

Abstract
Glucose-6-phosphatase-α (G6PC) is a key enzyme in glucose homeostasis that catalyzes the hydrolysis of glucose-6-phosphate to glucose and phosphate in the terminal step of gluconeogenesis and glycogenolysis. Mutations in the G6PC gene, located on chromosome 17q21, result in glycogen storage disease type Ia (GSD-Ia), an autosomal recessive metabolic disorder. GSD-Ia patients manifest a disturbed glucose homeostasis, characterized by fasting hypoglycemia, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, lactic acidemia, and growth retardation. G6PC is a highly hydrophobic glycoprotein, anchored in the membrane of the endoplasmic reticulum with the active center facing into the lumen. To date, 54 missense, 10 nonsense, 17 insertion/deletion, and 3 splicing mutations in the G6PC gene have been identified in more than 550 patients. Of these, 50 missense, 2 nonsense, and 2 insertion/deletion mutations have been functionally characterized for their effects on enzymatic activity and stability. While GSD-Ia is not more prevalent in any ethnic group, mutations unique to Caucasian, oriental, and Jewish populations have been described. Despite this, GSD-Ia patients exhibit phenotypic heterogeneity and a stringent genotype-phenotype relationship does not exist.
Introduction
Glycogen storage disease type I (GSD-I), also known as von Gierke disease, is a group of autosomal recessive disorders with an overall incidence of approximately 1 in 100,000 [Chou and Mansfield 1999; Chen 2001; Chou et al., 2002]. GSD-I was originally divided into four subtypes, corresponding to defects in the glucose-6-phosphatase-α (G6PC) catalytic unit, GSD-Ia (MIM232200); defects in the glucose-6-phosphate transporter (G6PT), GSD-Ib (MIM232220); defects in a putative phosphate transporter, GSD-Ic; and defects in a putative glucose transporter, GSD-Id [Chou and Mansfield 1999; Chen 2001; Chou et al., 2002]. GSD-Ia and GSD-Ib have been characterized at the molecular level. Both the G6PC [Lei et al., 1993] and G6PT [Gerin et al., 1997; Annabi et al., 1998] genes have been cloned and molecular genetic studies have confirmed that mutations inactivating G6PC cause GSD-Ia [Lei et al., 1993; 1994; 1995a; 1995b] and mutations inactivating G6PT cause GSD-Ib [Hiraiwa et al., 1999; Chen et al., 2000; 2002]. In contrast, GSD-Ic and Id have not been characterized at the molecular level. Clinical cases reported to represent GSD-Ic, GSD-I non-a, and GSD-Id, have been subjected to sequence analysis and shown to share deleterious mutations identified in the G6PT gene of GSD-Ib patients [Veiga-da-Cunha et al., 1998; 1999; Galli et al., 1999; Janecke et al., 2000]. This raises the possibility that G6PT is implicated in all cases of GSD-I non-a.
GSD-Ia is the most prevalent, representing over 80% of GSD-I cases. G6PC is a key enzyme for the maintenance of glucose homeostasis between meals, catalyzing the hydrolysis of glucose-6-phosphate (G6P) to glucose and phosphate in the terminal step of gluconeogenesis and glycogenolysis. GSD-Ia patients are unable to maintain glucose homeostasis and a hallmark of GSD-Ia is hypoglycemia following a short fast [Chou and Mansfield 1999; Chen 2001; Chou et al., 2002]. This loss of glucose homeostasis leads to the accumulation of elevated levels of G6P in the cytoplasm. This in turn stimulates the alternative metabolic pathways involving G6P. The primary impact, from which the name of the disease derives, is the synthesis and excessive accumulation of glycogen in the liver and kidney, which in turn promotes progressive hepatomegaly and nephromegaly. Other major metabolic consequences of elevated cytoplasmic G6P are hypercholesterolemia, hypertriglyceridemia, hyperuricemia, and lactic acidemia that also characterize the clinical pathophysiology of GSD-Ia. In addition to excessive glycogen deposition, additional accumulation of fats in the liver also contributes significantly to the hepatomegaly.
Prior to the cloning of the G6PC gene, GSD-Ia was diagnosed primarily by clinical and biochemical symptoms, supported by basic laboratory investigations, and confirmed by measurements of G6PC activity in liver biopsy samples. With the cloning of the G6PC gene, DNA-based diagnostic tests for GSD-Ia were developed by several laboratories. DNA testing for GSD-Ia, primarily in the form of targeted mutation analysis and exon sequencing of G6PC, is now available from several clinical testing laboratories which can be identified through the NIH funded GeneTests web site (http://www.geneclinics.org/servlet/access?prg=j&db=genetests&site=gt&id=8888891&fcn=c&qry=2678&res=nous&res=nointl&key=GnAY5RgtLYkBn&show_flag=c). GSD-Ia and GSD-Ib patients manifest a near identical metabolic phenotype but GSD-Ib patients also suffer from neutropenia, myeloid dysfunctions, and inflammatory bowl disease indistinguishable from the idiopathic Crohn's disease [Visser et al., 2000; Dieckgraefe et al., 2002; Chou and Mansfield 2003]. Therefore, flowcharts have been presented for the diagnosis of GSD-Ia in which the presence or absence of myeloid dysfunction determines whether to perform mutation analysis of the G6PC gene [Rake et al., 2000a]. However, neutropenia is not manifest by all GSD-Ib patients [Galli et al., 1999; Kure et al., 2000; Melis et al., 2005; Angaroni et al., 2006; Martens et al., 2006] and some GSD-Ia patients suffer from mild neutropenia [Weston et al., 2000]. Therefore, a clear diagnosis may still warrant mutational analysis of both G6PC and G6PT genes.
There is no cure for GSD-Ia and the current treatment for GSD-Ia is a dietary therapy augmented by various conventional drugs. For patients younger than 6 months old, the therapy typically consists of nocturnal nasogastric infusion of glucose to avoid hypoglycemia [Greene et al., 1976] because cornstarch is poorly tolerated due to immaturity of the gastrointestinal tract and lack of brush border enzymes [Weinstein and Wolfsdorf, 2002]. For patients older than 6 months, the therapy consists of a supplement of uncooked cornstarch, which serves as a slow release carbohydrate to prolong euglycemia between meals [Chen et al., 1984]. Many patients on a therapy of uncooked cornstarch fail to maintain “ideal plasma glucose levels” and manifest hyperlactataemia after midnight. A treatment regimen including nocturnal intragastric feeding of a glucose polymer during both childhood and adolescence to maintain blood glucose in the high normal range has been advocated to enable GSD-Ia patients to maintain normal urinary lactate levels and achieve optimal growth [Daublin et al., 2002]. In general, these dietary strategies enable patients to attain near normal growth and pubertal development, with fewer complications as they age. However, dietary therapy fails to completely prevent the occurrence of hyperlipidemia, hyperuricemia, lactic acidemia, hypercalciuria, and hypocitraturia [Weinstein et al., 2001; Rake et al., 2002]. As a result, osteoporosis, gout, pulmonary hypertension, kidney disease, and hepatic adenoma remain long term complications of GSD-Ia [Chou and Mansfield 1999; Chen 2001; Chou et al., 2002].
The human G6PC is a single copy gene that contains 5 exons [Lei et al., 1993] and spans 12.5 kb of DNA on chromosome 17q21 [Lei et al., 1994]. The expression of G6PC is tissue restricted, the predominant sites of expression being the liver, kidney, and intestine [Pan et al., 1998b]. The 3100-base G6PC transcript encodes a highly hydrophobic, endoplasmic reticulum (ER)-associated glycoprotein of 357 amino acids [Lei et al., 1993]. A G6PC isoform, G6PC3 (glucose-6-phosphatase-β) has recently been discovered and the G6PC3 gene also maps to chromosome 17q21 [Martin et al., 2002]. While similar in many respects to G6PC, G6PC3 contains an additional exon, spans only 5.4 kb of DNA, and most notably, is expressed ubiquitously [Martin et al., 2002]. The G6PC3 protein shares 36 % amino acid identity to G6PC [Martin et al., 2002; Shieh et al., 2003; Guionie et al., 2003] and is similar in many respect to G6PC. It too is a hydrophobic protein, though slightly shorter (346 amino acids) and shares topological structure, active center and kinetic properties with G6PC [Shieh et al., 2003; Ghosh et al., 2004]. However, mutations in G6PC3 are not associated with GSD-Ia, which is consistent with the primary role the liver, kidney and intestine play in inter-prandial glucose homeostasis for the body as a whole. The remainder of this review will focus primarily on G6PC and its relationship to GSD-Ia.
In this study, we summarize the reported G6PC mutations and review what mutagenesis studies have revealed about the structure and function of the G6PC catalytic unit. We also provide a summary table that unites the various nomenclatures used in the literature reporting mutations in G6PC in accordance with the guidelines of the Human Genome Variation Society (www.hgvs.org). Early reports of G6PC mutations used a nomenclature in which the transcription initiation site was designated nucleotide +1 [Lei et al., 1993]. These mutations were renamed in this study with the A of the ATG-translation initiation codon as nucleotide +1 [den Dunen and Antonarakis, 2000, 2001].
