

Abstract
We have employed recently developed blind modification search techniques to generate the most comprehensive map of post-translational modifications (PTMs) in human lens constructed to date. Three aged lenses, two of which had moderate cataract, and one young control lens were analyzed using multidimensional liquid chromatography mass spectrometry. A total of 491 modification sites in lens proteins were identified. There were 155 in vivo PTM sites in crystallins; 77 previously reported sites and 78 newly detected PTM sites. Several of these sites had modifications previously undetected by mass spectrometry in lens including carboxymethyl lysine (+58 Da), carboxyethyl lysine (+72 Da), and an arginine modification of +55 Da with yet unknown chemical structure. These new modifications were observed in all three aged lenses but were not found in the young lens. Several new sites of cysteine methylation were identified indicating this modification is more extensive in lens than previously thought. The results were used to estimate extent of modification at specific sites by spectral counting. We tested the longstanding hypothesis that PTMs contribute to age-related loss of crystallin solubility by comparing spectral counts between the water-soluble and water-insoluble fractions of the aged lenses, and found that the extent of deamidation was significantly increased in the water-insoluble fractions. Based on spectral counting, the most abundant PTMs in aged lenses were deamidations and methylated cysteines with other PTMs present at lower levels.
Keywords: proteomics, mass spectrometry, post-translational modification, spectral counting, modification abundance, human lens, crystallin, aging, cataract
Introduction
The human ocular lens is a particularly rich source of post-translationally modified proteins. This tissue is predominantly composed of a small number of highly soluble proteins known as crystallins. The proteins present in the mature lens fiber cells do not turnover, and the lens continues to grow throughout life with the center (the nucleus) of the lens containing the oldest proteins. Given the extreme age of most of the crystallin proteins, it is expected that a wide variety of post-translational modifications (PTMs) will accumulate at a large number of residues. Many modifications have already been identified including acetylation 1-7, carbamylation 4, 8, deamidation 3, 9-23, methylation 2, 3, 24, 25, oxidation 2, 3, 6, 11, 16, 21, 23, 26-28, phosphorylation 3, 6, 7, 10, 11, 28, 29, proteolysis 11, 16, 20, 21, 23, 25, 28, 30-35, and UV-filter adducts 36-38.
Tandem mass spectrometry (MS/MS) has played a major role in identifying PTMs in lens, however, most previous studies identified lens PTMs in a case-by-case fashion, and were usually limited to one or very few PTMs. More recently, advanced proteomic techniques such as multidimensional liquid chromatography separations (2-DLC) of complex lens digests have been used to identify large numbers of modified peptides from single lenses 3, 6. We have compiled a comprehensive list of nearly 200 lens PTM sites that have been previously reported (Supplemental Table 1). Despite the extensive list of modifications already documented in lens, there may be additional sites of known modifications such as deamidation or oxidation to be discovered, and cataractous lenses may exhibit novel PTMs that have not yet been identified. The lens tissue, therefore, represents a particularly challenging test for PTM identification algorithms.
Protein database search programs, such as Sequest 39 or Mascot 40, are routinely used for identification of unmodified peptides. However, these programs become less reliable and search times increase when analyzing spectra of modified or mutated peptides. The first approach to PTM identification was proposed by Yates et al., 41, 42 who advocated the enumeration and scoring of all possible modifications for each peptide from the database. This exhaustive search approach can only take into account a few modifications and is prohibitively slow for mutation detection. As a result, while both Sequest and Mascot have been extended for PTM detection,42, 43 modifications must be specified before searching, and detecting peptides with multiple modifications remains computationally expensive and of unknown reliability. A modification search against a greatly reduced number of proteins identified in an initial search without modifications is an efficient strategy to speed up exhaustive PTM searches, and has been implemented in X! Tandem 44 and was used by MacCoss, et al.6
To use Sequest, Mascot, or X! Tandem for PTM identification, a researcher has to postulate, in advance, which PTMs are present in the sample. As a result, the current practice is to perform a restrictive search for a small set of PTMs (such as phosphorylation) and ignore all other PTMs. This cannot identify modifications present in the sample that were not explicitly specified in the search, and there is also the possibility of incorrectly interpreting a peptide containing a modification that was not specified. The question arises whether one can design an unrestrictive PTM search algorithm that can identify all types of PTMs at once in a blind mode, i.e., without knowing which modifications exist in a sample.
