso (30mg/kg/day, intraperitoneal) were given for 7 days. Animals were then sacrificed and the heart was removed, fixed, and embedded in paraffin. To confirm specific labeling of cardiomyocytes, heart sections were labeled with alpha-actinin Ig (cardiomyocyte-specific marker). Only sections with >90% cardiomyocytes were used in experiments (see Supplemental Fig. 7). TUNEL staining was performed using In Situ cell death detection kits, TMR Red (Roche). Nuclei were co-stained with DAPI. An investigator blinded to the identity and treatment of the mice counted the number of total and TUNEL positive cells in each image.
Other sections were treated with either a general CaMKII antibody or oxidized CaMKII antiserum. Nuclei were co-stained with DAPI. Identity of cardiomyocytes was confirmed by co-stain with an antibody against α-actinin. Images were quantified for relative staining intensity using Image J (NIH, USA). Both the investigators and technical personnel assigned to immunostaining and quantification were blinded to the genetic identity and treatment of the mice in all studies.
Myocardial Infarction and echocardiography
Mice were anesthetized with ketamine/xylazine (87.5/12.5 mg/kg, respectively), and the left anterior descending (LAD) branch of the coronary artery was ligated using 8-0 ethilon suture (Ethicon) along the anterolateral border of the heart as close to the left atrial appendage as possible. Successful ligation of the artery is confirmed by blanching of the myocardium. Transthoracic echocardiograms were recorded in conscious sedated mice as described previously (Weiss et al., 2006), using a 15 MHz probe connected to a Sonos 5500 imager (Phillips Medical Systems, Bothell, WA). Images were acquired by an operator blinded to mouse genotype.
Statistical analysis
Statistical significance for mortality study was determined by chi-squares test. All other statistical significance was determined by One-Way ANOVA with post hoc Bonferonni tests. A p value of <0.05 was considered statistically significant. All results are presented as mean ± SEM.
Supplementary Material
Acknowledgments
The authors wish to acknowledge discussions with Dr Botond Bonfi, Dr Trudy Burns, Dr Johannes Hell, Dr David Murhammer, Dr Stefan Strack, and Dr Michael Welsh (University of Iowa) and technical contributions of Chantal Allamargot (University of Iowa Central Microscopy Research Facility). The authors also wish to acknowledge the graphic design contributions of Shawn Roach (University of Iowa). Mice lacking the MsrA gene were generously provided by Dr Earl Stadtman of NIH (Bethesda, MD). This work was funded by NIH R01 HL 079031, R01 HL 62494, and R01 HL 70250 (MEA); NIH R01 HL084583, R01 HL083422, and Pew Scholars Trust (PJM); NIH R01 GM57001 (MAS); NIH RR017369 (RMW); UI CVC Interdisciplinary Research Fellowship (JRE); UI Center for Biocatalysis and Bioprocessing Fellowship (SEO); and the University of Iowa Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF) Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol. 2004;14:679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Bers DM. Beyond beta blockers. Nat Med. 2005;11:379–380. doi: 10.1038/nm0405-379. [DOI] [PubMed] [Google Scholar]
- Colbran RJ. Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J Biol Chem. 1993;268:7163–7170. [PubMed] [Google Scholar]
- Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res. 2007;100:894–903. doi: 10.1161/01.RES.0000261657.76299.ff. [DOI] [PubMed] [Google Scholar]
- Engers R, Springer E, Kehren V, Simic T, Young DA, Beier J, Klotz LO, Clark IM, Sies H, Gabbert HE. Rac upregulates tissue inhibitor of metalloproteinase-1 expression by redox-dependent activation of extracellular signal-regulated kinase signaling. FEBS J. 2006;273:4754–4769. doi: 10.1111/j.1742-4658.2006.05476.x. [DOI] [PubMed] [Google Scholar]
- Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-Type Ca2+ Channel Facilitation Mediated by Phosphorylation of the [beta] Subunit by CaMKII. Mol. Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198–200. [PubMed] [Google Scholar]
- Hoshi T, Heinemann SH. Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531.1:1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CJ, Lahair MM, McCubrey JA, Franklin RA. Redox regulation of the calcium/calmodulin-dependent protein kinases. J Biol Chem. 2004;279:44573–44581. doi: 10.1074/jbc.M404175200. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, et al. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circ. 2006;114:1352–1359. doi: 10.1161/CIRCULATIONAHA.106.644583. [DOI] [PubMed] [Google Scholar]
- Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. doi: 10.1161/01.res.87.5.392. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Kumar RA, Koc A, Sun Z, Gladyshev VN. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc Natl Acad Sci. 2002;99:4245–4250. doi: 10.1073/pnas.072603099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou LL, Lloyd SJ, Schulman H. Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc Nat Acad Sci. 1986;83:9497–9501. doi: 10.1073/pnas.83.24.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology. 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, Laufs U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circ. 2003;108:1567–1574. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, Driskell IM, Schott JJ, Norris K, Leenhardt A, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circ. 2007;115:432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Keaney JF., Jr Are ACE inhibitors a “magic bullet” against oxidative stress? Circ. 2001;104:1571–1574. doi: 10.1161/hc3801.095585. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di LC, Beltrami CA, Krajewski S, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miller SG, Kennedy MB. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem. 1990;265:11204–11212. [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ, Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327:669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP, Solomon SD, Swedberg K, Van de WF, White H, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–1906. doi: 10.1056/NEJMoa032292. [DOI] [PubMed] [Google Scholar]
- Reynaert NL, van DV, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Ruan H, Tang XD, Chen ML, Joiner ML, Sun G, Brot N, Weissbach H, Heinemann SH, Iverson L, Wu CF, Hoshi T. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc Natl Acad Sci. 2002;99:2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Schwartz JH. Phosphorylation-dependent subcellular translocation of a Ca2+/calmodulin-dependent protein kinase produces an autonomous enzyme in Aplysia neurons. J Cell Biol. 1985;100:835–842. doi: 10.1083/jcb.100.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli LC, Wassef R, Heinemann SH, Hoshi T. Three methionine residues located within the regulator of conductance for K+ (RCK) domains confer oxidative sensitivity to large-conductance Ca2+-activated K+ channels. J Physiol. 2006;571:329–348. doi: 10.1113/jphysiol.2005.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman H, Greengard P. Ca2+-dependent protein phosphorylation system in membranes from various tissues, and its activation by “calcium-dependent regulator. Proc Nat Acad Sci. 1978;75:5432–5436. doi: 10.1073/pnas.75.11.5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma RV, Chapleau MW, Hajduczok G, Wachtel RE, Waite LJ, Bhalla RC, Abboud FM. Mechanical stimulation increases intracellular calcium concentration in nodose sensory neurons. Neuroscience. 1995;66:433–441. doi: 10.1016/0306-4522(94)00560-r. [DOI] [PubMed] [Google Scholar]
- Tojo A, Onozato ML, Kobayashi N, Goto A, Matsuoka H, Fujita T. Angiotensin II and oxidative stress in Dahl Salt-sensitive rat with heart failure. Hypertension. 2002;40:834–839. doi: 10.1161/01.hyp.0000039506.43589.d5. [DOI] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circ. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther WT, Matthews B, St John G, Nathan C, Brot N. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch Biochem Biophys. 2002;397:172–178. doi: 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]
- Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble RW, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circ. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291:H3065–H3075. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- Zangerle L, Cuenod M, Winterhalter KH, Do KQ. Screening of thiol compounds: depolarization-induced release of glutathione and cysteine from rat brain slices. J Neurochem. 1992;59:181–189. doi: 10.1111/j.1471-4159.1992.tb08889.x. [DOI] [PubMed] [Google Scholar]
- Zhabotinsky AM. Bistability in the Ca2+/calmodulin-dependent protein kinase- phosphatase system. Biophy J. 2000;79:2211–2221. doi: 10.1016/S0006-3495(00)76469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Thiel W, Guatimosim S, Song LS, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- Zhu WZ, Wang SQ, Chakir K, Yang DM, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng HP, Xiao RP. Linkage of beta(1)-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J of Clin Inv. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WZ, Woo AY, Yang DM, Cheng H, Crow MT, Xiao RP. Activation of CaMKII is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J of Biol Chem. 2007;282:10833–10839. doi: 10.1074/jbc.M611507200. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res. 2004;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.