Topology and Active Center of G6PC
G6PC contains 9 transmembrane domains that anchor it in the ER membrane [Pan et al., 1998a; 1998c]. The amino-terminus of the protein lies in the ER lumen and the carboxyl-terminus in the cellular cytoplasm (Fig. 1). Sequence analysis has identified a conserved motif, K-X6-RP-(X12-54)-PSGH-(X31-54)-SR-X5-H-X3-D, lying between residues 76 and 180 in G6PC, which aligns with the conserved phosphatase signature motif found in lipid phosphatases, acid phosphatases, and vanadium haloperoxidases [Hemrika and Wever, 1997; Stukey and Carman, 1997]. Based on the crystal structure of the vanadium haloperoxidase [Hemrika et al., 1997], the active center of G6PC is proposed to comprise Lys-76, Arg-83, His-119, Arg-170 and His-176 [Hemrika and Wever, 1997; Stukey and Carman, 1997]. During G6PC catalysis, an enzyme-phosphate intermediate is formed through a histidine residue [Feldman and Butler, 1972]. Using active site labeling and cyanogen bromide mapping, His-176 was shown to be the nucleophile that covalently bound the phosphate moiety forming the phosphohistidine-G6PC intermediate during catalysis [Ghosh et al., 2002].
Fig. 1.
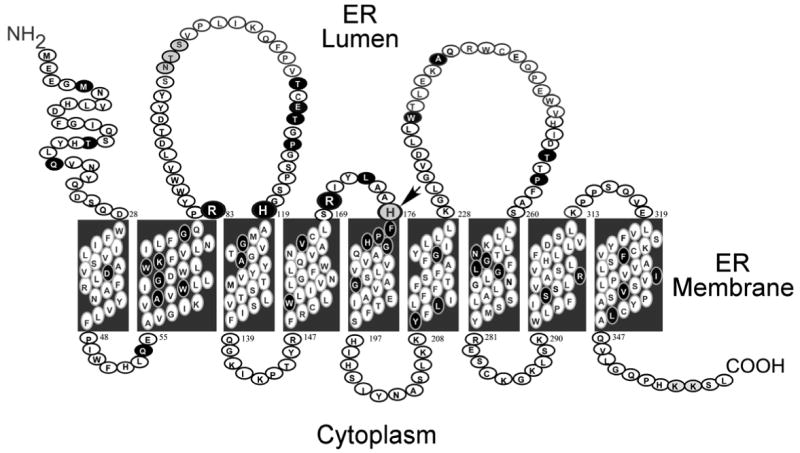
G6PC is anchored in the membrane of the ER by 9 transmembrane helices. The amino-terminus is located in the ER lumen and the carboxyl-terminus in the cellular cytoplasm. Missense and the p.F327del mutations identified in the G6PC gene of GSD-Ia patients are marked in black. Arg-83, His-119, Arg-170, and His-176, which contribute to the active center, are denoted by large circles. The phosphate acceptor His-176 is denoted by an arrow.
Mutations in four of the proposed active site residues, namely p.K76N, p.R83C, p.R83H, p.H119L, and p.R170Q have been identified in GSD-Ia patients and shown to completely abolish G6PC enzymatic activity [Lei et al., 1993; Shieh et al., 2002], consistent with their proposed role. Both conservative and non-conservative in vitro mutations of Arg-83 and His-119 have shown a loss of activity in G6PC catalysis further underlining the essential role of these 2 residues for catalytic activity [Lei et al., 1995c]. Surprisingly, mutations involving His-176 have not been identified in the G6PC gene of GSD-Ia patients. However, in vitro mutagenesis of His-176 to Ala (p.H176A), Ile (p.H176I), Lys (p.H176K), Met (p.H176M), Asn (p.H176N), Ser (p.H176S), or Arg (p.H176R) followed by transient expression analysis has shown these H176 mutants are devoid of G6P hydrolytic activity [Pan et al., 1998a] as would be expected for its role as the phosphate acceptor in G6PC. Sequence alignment indicates that G6PC and G6PC3 share the same active center structure and studies of G6PC3 support the conclusions about G6PC. In G6PC3 the active site residues include Lys-72, Arg-79, His-114, Arg-161, and His-167, and G6PC3 mutants R79A, H114A, or H167A are devoid of enzymatic activity [Shieh et al., 2003]. Importantly, His-167 in G6PC3 is also the phosphate acceptor forming a phosphohistindine-G6PC3 intermediate during catalysis [Ghosh et al., 2004].
Based on mutational and active site labeling studies, the current paradigm for the G6PC reaction mechanism is that His-176 is the nucleophile forming a phosphohistidine enzyme-intermediate, Arg-83 and Arg-170 donate hydrogen bonds to phosphate and stabilize the transition-state, and His-119 provides the proton needed to liberate the glucose moiety (Fig. 2). Of these residues, all but Arg-76 (in helix-2), are predicted to lie on the luminal side of the ER (Fig. 1), confirming the view that the active site of G6PC is not accessible from the cytoplasm [Nilsson et al., 1978]. Therefore, the generation of glucose from G6P hydrolysis is dependent upon the availability of G6P in the ER lumen. This places a second control on glucose homeostasis at the level of transport of cytoplasmic G6P across the ER membrane into the lumen. This process is mediated by the G6P transporter, G6PT [Chou et al., 2002; Chou and Mansfield 2003]. Deficiencies in G6PT cause a second subtype of GSD-I, GSD-Ib which represents approximately 20% of GSD-I cases. G6PC and G6PT are co-dependent, in that mutation of either leads to inactivation of the other [Lei et al., 1996; Hiraiwa et al., 1999] and, as expected, GSD-Ib patients exhibit identical metabolic abnormalities as GSD-Ia patients, although they also manifest the additional clinical complications of neutropenia, myeloid dysfunction, and inflammatory bowl disease [Visser et al., 2000; Dieckgraefe et al., 2002; Chou and Mansfield 2003].
Fig. 2.
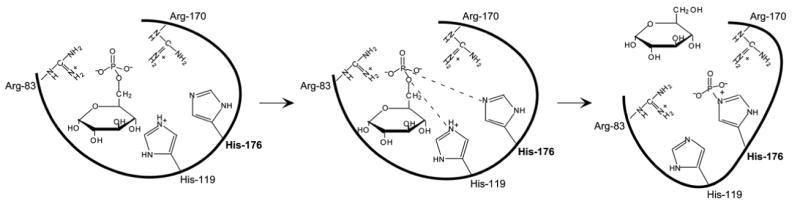
Proposed roles of Arg-83, His-119, Arg-170, and His-176 in the G6PC reaction mechanism. The single thick line represents the general backbone of the protein, which lies within the ER membrane.
Mutations of the G6PC Gene
To date, 84 separate G6PC gene mutations have been identified out of approximately 550 GSD-Ia patients (Table 1). These include 54 missense, 10 nonsense, 17 insertion/deletion, and 3 splicing mutations spread through the coding and exon-intron junction regions (Fig. 3). The initial mutation studies of G6PC, performed before antibodies against G6PC were available and the protein's secondary structure had been elucidated, focused on the catalytic impact of missense mutations [Lei et al., 1993, 1994, 1995a, 1995b; Bruni et al., 1999; Takahashi et al., 2000]. Subsequently, the topology of G6PC was established by Pan and coworkers [1998a, 1998b] showing that the protein is anchored in the ER by 9 transmembrane helices (Fig. 1). Using FLAG-tagged G6PC mutants and an anti-FLAG antibody, Shieh and coworkers [2002] functionally characterized 48 naturally occurring G6PC missense mutations along with the p.F327del mutation by enzymatic activity assays as well as by immunoblot analysis. They grouped these mutations into three categories – active site, helical and non-helical - based on their predicted topological position (Fig. 1). The active site mutations were assigned based on the designated catalytic site; the helical mutations were assigned based on the proposed transmembrane domains and the non-helical mutations were assigned based on the proposed luminal or cytoplasmic locations in G6PC (Fig. 4). Later, Ghosh et al. [2002] characterized the p.H176A active site mutant and Angaroni et al. [2004] characterized 2 additional missense mutations, p.T16R and p.Y209C in a similar fashion (Fig. 4).
Table 1.