The first blind approach to PTM identification was spectral alignment proposed by Pevzner et al.,45, 46 and there has been a renewed interest in blind PTM searches. OpenSea2 and SPIDER47 use approaches for blind PTM identification based on de novo interpretation of MS/MS spectra, while P-Mod48 and Interrogator49 have formulated blind PTM search algorithms limited to a single modification per peptide. Recently, Tsur et al. proposed a new blind approach to PTM identification, MS-Alignment,50 that extends the original spectral alignment algorithm45 and resulted in fast and reliable PTM identification. Spectral alignment eliminates dependence on accurate de novo interpretations by comparing a database peptide against every possible interpretation of an experimental spectrum, and the MS-Alignment algorithm resolved a number of open problems outlined in Pevzner et al.45 Instead of guessing which modifications might be present in an MS/MS sample, MS-Alignment produces a list of probable PTMs and their putative sites. The list of modifications found by MS-Alignment can be further used as input to restrictive database search programs that can rigorously score the putative PTM sites, and even find new ones that were missed by MS-Alignment. Since speed is an issue while searching with a large list of PTMs, we used the fast restrictive database search program InsPecT51 at this post-processing step.
The first applications of MS-Alignment50 led to the conclusion that the extent of modifications present in many samples may be much larger than previously thought. Recently, an alternative approach to blind PTM detection using FTMS52 (ModifiComb) has been described, and the results further support this conclusion. It appears that previous proteomic techniques may have underestimated the extent of modifications present in complex biological samples.
In this report, MS-Alignment/InsPecT was first used to identify modified peptides in four large 2-DLC datasets (over 43,000 MS/MS spectra) of 93-year old human cataractous lens that had been previously analyzed by OpenSea.3 The MS/MS spectra assigned to peptides containing PTMs were manually validated and many new modifications were confirmed. To establish the biological significance of these (and other) modifications, MS/MS data from seven additional 2-DLC separations of complex lens digests were analyzed. Three aged human lenses (70-year old normal, 70-year old cataract, and a repeat of the 93-year old cataract) and one young control human lens (3-day old) were analyzed. A total of 439,116 ion trap MS/MS spectra were collected and an alternative to manual validation was developed to verify the detected modifications. A data set of the automatically validated annotated MS/MS spectra identified in this study has been created to aid further developments in PTM analysis techniques.
Methods
Lens sample preparations
Lens proteins from a 93-year old human male with nuclear cataract (Grade III, Pirie scale) were separated into water-soluble and water-insoluble fractions, digested with trypsin, and 2-DLC MS/MS analysis performed as previously reported.3 Peptides were analyzed on two different instruments: an LCQ Classic ion trap and a Q-TOF hybrid mass spectrometer. This data set (43,518 spectra) is the same as that used by Searle, et al.3 and will be referred to as the “validation” data.
Three additional human lenses [3-day old, 70-year old normal, and 70-year old nuclear cataractous (Grade II, Pirie scale)] were obtained from the Lions Eye Bank of Oregon with IRB approval, dissected within 12 hours of death, graded for cataract severity, photographed, and frozen at -70°C until use. Individual lenses were homogenized on ice in a 20 mM phosphate, 1.0 mM EDTA buffer (pH 7.0) using 1.0 ml of buffer per lens, and the homogenate centrifuged at 20,000 g for 30 min to pellet the water-insoluble proteins. The water-insoluble pellet was resuspended once in the same volume of homogenizing buffer, and again pelleted. The resulting pellet was re-suspended by brief sonication (5 sec × 2) on ice, and the protein content of both soluble and insoluble fractions determined in triplicate using a BCA assay (Pierce Biotechnology, Inc., Rockford, IL, USA). 2.5 mg portions of the samples were dried by vacuum centrifugation and stored at -70°C until analysis.
Dried water-soluble or water-insoluble lens samples (from the three lenses mentioned above and additional samples from the 93-year old cataractous lens that was previously analyzed) were dissolved in buffer containing 8M de-ionized urea, 0.8 M Tris, 0.08 M methylamine, and 8 mM CaCl2 (pH 8.5). Cysteine residues were reduced and alkylated by successive treatment with DTT and iodoacetamide, and sequencing grade modified trypsin (Trypsin Gold from Promega, Madison, WI, USA) added at a ratio of 1:25 protease to substrate, resulting in a dilution of the urea to a final 2M concentration. Digestion occurred during an 18 hour incubation at 37°C with shaking. Following digestion, formic acid was added to a final concentration of 5%, and peptides solid phase extracted using Sep-Pak Light cartridges (Millipore, Billerica, MA, USA).