G6PC mutations accounting for GSD-Ia
| Mutation type | Nucleotide change | Amino acid or other change | Exon | Domain | Reference |
|---|---|---|---|---|---|
| Missense | c.14T>G | p.M5R | I | NH2 | Trioche et al. [2000] |
| c.46A>G | p.T16A | I | NH2 | Wu et al. [2000a] | |
| c.47C>G | p.T16R | I | NH2 | Angaroni et al. [2004] | |
| c.59A>G | p.Q20R | I | NH2 | Seydewitz and Matern [2000] | |
| c.113A>T | p.D38V | I | H1 | Chevalier-Porst et al. [1996]; Parvari et al. [1997b] | |
| c.161A>C | p.Q54P | I | C1 | Trioche et al. [2000] | |
| c.187T>C | p.W63R | I | H2 | Stroppiano et al. [1999] | |
| c.193G>C | p.A65P | I | H2 | Matern et al. [2002] | |
| c.202G>A | p.G68R | I | H2 | Reis et al. [2001] | |
| c.228G>C | p.K76N | I | H2 | Kozek et al. [2000] | |
| c.229T>C | p.W77R | I | H2 | Chevalier-Porst et al. [1996] | |
| c.241G>C | p.G81R | II | H2 | Seydewitz and Matern [2000] | |
| c.247C>T | p.R83C | II | AS | Lei et al. [1993]; Parvari et al. [1995] | |
| c.248G>A | p.R83H | II | AS | Lei et al. [1995a]; Hwu et al. [1995] | |
| c.323C>T | p.T108I | II | L1 | Trioche et al. [2000] | |
| c.328G>A | p.E110K | II | L1 | Chevalier-Porst et al. [1996] | |
| c.328G>C | p.E110Q | II | L1 | Parvari et al. [1997b] | |
| c.332C>T | p.T111I | II | L1 | Trioche et al [2000] | |
| c.338C>T | p.P113L | II | L1 | Seydewitz and Matern [2000] | |
| c.356A>T | p.H119L | III | AS | Wu et al. [2000b] | |
| c.365G>A | p.G122D | III | H3 | Akanuma et al. [2000]; Goto et al. [2000] | |
| c.370G>A | p.A124T | III | H3 | Chevalier-Porst et al. (1996) | |
| c.466G>T | p.W156L | IV | H4 | Seydewitz and Matern [2000] | |
| c.497T>C | p.V166A | IV | H4 | Kozek et al. [2000] | |
| c.497T>G | p.V166G | IV | H4 | Parvari et al. [1995; 1997a] | |
| c.509G>A | p.R170Q | IV | AS | Rake et al. [2000] | |
| c.518T>C | p.L173P | IV | L2 | Li et al. [2007] | |
| c.530T>G | p.F177C | IV | H5 | Matern et al. [2002] | |
| c.532C>G | p.P178A | IV | H5 | Ki et al. [2004] | |
| Missense | c.532C>T | p.P178S | IV | H5 | Lei et al. [1995a] |
| c.536A>C | p.H179P | IV | H5 | Akanuma et al. [2000] | |
| c.551G>A | p.G184E | IV | H5 | Chevalier-Porst et al. (1996) | |
| c.551G>T | p.G184V | IV | H5 | Stroppiano et al. [1999] | |
| c.563G>T | p.G188D | IV | H5 | Seydewitz and Matern [2000] | |
| c.563G>C | p.G188R | IV | H5 | Chevalier-Porst et al. [1996] | |
| c.563G>A | p.G188S | IV | H5 | Lei et al. (1995a) | |
| c.616 A>G | p.Y209C | V | H6 | Angaroni et al. [2004] | |
| c.632T>C | p.L211P | V | H6 | Chevalier-Porst et al. [1996] | |
| c.664G>A | p.G222R | V | H6 | Stroppiano et al [1999] | |
| c.664G>C | p.G222R | V | H6 | Lei et al. [1995b]; Parvari et al. [1997b] | |
| c.706T>A | p.W236R | V | L3 | Lei et al. [1995a] | |
| c.721G>A | p.A241T | V | L3 | Trioche et al [2000] | |
| c.764C>T | p.T255I | V | L3 | Chou et al. [2002] | |
| c.770C>T | p.P257L | V | L3 | Akanuma et al. [2000] | |
| c.792C>A | p.N264K | V | H7 | Keller et al. [1998] | |
| c.794T>C | p.L265P | V | H7 | Seydewitz and Matern [2000] | |
| c.797G>T | p.G266V | V | H7 | Rake et al. [2000] | |
| c.808G>C | p.G270R | V | H7 | Trioche et al [2000] | |
| c.809G>T | p.G270V | V | H7 | Lei et al. [1995a]; Chevalier-Porst et al. [1996] | |
| c.883C>T | p.R295C | V | H8 | Lei et al. [1993] | |
| c.892T>C | p.S298P | V | H8 | Stroppiano et al. [1999] | |
| c.964T>C | p.F322L | V | H9 | Trioche et al [2000] | |
| c.1012G>T | p.V338F | V | H9 | Stroppiano et al. [1999] | |
| c.1022T>A | p.I341N | V | H9 | Lee et al. [1996] | |
| c.[1034T>G; 1035C>A] | p.L345R | V | H9 | Lei et al. [1995a] | |
| Nonsense | c.70C>T | p.Q24X | I | NH2 | Rocha et al [2000] |
| c.149G>A | p.W50X | I | C1 | Seydewitz and Matern [2000] | |
| c.189G>A | p.W63X | I | H2 | Lei et al. [1995a] | |
| c.209G>A | p.W70X | I | H2 | Trioche et al. [2000] | |
| c.310C>T | p.Q104X | II | L1 | Wong et al. [2001] | |
| c.384C>A | p.Y128X | III | H3 | Ki et al. [2004] | |
| c.508C>T | p.R170X | IV | AS | Akanuma et al. [2000]; Rake et al. [2000] | |
| c.516C>A | p.Y172X | IV | L2 | Stroppiano et al. [1999] | |
| c.724C>T | p.Q242X | V | L3 | Lei et al. [1995a] | |
| c.1039C>T | p.Q347X | V | COOH | Lei et al. [1994]; Chevalier-Porst et al. [1996] | |
| Insertion/Deletion | c.18_19insTGAA | p.V7Xfs | I | Stroppiano et al. [1999] | |
| c.79delC | p.Q27Xfs | I | Lei et al. [1995a]; Chevalier-Porst et al. [1996] | ||
| c.96_97delGG | p.L32Xfs | I | Rake et al. [2000] | ||
| c.150_151delGT | p.W50Xfs | I | Meaney et al. [2001] | ||
| c.262delG | p.V88Xfs | II | Lam et al. [1998] | ||
| c.380_381insTA | p.Y127Xfs | III | Lei et al. [1995b] | ||
| c.[439_440delAinsTG] | p.R147Xfs | III | Stroppiano et al. [1999] | ||
| c.461_465delTTTTG | p.I154Xfs | IV | Kozek et al. [2000] | ||
| c.537delT | p.H179Xfs | IV | Goto et al. [2000] | ||
| c.714delG | p.L238Xfs | V | Trioche et al [2000] | ||
| c.734_735insG | p.C245Xfs | V | Chevalier-Porst et al. [1996] | ||
| c.[734_735insC; 743delC] | p.W244_E249del CEQPinsWRAA | V | Lei et al. [1995a] | ||
| c.757delA | p.I253fs | V | Chou et al. [2002] | ||
| c.788delA | p.K263Xfs | V | Rake et al. [2000] | ||
| c.793delC | p.L265Xfs | V | Trioche et al. [2000] | ||
| c.854_855insAA | p.K285fs | V | Chiang et al. [2000] | ||
| c.979_981delTTC | p.F327del | V | Lei et al. [1995b]; Chevalier-Porst et al. [1996] | ||
| Splicing | c.230+4A>G | IVS1+4A>G | Intron 1 | Chevalier-Porst et al. [1996] | |
| c.231-1G>A | IVS1-1G>A | Intron 1 | Akanuma et al. [2000] | ||
| c.648G>T | c.563_653 del91 frameshift | V | Kajihara et al. [1995]; Okubo et al. [1997]; Lam et al. [1998] |
Numbering according to cDNA sequence in Lei et al. [1993], GeneBank Accession Number U01120. Nucleotide +1 is the A of the ATG initiation codon (which is codon +1). The domains for missense and nonsense mutations are based on the finding that human G6PC is anchored in the ER by 9 transmambrane helices [Pan et al., 1998a, 1998c]. NH2, amino terminal domain; H, helical transmembrane domain; C, cytoplasmic loop; L, luminal loop; AS, active site; COOH, carboxyl terminal domain.
Fig. 3.

Mutations identified in the G6PC gene of GSD-Ia patients. The G6PC gene is shown as a line diagram with the 5 exons marked as boxes I to V. Black boxes represent coding regions, white boxes the 5′ and 3′ untranslated regions of the G6PC transcript. The positions of all known mutations are listed from left to right as insertion/deletion, nonsense, splicing, and missense mutations.
Fig. 4.

A summary of mutations of G6PC that affect phosphohydrolase activity. The G6PC protein is represented by a line diagram, with the 9 helical transmembrane domains marked as boxes H1 to H9. Protein mutations that destroy G6PC activity are listed. Mutants retaining some residual activity are listed with the percent of wild-type enzymatic activity retained in parentheses. The active site mutant H176A, show in italic, is not naturally occurring.