2-DLC separations and MS/MS acquisition
The Sep-Pak cleaned digests of lens proteins were injected onto a 100 × 2.1 mm polysulfoethyl A cation exchange column (The Nest Group, Inc., Southborough, MA, USA) at a 200 μl/min flow rate. Mobile phase A contained 10 mM sodium phosphate (pH 3.0) and 25% acetonitrile, and mobile phase B was identical, except it contained 350 mM KCl. Following 5 minutes of loading and washing in mobile phase A, peptides were eluted using a linear gradient of 0-50% B over 45 min, followed by a linear gradient of 50-100% B over 20 min. One-minute fractions were collected, dried by vacuum centrifugation, and re-dissolved by shaking in 100 μl of 5% formic acid. Fractions at the beginning or end of the salt gradient were combined, based on UV intensities, to reduce the number of fractions to about 40 per lens sample. Portions of each fraction (10 μl) were analyzed by LC/MS using an Agilent 1100 series capillary LC system and an LCQ Classic ion trap mass spectrometer (Thermo Electron, San Jose, CA, USA) using a standard electrospray source fitted with a 34 gauge metal needle (ThermoFinnigan, Cat. No. 97144-20040). Samples were applied at 20 μl/min to a trap cartridge, and then switched onto a 0.5 X 250 mm Zorbax SB-C18 column (Agilent Technologies, Palo Alto, CA, USA) using a mobile phase A containing 0.2% acetic acid. The gradient was 7-35% acetonitrile over 90 minutes at a 10 μl/min flow rate. This shallow reverse phase gradient was capable of completely resolving unmodified and deamidated peptide forms, verified with several pairs of synthetic peptides where Asn or Gln residues were replaced by Asp or Glu, respectively. Data dependent collection of MS/MS spectra used the dynamic exclusion feature of the instrument's control software (repeat count equal to 1, exclusion duration of 3 min, list size of 25) to obtain MS/MS spectra of the three most abundant parent ions following each survey scan. DTA files were created using extract_msn (ThermoFinnigan) from 400 to 4000 Da, with a minimum peak count of 25, and charge state analysis (ZSA, ThermoFinnigan) was performed to assign higher charge states and greatly reduce the number of duplicate DTA files. Each lens fraction was analyzed identically, and approximately 60,000 MS/MS spectra were collected per lens sample for a total of 439,116 MS/MS spectra.
Identification of modified peptides
Spectra were searched against a database of crystallins and other abundant lens proteins (35 proteins; 12,000 amino acids). The MS-Alignment algorithm50 was used to generate a list of putative modifications in the sample of the form (aa, Δ), where Δ is a characteristic offset of PTM on amino acid aa. We examined each modification that occurred in at least 100 out of the 439,116 spectra with match quality score greater than zero. We rejected modifications that were judged to be artifacts (addition or subtraction of amino acids at peptide termini, or abundant modifications incorrectly assigned to an adjacent amino acid, etc.), and compiled a list of putative modification sites in the lens proteins.
To refine the list of modifications, we performed restrictive searches using the list of putative modifications provided by MS-Alignment. Sometimes “new” modifications could be explained by combinations of previously reported modifications. For instance, the modification (M, +58) resulted from an oxidation (+16) of an N-terminal methionine residue that was acetylated (+42). To screen for sites that could be explained in this way, we used InsPecT51 to perform the searches allowing up to three modifications per peptide from the restricted list of modifications. Parent ion mass corrections and a fragment ion mass tolerance of 0.5 Da were used for ion trap data. Fragment ion mass tolerances were 0.1 Da and no parent ion mass corrections were performed for QTOF data.