These studies revealed six key findings. Firstly, mutations in the active site residues, namely p.R83C, p.R83H, p.H119L, p.R170Q, and p.H176A completely abolish G6PC enzymatic activity, consistent with their proposed role. Secondly, Western blot analysis showed that the active site mutants supported the synthesis of similar levels of G6PC protein compared to the wild-type enzyme [Shieh et al., 2002; Ghosh et al., 2002], indicating that the active site residues played no essential role in the stability of the enzyme. Thirdly, among the 32 helical mutations, 23 (72%) completely abolish G6PC activity, 7 retain residual activity, while two (p.G122D in helix-3, 28.2%; p.F322L in helix-9, 17.4%) retain significant activity. The p.G122D and p.F322L mutants have similar stability to wild-type G6PC, suggesting that helix-3 is more flexible and tolerant of changes. Fourthly, the majority (61%) of helical mutants, including p.D38V (helix-1), p.W63R/p.G68R (helix-2), p.V166A (helix-4), p.G188D/p.G188S/p.G188R (helix-5), p.Y209C/p.L211P/p.G222R (helix-6), p.N264K/p.L265P/p.G266V/p.G270V/p.G270R (helix-7), p.R295C/p.S298P (helix-8), and p.F327del/p.V338F/p.I341N/p.L345R (helix-9) supported the synthesis of reduced levels of G6PC protein compared to the wild-type enzyme, indicating these mutations destabilize G6PC [Shieh et al., 2002; Angaroni et al., 2004]. The studies also show that the structural integrity of transmembrane helices is critical for the correct folding, stability and enzymatic activity of G6PC. Fifthly, eight (57%) of the 14 non-helical mutations retain residual G6PC activity with 4 mutants retaining 15% or more of wild-type G6PC activity, implying the non-helical regions of the lumen and cytoplasm are less critical to activity. Finally, the non-helical mutations play no essential role in the stability of the enzyme [Shieh et al., 2002], with the exception of the T16R mutation [Angaroni et al., 2004], which does decrease stability. Presently, 4 of the naturally occurring missense mutations, p.A65P [Matern et al., 2002], p.L173P [Li et al., 2007], p.F177C [Matern et al., 2002], and p.P178A [Ki et al., 2004] remain to be characterized functionally.
Among the 10 naturally occurring nonsense mutations identified, p.R170X [Takahashi et al., 2000] and p.Q347X [Lei et al., 1994] have been functionally characterized and both completely inactive the G6PC enzyme. Since the p.Q347X mutant lacking 11 amino acids at the carboxyl terminus is devoid of enzymatic activity, this suggests that the remaining 8 nonsense mutations in G6PC (Table 1) predicted to encode polypeptides of 241 amino acids or shorter should also be null mutations. Progressive deletions in the form of p.K355X, p.H353X, p.Q351X, p.G350X, and p.L349X mutations lacking, respectively, 3, 5, 7, 8, and 9 amino acids at the C-terminus retain 65%, 60%, 43%, 41.5% and 5.3%, respectively, of wild-type G6PC activity [Lei et al., 1995c]. The consistent decline with the severity of deletion for the first 8 residues suggests they contribute moderately to activity. Within these residues lies the ER retention motif, KKSL [Jackson et al., 1990] between residues 354 to 357. Since the G6PC mutants p.G350X, p.Q351X, and p.H353X exhibit latency and thermal stability indistinguishable from that of the wild-type enzyme [Lei et al., 1995c], these results suggest that the ER retention signal is not required for ER localization of G6PC or enzymatic activity. This is also true for G6PC3 which is localized to the ER membrane despite the absence of an apparent ER retention motif [Shieh et al., 2003]. The sudden drop in activity upon deletion of Leu-349 in G6PC however, suggests it is a more critical residue.
Two naturally occurring insertion/deletion mutations, p.W244_E249delCEQPinsWRAA and p.F327del have been functionally characterized and shown to be devoid of enzymatic activity [Lei et al., 1995a, 1995b]. The remaining 15 insertion/deletion mutations are frameshift mutations (Table 1) predicted to encode polypeptides of 6 to 300 amino acids which are expected to be devoid of G6PC enzymatic activity, although these remain to be tested experimentally.
Three natural occurring splicing mutations, c.230+4A>G, c.231-1G>A, and c.648G>T have been identified (Table 1). The c.231-1G>A mutation causes exon 2 skipping [Akanauma et al., 2000] and the c.648G>T mutation results in a 91-nt deletion in exon 5 [Kajihara et al., 1995] encoding a severely truncated polypeptide of 201 amino acids [Okubo et al., 1997]. Both mutations are predicted to inactivate G6PC activity, but have yet to be tested experimentally.
While GSD-Ia is not restricted to any one ethnic group, mutations unique to Caucasian/Hispanic [Chevalier-Porst et al., 1996; Hinds and Burchell, 1996; Matern et al., 1996; Parvari et al., 1997b, 1999; Keller et al., 1998; Huner et al., 1998; Bruni et al., 1999; Linnebank et al., 1999; Stroppiano et al., 1999; Kozak et al., 2000; Rake et al., 2000a, 2000b; Rocha et al., 2000; Seydewitz and Matern, 2000; Trioche et al., 2000; Weston et al., 2000; Meaney et al., 2001; Reis et al., 2001; Terzioglu et al., 2001; Miltenberger-Miltenyi et al., 2005], Chinese [Hwu et al., 1995, 1999; Lee et al., 1996; Lam et al., 1998; Chiang et al., 2000; Wu et al., 2000a, 2000b; Wong et al., 2001; Qiu et al., 2003; Li et al., 2007], Japanese [Kajihara et al., 1995; Okubo et al., 1997; Karasawa et al., 1998; Akanuma et al., 2000; Takahashi et al., 2000; Nakamura et al., 2001], Korean [Goto et al., 2000; Ki et al., 2004], and Jewish [Lei et al., 1995a; Parvari et al., 1995, 1997a; Ekstein et al., 2004] GSD-Ia patients have been described (Table 2). The prevalent mutations identified in the 676 alleles from Caucasian GSD-Ia patients are p.R83C (33%) and p.Q347X (18%). Of the 112 alleles characterized from Chinese patients the prevalent mutations are p.R83H (26%) and c.648G>T (54%). Amongst the 172 Japanese alleles and 28 Korean alleles from GSD-Ia patients, there is a common prevalent mutation, c.648G>T, accounting for 91% and 75% of the mutations, respectively. The common 648G>T mutation found in Chinese, Japanese, and Korean might reflect the genetic relationship between these three races. The prevalent mutation in the 24 Hispanics alleles characterized is c.380_381insTA, representing 54% of the mutations. Out of 94 Jewish alleles characterized, all but two are p.R83C, with p.Q347X making up the balance. Indeed, GSD-Ia is particularly common in the Ashkenazi Jewish population where the carrier frequency for the p.R83C mutation is 1.4% [Ekstein et al., 2004].
Table 2.
Prevalent G6PC mutations in GSD-Ia patients
| Caucasian | Chinese | Japanese | Korean | Hispanic | Jewish |
|---|---|---|---|---|---|
| p.R83C (33%) | p.R83H (26%) | c.648G>T (91%) | c.648G>T (75%) | c.380_381insTA (54%) | p.R83C (98%) |
| p.Q347X (18%) | c.648G>T (54%) | p.Q347X (2%) |
Genogype-Phenotype Correlation
GSD-Ia patients exhibit phenotypic heterogeneity and there is little evidence for a stringent genotype-phenotype relationship for each GSD-Ia gene mutation. However, several reports suggest some mutations are more commonly associated with more or less severe phenotypes. For example, Nakamura et al. [2001] reported a patient first diagnosed as GSD-Ia when 40 years old, who presented with “liver dysfunction and a liver tumour.” Just two years later the patient had hepatocellular carcinoma. Other causes for hepatocellular carcinoma, such as alcohol abuse, hepatitis B, hepatitis C, and loss of heterozygosity for the p53 gene were excluded. Genotyping revealed that the patient was homozygous for the c.648G>T splicing mutation which led the authors to speculate that this particular mutation is associated with an increased risk for hepatocellular carcinoma. A Chinese female patient with a similarly mild disease history and homozygous for c.648G>T was diagnosed with an incurable hepatocellular carcinoma when 43 years old [Matern et al., 2002] which would support this view. However, in a larger study, genotype-phenotype correlation was carefully examined in 40 GSD-Ia patients homozygous for the c.648G>T mutation [Akanuma et al., 2000]. They showed that the age of disease onset, the severity of the symptoms, and complications including the appearance of hepatic adenoma and carcinoma varied greatly among the 40 patients, indicating that the genotype of the G6PC is not the sole determinant of clinical severity. However, the authors noted none of the 40 patients with this genotype had suffered from severe hypoglycemia in infancy, suggesting that the c.648G>T splicing mutation, common in Eastern Asian, may confer a milder GSD-Ia phenotype with respect to recurrent hypoglycemia. A milder phenotype would, on the surface appear a surprising characteristic for this mutation because it is predicted to encode a severely truncated polypeptide of 201 amino acids which should be devoid of G6PC activity. One possible explanation could lie in the observation that many splicing mutations are not efficient, and can allow leaky expression of the enzymatically active native protein. Arguing against this, however is the observation that in at least one homozygous GSD-Ia c.648G>T patient, PCR amplification of liver biopsy RNA failed to detect the wild-type G6PC transcript, which would suggest there was no hepatic G6PC activity in this patient [Kajihara et al., 1995].