Validation of identified modifications
While existing MS/MS database search tools rigorously assess statistical significance of peptide interpretations, evaluating reliability of MS/MS interpretations of modified peptides remains an open problem. We chose a conservative approach to evaluate the reliability of identified modified peptides and performed manual validation on all four of the “validation” data sets. The lists of modified peptides from InsPecT for each of the four fractions were converted to data formats that could be read by the MS/MS spectrum viewer from OpenSea 3. The OpenSea spectrum viewer was configured to highlight y-ions, b-ions, a-ions, and loss of water or ammonia from major ions. For visualization purposes, fragment ions were highlighted if they were within 0.25 or 0.40 AMU of the calculated masses for the QTOF or ion trap, respectively. Modification sites, modification mass shifts, and the mass differences between the measured parent ion and the calculated modified sequence were displayed along with the spectra. Interactive visualization tools allowed zooming regions of spectra, measurement of the mass differences between spectral peaks, and simultaneous viewing of paired spectra of modified and unmodified peptides.
Manual validation accepted modified spectra satisfying the following criteria:
Correct parent ion mass shift values,
Appropriately shifted y- and b-ions confirming the modification mass and site,
Consistent fragment ion pattern between unmodified peptides and modified peptides (keeping point #5 below in mind),
Modifications located at peptide termini (or beyond the low and high mass cutoffs for the ion trap) were rejected if there was any potential assignment ambiguity,
-
The presence (or absence) of known modification specific fragmentation ion spectral features:
Neutral loss of 43 Da associated with carbamylation
Neutral loss of 98 Da from phosphorylated serine
Neutral loss of 64 Da from oxidized methionine
Enhanced cleavage C-terminal to deamidated asparagines,
Higher mass fragment ions were required to have realistic isotopic distributions,
Missed trypsin cleavages were required for modified arginine or lysine residues,
Peptides containing modified basic sites (K, R, N-terminus) were required to elute in an earlier SCX fraction (if the corresponding unmodified peptides from missed trypsin cleavage could be identified)
Manual validation would have taken too much time to be practical for analyses of additional lenses so an alternative automated validation strategy was developed. OpenSea, X! Tandem, and InsPecT searches of the same data sets were performed and spectral annotations compared on a spectrum by spectrum basis.
X! Tandem searches were configured using the 24 most frequent mass shifts and affected amino acids reported from MS-Alignment determined during manual validation. Due to the large number of potential modifications necessary to account for the InsPecT results, only peptides with tryptic termini were allowed in the X! Tandem searches. In addition, “blind” OpenSea analyses3 of the data sets were also performed using previously reported parameters for ion traps3 without amino acid substitutions allowed. Both X! Tandem and OpenSea used a human species subset of the current Swiss-Prot protein database (12,077 entries). Without invoking any scoring thresholds, the interpretations where all three programs agreed on modification and site were considered correctly assigned (“SameSite” consensus). A diagram of the analysis workflow is shown in Figure 1.
Figure 1.

Workflow to identify post-translational modifications in lens proteins. Samples were analyzed with an unrestrictive (“blind”) search (MS-Alignment) in order to discover novel chemical modifications. A restrictive search (InsPecT) was then performed to increase sensitivity. Modifications were validated by looking for consensus across three search tools (InsPecT, OpenSea, and X! Tandem), and quantified using spectral counting.
Integrated ion current measurements
Theoretical monoisotopic masses of unmodified and deamidated peptides were used to extract single-ion chromatograms using QualBrowser (ThermoFinnigan) for a 1.0 Da range centered on the specified mass values. Chromatogram peaks were integrated and summed across all SCX fractions (typically 3 or 4 fractions) where the peptides of interest were observed. The total ion current of each peptide form was used to estimate the extent of deamidation (deamidated ion current divided by sum of both unmodified and deamidated ion currents), expressed as a percentage.
Results
Analysis of previously acquired 93-year old human cataractous lens data sets
MS-Alignment was configured to find blind modifications of masses from -200 to +200 Da, and used to analyze the “validation” data set. A list of putative modifications (mass shifts and affected amino acids) was used by InsPecT in a restrictive search allowing up to three modifications per peptide. A total of 378 distinct modification sites were identified from analysis of the 43,518 spectra50 (four separate 2-DLC analyses). Manual validations of all modified peptides were performed as described above. A site was considered valid in a particular 2-DLC run if at least one spectrum contained sufficient evidence to confirm peptide sequence and localize the modification. Examinations of all spectra associated with a specific modification were used to classify the site as “yes”, “no”, or “maybe”. Combined evidence across the four data sets was used to access whether or not the site was considered confidently identified. Manual validation results are listed in Supplemental Table 2.
Modifications were grouped into likely in vivo modifications and known in vitro artifacts (N-terminal carbamylation, N-terminal pyroglutamic acid, potassium adducts, and parent ion loss of water and ammonia). Of the 378 modification sites present in all lens proteins, there were 219 putative in vivo PTM sites. The number of those sites passing manual validation was 149, and 101 sites were in crystallins. We emphasize that these sites are the most reliable PTMs and it is likely that some of the rejected sites are actually false negatives.
The automated validation strategy was tested on the “validation” data. The total number of modification sites reported by MS-Alignment/InsPecT was 378, there were 228 sites (either in vivo or in vitro) that passed manual validation, 196 sites had “SameSite” consensus status (X! Tandem, OpenSea, and InsPecT predictions agreed), and 178 of the consensus sites also passed manual validation. The consensus validation procedure had slightly lower sensitivity compared to manual validation (196/228=0.86), had a reasonable specificity (178/196=0.91), and, most importantly, could be fully automated for high-throughput PTM studies. The consensus results are also listed in Supplemental Table 2.
Analyses of additional human lenses
To verify that previously unreported modifications were age-related and not in vitro artifacts, 2-DLC analyses of three additional lenses (3-day old, 70-year old normal, and 70-year old cataractous) and repeat analyses of the 93-year old cataractous lens were performed. Due to the small number of lenses analyzed, no attempt was made to find modifications unique to cataracts. For each of the three aged lenses, proteins were separated into water-soluble and water-insoluble fractions. The young lens contained such a small amount of water-insoluble protein that only the water-soluble fraction could be analyzed. The seven ion trap 2-DLC runs produced 439,116 MS/MS spectra which were searched for PTMs using MS-Alignment/InsPecT. A total of 63,936 spectra had MQScores exceeding 1.0 and 18,719 spectra contained modifications (29%).
Although replicate analyses of each lens were not performed to access reproducibility of the reported results, we adopted several methods to ensure that PTM sites were reliably identified. The large number of SCX fraction for a less complex mixture, such as the lens, resulted in frequent redundant observation of modified peptides, and we required a minimum of two modified spectra for each candidate PTM site. On average, approximately 3 distinct peptide forms (tryptic, semi-tryptic, and missed cleavage peptides) were observed per reported PTM site, increasing the confidence in the detected sites. Modifications were individually tabulated for each lens, and only modifications that were observed in at least two of the three aged lenses were reported. Agreement of results from different analysis software can reduce random identifications, and all annotations having an MQScore greater than 1.0 were tested for “SameSite” consensus by three different analysis programs.
We used the young 3-day old lens as a control to classify the PTMs into likely in vitro modifications and putative in vivo PTMs. Many types of modifications were seen with high frequency in the 3-day old lens and in the aged lenses, and were classified as in vitro artifacts (carbamylation, salt adducts, loss of water, loss of ammonia, and pyroglutamic acid). Since some proportion of the detected deamidations could be introduced during sample processing or be non-deamidated forms of the peptides misidentified by the ion trap, deamidation abundance estimates (described below) for the aged lenses were compared to the corresponding abundance estimates for the 3-day old control lens, and results filtered to remove potentially misidentified deamidation sites. Carbamylated lysine residues were treated in a similar fashion.
A total of 491 putative PTM sites passed “SameSite” consensus and were seen in at least two of the aged lenses (Supplemental Table 3). After excluding likely in vitro artifacts, the remaining number was 352 sites. The number of PTM sites in major crystallin proteins was 220, which included 99 potential deamidation sites. Based on the number of assigned spectra, a chi-square test with one degree of freedom was used to identify the 46 deamidation sites that were more abundant (P>0.05) in the aged lenses compared to the 3-day old control. In the chi-square test, observed spectral counts were compared to expected values derived from average PTM rates in the two sets of data being compared. A p>0.05 would correspond to a chi square value in excess of 3.84. Similarly, 4 of 16 carbamylated lysine sites were more abundant in the aged lenses. Following this analysis, the final tally of identified aged human lens PTM sites was 155. The in vivo PTM sites for the nine major crystallins are shown in Figure 2 (α crystallins), Figure 3 (β crystallins), and Figure 4 (γ crystallins). There were four deamidation sites (N49, Q66, Q67, and N160) in γD seen in all three aged lenses with relative modification rates in excess of 10% that did not pass the chi-square test due to the low abundance of γD in the young lens. There were also 4 known sites of protein N-terminal acetylation in βA1/A3, βA4, and βB3 that were detected in the young control lens but did not pass the filtering criteria. The relatively low abundances of βA2, βB3, and γB resulted in too few modification sites (four) to warrant inclusion in the figures.