A similar, inconsistent observation has been reported for a Japanese patient homozygous for the p.P257L mutation which retains only 6.1% of wild-type G6PC activity. The patient is reported to never experience a symptomatic hypoglycemic episode and not require dietary therapy [Takahashi et al., 2000]. Similarly, a compound heterozygous patient having p.R83C and p.N264K null mutations does not manifest significant fasting hypoglycemia [Keller et al., 1998]. In contrast, a compound heterozygous patient with p.E110Q and p.G222R mutations that retain 16% and 2.6%, respectively of wild-type G6PC activity manifest typical severe symptoms of GSD-Ia [Parvari et al., 1997b]. Rake et al. [2000] also reported variable phenotypes among affected siblings with the same G6PC genotype. This may point either to the inadequacy of the in vitro expression systems used to assess function, or to the presence of modifiers that can compensate for or stabilize low level expression in vivo.
There is a report of GSD-Ia patients exhibiting the myeloid dysfunction usually considered a characteristic phenotype of GSD-Ib, not GSD-Ia [Weston et al., 2000]. The patients carry a homozygous p.G188R null mutation in the G6PC gene yet manifest mild neutropenia. When isolated, their neutrophils exhibit decreased oxidative burst activity, impaired chemotaxis, and defective killing of E. coli, all phenotypical of GSD-Ib [Chen 2001; Chou et al., 2002; Chou and Mansfield 2003]. Mutations in the G6PT gene were not detected and this unusual phenotype is not encountered in compound heterozygous GSD-Ia patients carrying p.G188R. Again, the cause for this unusual phenotype is currently unknown, but may lie in the functional co-dependence of the G6PC and G6PT (Lei et al., 1996; Hiraiwa et al., 1999].
Animal Models
There are mouse [Lei et al., 1996] and dog [Kishnani et al., 1997] models of GSD-Ia. Both animals are physiologically similar to humans with respect to G6P metabolism. The G6PC-deficient mice, generated by gene targeting, manifest all of the symptoms of human GSD-Ia - hypoglycemia, growth retardation, hepatomegaly, nephromegaly, hyperlipidemia, and hyperuricemia, and mild lactic acidemia [Lei et al., 1996]. The GSD-Ia dog identified originally was a Maltese breed carrying a natural p.M121I G6PC mutation that retains 6.6% of wild-type activity when measured by transient expression assays [Kishnani et al., 1997]. However, because the Maltese breed is small in size, exhibits low survival rate of newborns, and has a small litter size, a new dog model was established by crossbreeding a carrier Maltese with Beagles [Kishnani et al., 2001]. The two canine models manifest all the typical symptoms of the human disorder, including hyperlactacidemia [Kishnani et al., 1997; 2001]. Both mouse and canine models are being used to further our understanding of the biology and pathophysiology of GSD-Ia, to develop novel therapies for this disorder, and to monitor the long-term complications of GSD-Ia. The phenotypic similarity between the GSD-Ia mice, carrying a null mutation, and the GSD-Ia dog carrying a mutation conferring 6.6% of normal G6PC activity indicates that to correct the GSD-Ia disorder, more than 7% of normal G6PC activity must be restored in the liver. Consistent with this, our recent gene therapy study has shown that sustained restoration of approximately 11% of normal hepatic G6PC activity is adequate to maintain glucose homeostasis in GSD-Ia mice [Ghosh et al., 2006]. However, liver and kidney recovery differ. The study also showed that sustained restoration of 7% of normal renal G6PC activity could not prevent the development of kidney disease in GSD-Ia mice [Ghosh et al., 2006]. For the study of hepatic adenomas, which develop in 50% of GSD-Ia patients in the second or third decade of life [Bianchi 1993; Labrune et al., 1997; Lee 2002], the existing animal models have not been useful, primarily because the dietary therapies used to sustain the life of GSD-Ia animals have not yet been refined to prevent premature death of these animals from hypoglycemia seizures.
Future Prospects
Despite many advances at the molecular genetic level, there are still a number of inconsistencies in GSD-Ia that remain to be resolved. The phenotypic heterogeneity and the lack of a stringent genotype-phenotype in GSD-Ia raise the possibility of the existence of genetic modifiers which may modify the phenotype. However, clear experimental evidence for the presence of such modifiers, and their identities remain to be addressed. Moreover, even with our current understanding of the mutations and their consequences, there are no alternatives to the current standard of care. The primary complications of GSD-Ia are a loss of glucose homeostasis due to the loss of functional G6PC in the liver and kidney, followed by secondary renal and liver disease. However, the etiology of renal and liver disease in GSD-Ia remains unclear. The renal biopsies of GSD-Ia patients reveal interstitial fibrosis, tubular atrophy, and focal segmental glomerulosclerosis [Chen et al., 1988; Baker et al., 1989]. Most hepatic adenomas seen in GSD-Ia patients tend to be small, multiple, and nonencapsulated, but in approximately 10% patients these benign lesions can undergo malignant transformation [Bianchi 1993; Labrune et al., 1997; Lee 2002]. The animal models of GSD-Ia are now being used to delineate the molecular mechanisms responsible for the pathogenesis of this disorder. Using GSD-Ia mice, we have recently shown that at least one mechanism underlying the nephropathy occurring in GSD-Ia involves angiotensin system-mediated renal fibrosis [Yiu et al., 2008]. The animal models of GSD-Ia also offer the opportunity to explore somatic gene therapy. While promising in animals [Beaty et al., 2002; Sun et al., 2002; Ghosh et al., 2006; Koeberl et al., 2006], gene therapy still remains distant for human patients.
Acknowledgments
This research was supported in part by the Intramural Research Programs of the NICHD, NIH.
References
- Akanuma J, Nishigaki T, Fujii K, Matsubara Y, Inui K, Takahashi K, Kure S, Suzuki Y, Ohura T, Miyabayashi S, Ogawa E, Iinuma K, Okada S, Narisawa K. Glycogen storage disease type Ia: molecular diagnosis of 51 Japanese patients and characterization of slicing mutations by analysis of ectopically transcribed mRNA from lymphoblastoid cells. Am J Med Genet. 2000;91:107–112. [PubMed] [Google Scholar]
- Angaroni CJ, Dodelson-Kremer R, Argaraña CE, Paschini-Capra AE, Giner-Ayala AN, Pezza RJ, Pan CJ, Chou JY. Glycogen Storage Disease Type Ia in Argentina. Two novel Glucose-6-phosphatase mutations affecting protein stability. Mol Genet Metab. 2004;83:276–279. doi: 10.1016/j.ymgme.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Angaroni CJ, Labrune P, Petit F, Sastre D, Capra AE, Dodelson de Kremer R, Argaraña CE. Glycogen storage disease type Ib without neutropenia generated by a novel splice-site mutation in the glucose-6-phosphate translocase gene. Mol Genet Metab. 2006;88:96–99. doi: 10.1016/j.ymgme.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Annabi B, Hiraiwa H, Mansfield BC, Lei KJ, Ubagai T, Polymeropoulos MH, Moses SW, Parvari R, Hershkovitz E, Mandel H, Frydman M, Chou JY. The gene for glycogen storage disease type 1b maps to chromosome 11q23. Am J Hum Genet. 1998;62:400–405. doi: 10.1086/301727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L, Dahlem S, Goldfarb S, Kern EF, Stanley CA, Egler J, Olshan JS, Heyman S. Hyperfiltration and renal disease in glycogen storage disease, type I. Kidney Int. 1989;35:1345–1350. doi: 10.1038/ki.1989.133. [DOI] [PubMed] [Google Scholar]
- Beaty RM, Jackson M, Peterson D, Bird A, Brown T, Benjamin DK, Jr, Juopperi T, Kishnani P, Boney A, Chen YT, Koeberl DD. Delivery of glucose-6-phosphatase in a canine model for glycogen storage disease, type Ia, with adeno-associated virus (AAV) vectors. Gene Ther. 2002;9:1015–1022. doi: 10.1038/sj.gt.3301728. [DOI] [PubMed] [Google Scholar]
- Bianchi L. Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr. 1993;52 1:S63–S70. doi: 10.1007/BF02072092. [DOI] [PubMed] [Google Scholar]
- Bruni N, Rajas F, Montano S, Chevalier-Porst F, Maire I, Mithieux G. Enzymatic characterization of four new mutations in the glucose-6-phosphatae (G6PC) gene which cause glycogen storage disease type Ia. Ann Hum Genet. 1999;63:141–146. doi: 10.1046/j.1469-1809.1999.6320141.x. [DOI] [PubMed] [Google Scholar]
- Chen LY, Lin B, Pan CJ, Hiraiwa H, Chou JY. Structural requirements for the stability and microsomal transport activity of the human glucose-6-phosphate transporter. J Biol Chem. 2000;275:34280–34286. doi: 10.1074/jbc.M006439200. [DOI] [PubMed] [Google Scholar]
- Chen LY, Pan CJ, Shieh JJ, Chou JY. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Hum Mol Genet. 2002;11:3199–3207. doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 1521–1551. [Google Scholar]
- Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen storage disease. N Engl J Med. 1984;310:171–175. doi: 10.1056/NEJM198401193100306. [DOI] [PubMed] [Google Scholar]
- Chen YT, Coleman RA, Scheinman JI, Kolbeck PC, Sidbury JB. Renal disease in type I glycogen storage disease. N Engl J Med. 1988;318:7–11. doi: 10.1056/NEJM198801073180102. [DOI] [PubMed] [Google Scholar]
- Chevalier-Porst F, Bozon D, Bonardot AM, Bruni N, Mithieux G, Mathieu M, Maire I. Mutation analysis in 24 French patients with glycogen storage disease type 1a. J Med Genet. 1996;33:358–360. doi: 10.1136/jmg.33.5.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SC, Lee YM, Chang MH, Wang TR, Ko TM, Hwu WL. Glucosee-6-phosphatase gene mutations in Taiwan Chinese patients with glycogen storage disease type Ia. J Hum Genet. 2000;45:197–199. doi: 10.1007/s100380070026. [DOI] [PubMed] [Google Scholar]
- Chou JY, Mansfield BC. Molecular genetics of type 1 glycogen storage diseases. Trend Endocrinol Metab. 1999;10:104–113. doi: 10.1016/s1043-2760(98)00123-4. [DOI] [PubMed] [Google Scholar]
- Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- Chou JY, Mansfield BC. Glucose-6-phosphate transporter: the key to glycogen storage disease type Ib. In: Broer S, Wagner CA, editors. Membrane Transporter Diseases. New York: Kluwe Academic/Plenum Publishers; 2003. pp. 191–205. [Google Scholar]
- Daublin G, Schwahn B, Wendel U. Type I glycogen storage disease: favorable outcome on a strict management regimen avoiding increased lactate production during childhood and adolescence. Eur J Pediatr. 2002;161 1:S40–S45. doi: 10.1007/s00431-002-1001-1. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109:121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- Dieckgraefe BK, Korzenik JR, Husain A, Dieruf L. Association of glycogen storage disease 1b and Crohn disease: results of a North American survey. Eur J Pediatr. 2002;161 1:S88–S92. doi: 10.1007/s00431-002-1011-z. [DOI] [PubMed] [Google Scholar]
- Ekstein J, Rubin BY, Anderson SL, Weinstein DA, Bach G, Abeliovich D, Webb M, Risch N. Mutation frequencies for glycogen storage disease Ia in the Ashkenazi Jewish population. Am J Med Genet. 2004;129A:162–164. doi: 10.1002/ajmg.a.30232. [DOI] [PubMed] [Google Scholar]
- Feldman F, Butler LG. Protein-bound phosphoryl histidine: a probable intermediate in the microsomal glucose-6-phosphatase-inorganic pyrophosphatase reaction. Biochim Biophys Acta. 1972;268:698–710. doi: 10.1016/0005-2744(72)90274-4. [DOI] [PubMed] [Google Scholar]
- Galli L, Orrico A, Marcolongo P, Fulceri R, Burchell A, Melis D, Parini R, Gatti R, Lam C, Benedetti A, Sorrentino V. Mutations in the glucose-6-phosphate transporter (G6PT) gene in patients with glycogen storage diseases type 1b and 1c. FEBS Lett. 1999;459:255–258. doi: 10.1016/s0014-5793(99)01248-x. [DOI] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF, Van Schaftingen E. Sequence of a putative glucose-6-phosphate translocase, mutated in glycogen storage disease type 1b. FEBS Lett. 1997;419:235–238. doi: 10.1016/s0014-5793(97)01463-4. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh JJ, Pan CJ, Sun MS, Chou JY. The catalytic center of glucose-6-phosphatase: His176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2002;277:32837–32842. doi: 10.1074/jbc.M201853200. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh JJ, Pan CJ, Chou JY. Histidine-167 is the phosphate acceptor in glucose-6-phosphatase-β forming a phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2004;279:12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Allamarvdasht M, Pan CJ, Sun MS, Mansfield BC, Byrne BJ, Chou JY. Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer. Gene Ther. 2006;13:321–329. doi: 10.1038/sj.gt.3302650. [DOI] [PubMed] [Google Scholar]
- Goto M, Taki T, Sugie H, Miki Y, Kato H, Hayashi Y. A novel mutation in the glucose-6-phosphatase gene in Korean twins with glycogen storage disease type Ia. J Inherit Metab Dis. 2000;23:851–852. doi: 10.1023/a:1026777106212. [DOI] [PubMed] [Google Scholar]
- Greene HL, Slonim AE, O'Neill JA, Jr, Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N Engl J Med. 1976;294:423–425. doi: 10.1056/NEJM197602192940805. [DOI] [PubMed] [Google Scholar]
- Guionie O, Clottes E, Stafford K, Burchell A. Identification and characterization of a new human glucose-6-phosphatase isoform. FEBS Lett. 2003;551:159–164. doi: 10.1016/s0014-5793(03)00903-7. [DOI] [PubMed] [Google Scholar]
- Hemrika W, Wever R. A new model for the membrane topology of glucose-6-phosphatase: the enzyme involved in von Gierke disease. FEBS Lett. 1997;409:317–319. doi: 10.1016/s0014-5793(97)00530-9. [DOI] [PubMed] [Google Scholar]
- Hemrika W, Renirie R, Dekker HL, Barnett P, Wever R. From phosphatases to vanadium peroxidases: a similar architecture of the active site. Proc Natl Acad Sci USA. 1997;94:2145–2149. doi: 10.1073/pnas.94.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds CJ, Burchell A. The inheritance of a novel mutation in glycogen storage disease type 1a. Biochem Soc Trans. 1996;24:232S. doi: 10.1042/bst024232s. [DOI] [PubMed] [Google Scholar]
- Hiraiwa H, Pan CJ, Lin B, Moses SW, Chou JY. Inactivation of the glucose-6-phosphate transporter causes glycogen storage disease type 1b. J Biol Chem. 1999;274:5532–5536. doi: 10.1074/jbc.274.9.5532. [DOI] [PubMed] [Google Scholar]
- Huner G, Podskarbi T, Schutz M, Baykal T, Sarbat G, Shin YS, Demirkol M. Molecular aspects of glycogen storage disease type Ia in Turkish patients: a novel mutation in the glucose-6-phosphatase gene. J Inherit Metab Dis. 1998;21:445–446. doi: 10.1023/a:1005339616074. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Chuang SC, Tsai LP, Chang MH, Chuang SM, Wang TR. Glucose-6-phosphatase gene G327A mutation is common in Chinese patients with glycogen storage disease type 1a. Hum Mol Genet. 1995;4:1095–1096. doi: 10.1093/hmg/4.6.1095. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Chuang SC, Huaang SF, Chang MH, Wen WH, Wang TR. Hypercalcemia in glycogen storage disease type Ia: a case with R83C and 341delG mutations. J Inher Metab Dis. 1999;22:937–938. doi: 10.1023/a:1005651809892. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990;9:3153. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecke AR, Linder M, Erdel M, Mayatepek E, Moslinger D, Podskarbi T, Fresser F, Stockler-Ipsirogly S, Hoffmann GF, Utermann G. Mutation analysis in glycogen storage disease type 1 non-a. Hum Genet. 2000;107:285–289. doi: 10.1007/s004390000371. [DOI] [PubMed] [Google Scholar]
- Kajihara S, Matsuhashi S, Yamamoto K, Kido K, Tsuji K, Tanae A, Fujiyama S, Itoh T, Tanigawa K, Uchida M, Setoguchi Y, Motomura M, Mizuta T, Sakai T. Exon redefinition by a point mutation within exon 5 of the glucose-6-phosphatase gene is the major cause of glycogen storage disease type 1a in Japan. Am J Hum Genet. 1995;57:549–555. [PMC free article] [PubMed] [Google Scholar]
- Karasawa Y, Kobayashi M, Nakano Y, Aoki Y, Kawa S, Kiyosawa K, Seki H, Kawasaki S, Furihata K, Itoh N. A case of glycogen storage disease type Ia with multiple hepatic adenomas and G727T mutation in the glucose-6-phosphatase gene, and a comparison with other mutations previously reported. Am J Gastroenterol. 1998;93:1550–1553. doi: 10.1111/j.1572-0241.1998.00480.x. [DOI] [PubMed] [Google Scholar]
- Keller KM, Schutz M, Podskarbi T, Bindl L, Lentze MJ, Shin YS. A new mutation of the glucose-6-phosphatase gene in a 4-year-old girl with oligosymptomatic glycogen storage disease type 1a. J Pediat. 1998;132:360–361. doi: 10.1016/s0022-3476(98)70463-9. [DOI] [PubMed] [Google Scholar]
- Ki CS, Han SH, Kim HJ, Lee SG, Kim EJ, Kim JW, Choe YH, Seo JK, Chang YJ, Park JY. Mutation spectrum of the glucose-6-phosphatase gene and its implication in molecular diagnosis of Korean patients with glycogen storage disease type Ia. Clin Genet. 2004;65:487–489. doi: 10.1111/j.1399-0004.2004.00260.x. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Bao Y, Wu JY, Brix AE, Lin JL, Chen YT. Isolation and nucleotide sequence of canine glucose-6-phosphatase mRNA: identification of mutation in puppies with glycogen storage disease type 1a. Biochem Mol Med. 1997;61:168–177. doi: 10.1006/bmme.1997.2600. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Faulkner E, VanCamp S, Jackson M, Brown T, Boney A, Koeberl D, Chen YT. Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia) Vet Pathol. 2001;38:83–91. doi: 10.1354/vp.38-1-83. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Sun BD, Damodaran TV, Brown T, Millington DS, Benjamin DK, Jr, Bird A, Schneider A, Hillman S, Jackson M, Beaty RM, Chen YT. Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia. Gene Ther. 2006;13:1281–1289. doi: 10.1038/sj.gt.3302774. [DOI] [PubMed] [Google Scholar]
- Kozak L, Francova H, Hirabincova E, Stastna S, Peskova K, Elleder M. Identification of mutations in the glucose-6-phosphatase gene in Czech and Slovak patients with glycogen storage disease type Ia, including novel mutations K76N, V166A, and 540del5. Hum Mutat. 2000;16:89. doi: 10.1002/1098-1004(200007)16:1<89::AID-HUMU17>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Kure S, Hou DC, Suzuki Y, Yamagishi A, Hiratsuka M, Fukuda T, Sugie H, Kondo N, Matsubara Y, Narisawa K. Glycogen storage disease type Ib without neutropenia. J Pediatr. 2000;137:253–256. doi: 10.1067/mpd.2000.107472. [DOI] [PubMed] [Google Scholar]
- Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276–279. doi: 10.1097/00005176-199703000-00008. [DOI] [PubMed] [Google Scholar]
- Lam CW, But WM, Shek CC, Tong SF, Chan YS, Choy KW, Tse WY, Pang CP, Hjelm NM. Glucose-6-phosphatase gene (727G→T) splicing mutation is prevalent in Hong Kong Chinese patients with glycogen storage disease type 1a. Clin Genet. 1998;53:184–190. doi: 10.1111/j.1399-0004.1998.tb02674.x. [DOI] [PubMed] [Google Scholar]
- Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161 1:S46–S49. doi: 10.1007/s00431-002-1002-0. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Lee HM, Chi CS, Shu SG, Lin LY, Lin WH. Genetic analysis of the glucose-6-phosphatase mutation of type 1a glycogen storage disease in a Chinese family. Clin Genet. 1996;50:206–211. doi: 10.1111/j.1399-0004.1996.tb02627.x. [DOI] [PubMed] [Google Scholar]
- Lei KJ, Shelly LL, Pan CJ, Sidbury JB, Chou JY. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science. 1993;262:580–583. doi: 10.1126/science.8211187. [DOI] [PubMed] [Google Scholar]
- Lei KJ, Pan CJ, Shelly LL, Liu JL, Chou JY. Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. J Clin Invest. 1994;93:1994–1999. doi: 10.1172/JCI117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei KJ, Chen YT, Chen H, Wong LJC, Liu JL, McConkie-Rosell A, Van Hove JLK, Ou HCY, Yeh NJ, Pan LY, Chou JY. Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet. 1995a;57:766–771. [PMC free article] [PubMed] [Google Scholar]
- Lei KJ, Shelly LL, Lin B, Sidbury JB, Chen YT, Nordlie RC, Chou JY. Mutations in the glucose-6-phosphatase gene are associated with glycogen storage disease type 1a and 1aSP but not 1b and 1c. J Clin Invest. 1995b;95:234–240. doi: 10.1172/JCI117645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei KJ, Shelly LL, Pan CJ, Liu JL, Chou JY. Structure-function analysis of human glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. J Biol Chem. 1995c;270:11882–11886. doi: 10.1074/jbc.270.20.11882. [DOI] [PubMed] [Google Scholar]
- Lei KJ, Chen H, Pan CJ, Ward JM, Mosinger B, Lee EJ, Westphal H, Mansfield BC, Chou JY. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat Genet. 1996;13:203–209. doi: 10.1038/ng0696-203. [DOI] [PubMed] [Google Scholar]
- Li DZ, Liao C, Tang XW. Prenatal diagnosis of glycogen storage disease type Ia, presenting a new mutation in the glucose-6-phosphatase gene. Prenat Diagn. 2007;27:685–686. doi: 10.1002/pd.1764. [DOI] [PubMed] [Google Scholar]
- Linnebank M, Rapp B, Homberger A, Winter C, Marquardt T, Kunzel U, Koch HG. Homozygous state for a new single bp-deletion (g.787delA) in exon 5 of the glucose-6-phosphatase gene in a patient with early onset of glycogen storage disease type 1a. Hum Mut. 1999;13:414. [Google Scholar]
- Martens DH, Kuijpers TW, Maianski NA, Rake JP, Smit GP, Visser G. A patient with common glycogen storage disease type Ib mutations without neutropenia or neutrophil dysfunction. J Inherit Metab Dis. 2006;29:224–225. doi: 10.1007/s10545-006-0146-x. [DOI] [PubMed] [Google Scholar]
- Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC, O'Brien RM. Identification and characterization of a human cDNA and gene encoding a ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol. 2002;29:205–222. doi: 10.1677/jme.0.0290205. [DOI] [PubMed] [Google Scholar]
- Matern D, Niederhoff H, Brandis M, Chou JY. Verification of diagnosis in a 17-year old boy with clinical glycogen storage disease type 1a by mutational screening. J Inher Metab Dis. 1996;19:205–208. doi: 10.1007/BF01799430. [DOI] [PubMed] [Google Scholar]
- Matern D, Seydewitz HH, Bali D, Lang C, Chen YT. Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. Eur JPediatr. 2002;161 1:S10–S19. doi: 10.1007/s00431-002-0998-5. [DOI] [PubMed] [Google Scholar]
- Meaney C, Cranston T, Lee P, Genet S. A common 2 bp deletion mutation in the glucose-6-phosphatase gene in Indian patients with glycogen storage disease type Ia. J Inherit Metab Dis. 2001;24:517–518. doi: 10.1023/a:1010598109582. [DOI] [PubMed] [Google Scholar]
- Melis D, Fulceri R, Parenti G, Marcolongo P, Gatti R, Parini R, Riva E, Della Casa R, Zammarchi E, Andria G, Benedetti A. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr. 2005;164:501–508. doi: 10.1007/s00431-005-1657-4. [DOI] [PubMed] [Google Scholar]
- Miltenberger-Miltenyi G, Szonyi L, Balogh L, Utermann G, Janecke AR. Mutation spectrum of type I glycogen storage disease in Hungary. J Inherit Metab Dis. 2005;28:939–944. doi: 10.1007/s10545-005-0186-7. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ozawa T, Kawasaki T, Nakamura H, Sugimura H. Glucose-6-phosphatase gene mutations in 20 adult Japanese patients with glycogen storage disease type 1a with reference to hepatic tumors. J Gastroenterol Hepatol. 2001;16:1402–1408. doi: 10.1046/j.1440-1746.2001.02645.x. [DOI] [PubMed] [Google Scholar]
- Nilsson OS, Arion WJ, Depierre JW, Dallner G, Ernster L. Evidence for the involvement of a glucose-6-phosphate carrier in microsomal glucose-6-phosphatase activity. Eur J Biochem. 1978;82:627–634. doi: 10.1111/j.1432-1033.1978.tb12059.x. [DOI] [PubMed] [Google Scholar]
- Okubo M, Aoyama Y, Kishimoto M, Shishiba Y, Murase T. Identification of a point mutation (G727T) in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type 1a, and carrier screening in healthy volunteers. Clin Genet. 1997;51:179–183. doi: 10.1111/j.1399-0004.1997.tb02449.x. [DOI] [PubMed] [Google Scholar]
- Pan CJ, Lei KJ, Annabi B, Hemrika W, Chou JY. Transmembrane topology of glucose-6-phosphatase. J Biol Chem. 1998a;273:6144–6148. doi: 10.1074/jbc.273.11.6144. [DOI] [PubMed] [Google Scholar]
- Pan CJ, Kei KJ, Chen H, Ward JM, Chou JY. Ontogeny of the murine glucose-6-phosphatase system. Arch Biochem Biophys. 1998b;358:17–24. doi: 10.1006/abbi.1998.0849. [DOI] [PubMed] [Google Scholar]
- Pan CJ, Lei KJ, Chou JY. Asparagine-linked oligosaccharides are localized to a luminal hydrophilic loop in human glucose-6-phosphatase. J Biol Chem. 1998c;273:21658–21662. doi: 10.1074/jbc.273.34.21658. [DOI] [PubMed] [Google Scholar]
- Parvari R, Moses S, Hershkovitz E, Carmi R, Bashan N. Characterization of the mutations in the glucose-6-phosphatase gene in Israeli patients with glycogen storage disease type 1a: R83C in six Jews and a novel V166G mutation in a Muslim Arab. J Inher Metab Dis. 1995;18:21–27. doi: 10.1007/BF00711368. [DOI] [PubMed] [Google Scholar]
- Parvari R, Lei KJ, Bashan N, Hershkovitz E, Stanley HK, Barash V, Lerman-Sagie T, Mandel H, Chou JY, Moses S. Glycogen storage disease type 1a in Israel. Biochemical, clinical, and mutational studies. Am J Med Genet. 1997a;72:286–290. doi: 10.1002/(sici)1096-8628(19971031)72:3<286::aid-ajmg6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Parvari R, Lei KJ, Szonyi L, Narkis G, Moses S, Chou JY. Two new mutations in the glucose-6-phosphatase gene cause glycogen storage disease in Hungarian patients. Euro J Hum Genet. 1997b;5:191–195. [PubMed] [Google Scholar]
- Parvari R, Isam J, Moses SW. Glycogen storage disease type 1a in three siblings with the G270V mutation. J Inher Metab Dis. 1999;22:149–154. doi: 10.1023/a:1005445802822. [DOI] [PubMed] [Google Scholar]
- Qiu WJ, Gu XF, Ye J, Han LSh, Zhang YF, Liu XQ. Molecular genetic analysis of glycogen storage disease type Ia in 26 Chinese patients. J Inherit Metab Dis. 2003;26:811–812. doi: 10.1023/b:boli.0000009992.78840.77. [DOI] [PubMed] [Google Scholar]
- Rake JP, ten Berge AM, Verlind E, Visser G, Verlind E, Niezen-Koning KE, Buys CH, Smit GPA, Scheffer H. Glycogen storage disease type 1a: recent experience with mutation analysis, a summary of mutations reported in the literature and a newly developed diagnostic flowchart. Eur J Pediatr. 2000a;159:322–330. doi: 10.1007/s004310051281. [DOI] [PubMed] [Google Scholar]
- Rake JP, ten Berge AM, Visser G, Verlind E, Niezen-Koning KE, Buys CH, Smit GPA, Scheffer H. Identification of a novel mutation (867delA) in the glucose-6-phosphatase gene in two siblings with glycogen storage disease type Ia with different phenotypes. Hum Mutat. 2000b;15:381. doi: 10.1002/(SICI)1098-1004(200004)15:4<381::AID-HUMU13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161 1:S20–S34. doi: 10.1007/s00431-002-0999-4. [DOI] [PubMed] [Google Scholar]
- Reis FC, Caldas HC, Norato DY, Schwartz IV, Giugliani R, Burin MG, Sartorato EL. Glycogen storage disease type Ia: molecular study in Brazilian patients. J Hum Genet. 2001;46:146–149. doi: 10.1007/s100380170102. [DOI] [PubMed] [Google Scholar]
- Rocha H, Cabral A, Vilarinho L. Identification of a novel mutation (Q24X) in the glucose-6-phosphatase gene of a Portuguese patient with GSD-Ia. Hum Mutat. 2000;16:449. doi: 10.1002/1098-1004(200011)16:5<449::AID-HUMU25>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Seydewitz HH, Matern D. Molecular genetic analysis of 40 patients with glycogen storage disease type Ia: 100% mutation detection rate and 5 novel mutations. Hum Mutat. 2000;15:115–116. doi: 10.1002/(SICI)1098-1004(200001)15:1<115::AID-HUMU23>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Shieh JJ, Terizioglu M, Hiraiwa H, Marsh J, Pan CJ, Chen LY, Chou JY. The molecular basis of glycogen storage disease type 1a: structure and function analysis of mutations in glucose-6-phosphatase. J Biol Chem. 2002;277:5047–5053. doi: 10.1074/jbc.M110486200. [DOI] [PubMed] [Google Scholar]
- Shieh JJ, Pan CJ, Mansfield BC, Chou JY. Glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem. 2003;278:47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- Stroppiano M, Regis S, DiRocco M, Caroli F, Gandullia P, Gatti R. Mutations in the glucose-6-phosphatase gene of 53 Italian patients with glycogen storage disease type 1a. J Inher Metab Dis. 1999;22:43–49. doi: 10.1023/a:1005495131118. [DOI] [PubMed] [Google Scholar]
- Stukey J, Carman GM. Identification of a novel phosphatase sequence motif. Protein Sci. 1997;6:469–472. doi: 10.1002/pro.5560060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MS, Pan CJ, Shieh JJ, Ghosh A, Chen LY, Mansfield BC, Ward JM, Byrne BJ, Chou JY. Sustained hepatic and renal glucose-6-phosphatase expression corrects glycogen storage disease type Ia in mice. Hum Mol Genet. 2002;11:2155–2164. doi: 10.1093/hmg/11.18.2155. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Akanuma J, Matsubara Y, Fujii K, Kure S, Suzuki Y, Wataya K, Sakamoto O, Aoki Y, Ogasawara M, Ohura T, Miyabayashi S, Narisawa K. Heterogeneous mutations in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type Ia. Am J Med Genet. 2000;92:90–94. [PubMed] [Google Scholar]
- Terzioglu M, Emre S, Ozen H, Saltik IN, Kocak N, Ciliv G, Yuce A, Gurakan F. Glucose-6-phosphatase gene mutations in Turkish patients with glycogen storage disease type Ia. J Inherit Metab Dis. 2001;24:881–882. doi: 10.1023/a:1013956611607. [DOI] [PubMed] [Google Scholar]
- Trioche P, Francoual J, Chalas J, Capel L, Lindenbaum A, Odievre M, Labrune P. Genetic heterogeneity of glycogen storage disease type 1a in France: a study of 48 patients. Hum Mutat. 2000;16:444. doi: 10.1002/1098-1004(200011)16:5<444::AID-HUMU10>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen YT, de Barsy T, de Lonlay P, Dionisi-Vici C, Fenske CD, Lee PJ, Leonard JV, Maire I, McConkie-Rosell A, Schweitzer S, Vikkula M, Van Schaftingen E. A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. Am J Hum Genet. 1998;63:976–983. doi: 10.1086/302068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen YT, Lee PJ, Leonard JV, Maire I, Wendel U, Vikkula M, Van Schaftingen E. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur J Hum Genet. 1999;7:717–723. doi: 10.1038/sj.ejhg.5200366. [DOI] [PubMed] [Google Scholar]
- Visser G, Rake JP, Fernandes J, Labrune P, Leonard JV, Moses S, Ullrich K, Smit GP. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I. J Pediatr. 2000;137:187–191. doi: 10.1067/mpd.2000.105232. [DOI] [PubMed] [Google Scholar]
- Weinstein DA, Somers MJ, Wolfsdorf JI. Decreased urinary citrate excretion in type 1a glycogen storage disease. J Pediatr. 2001;138:378–382. doi: 10.1067/mpd.2001.111322. [DOI] [PubMed] [Google Scholar]
- Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr. 2002;161 1:S35–S39. doi: 10.1007/s00431-002-1000-2. [DOI] [PubMed] [Google Scholar]
- Weston BW, Lin JL, Muenzer J, Cameron HS, Arnold RR, Seydewitz HH, Mayatepek E, Van Schaftingen E, Veiga-da-Cunha M, Matern D, Chen YT. Glucose-6-phosphatase mutation G188R confers an atypical glycogen storage disease type 1b phenotype. Pediatr Res. 2000;48:329–334. doi: 10.1203/00006450-200009000-00011. [DOI] [PubMed] [Google Scholar]
- Wong LJC, Hwu WL, Dai P, Chen TJ. Molecular genetics of glycogen storage disease type 1a in Chinese patients of Taiwan. Mol Genet Metab. 2001;72:175–180. doi: 10.1006/mgme.2000.3129. [DOI] [PubMed] [Google Scholar]
- Wu MC, Tsai FJ, Le CC, Lin SP, Wu JY. Identification of a novel missense mutation (T16A) in the glucose-6-phosphatase gene in a Taiwan Chinese patient with glycogen storage disease Ia. Hum Mut. 2000a;15:390. doi: 10.1002/(SICI)1098-1004(200004)15:4<390::AID-HUMU32>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Wu MC, Tsai FJ, Lee CC, Tsai CH, Wu JY. A novel missense mutation (H119L) identified in a Taiwan Chinese family with glycogen storage disease type 1a (von Gierke disease) Human Mutat. 2000b;16:447. doi: 10.1002/1098-1004(200011)16:5<447::AID-HUMU17>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Yiu WH, Pan CJ, Ruef RA, Peng WT, Starost MF, Mansfield BC, Chou JY. The angiotensin system mediates renal fibrosis in glycogen storage disease type Ia nephropathy. Kid Internal. 2007 doi: 10.1038/sj.ki.5002718. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]