Figure 2.

Modification sites identified in (A) αA crystallin and (B) αB crystallin. Modified residues are bolded and the mass shifts of the modifications are specified above the residues.
Figure 3.

Modification sites identified in (A) βA3/A1 crystallin, (B) βA4 crystallin, (C) βB1 crystallin, and (D) βB2 crystallin. Modified residues are bolded and the mass shifts of the modifications are specified above the residues.
Figure 4.

Modification sites identified in (A) γS crystallin, (B) γC crystallin, and (C) γD crystallin. Modified residues are bolded and the mass shifts of the modifications are specified above the residues.
We detected several novel PTMs in the aged lenses that were absent in the 3-day old control lens. We identified numerous lysine modifications (carbamylation, carboxymethylation, and carboxyethylation) at many sites, and one example is shown for βB1 K117 in Figure 5. Novel arginine +55 Da modifications were observed at 11 sites and MS/MS spectra from three different peptides are shown in Figure 6. The lysine and arginine modifications occurred at sites of missed trypsin cleavage and, in some cases, the unmodified peptide was also observed. If a basic site in a peptide was modified, such as N-terminal carbamylation, there was a corresponding shift to earlier SCX elution times. In all cases where the unmodified missed cleavage peptide was observed, the lysine or arginine modifications caused a large (approximately 10 min) change to earlier SCX elution time indicating a modified basic site (data not shown).
Figure 5.

Tandem mass spectra used to assign the carboxymethyl lysine modification to K117 in βB1 peptide GEMFILEKGEYPR. The matched y-ions (in blue) and b-ions (in red) are highlighted and labeled. Panel A shows the unmodified, missed-cleavage peptide, which eluted in SCX fraction 42, for the 70-year old normal human water-insoluble lens sample. Panel B is the modified peptide (GEMFILEK+58GEYPR) from the same lens sample, which eluted in the earlier SCX fraction 33 indicating a modified basic site. Panel C is the same modified peptide from the 93-year old cataractous water-soluble lens sample observed with the QTOF instrument. The modification mass shift was 57.99 Da. In panels B and C, all y-ions (shown in blue) starting with y6 are clearly shifted by +58, and the modification can be unambiguously assigned to K117.
Figure 6.

Ion trap tandem mass spectra of peptides containing the arginine +55 Da modifications are shown for (A) αA R12 in peptide R+55TLGPFYPSR, (B) γS R78 in peptide WMGLNDR+55LSSCR, and (C) βB1 R85 in peptide AEFSGECSNLADR+55GFDR. The spectra are from the 70-year old normal lens, the 70-year old cataractous lens, and the 93-year old cataractous lens, respectively. In all cases the modification caused a missed trypsin cleavage. Matched y- (in blue) and b-ions (in red) are highlighted and labeled.
PTM abundance estimates from spectral counting
The large numbers of modified spectra observed in this study suggested that PTM abundances could be estimated by spectral counting.53-55 In this initial study, we have assumed that the relative ion currents of modified peptides would be similar to unmodified peptides, and that the selection process of ions for fragmentation would, therefore, be similar for all peptides. Use of synthetic peptides during chromatography testing (see Methods section) indicated that this assumption was valid for deamidated peptides in simple mixtures. We have also assumed that modified peptides undergo similar fragmentation to their unmodified forms and that identification rates by the analysis software would also be the same. However, more labile modifications such as phosphorylated serines (relatively less abundant in lens) would likely have suppressed spectral counts due to poorer fragmentation and identification. It is possible that absolute PTM abundances based on spectral counts could differ from their true abundances if these assumptions do not hold true and we have, therefore, compared relative abundances between lens samples processed under identical conditions to determine changes associated with aging and loss of crystallin solubility.
For each amino acid residue, the numbers of spectra matched to sequences containing that amino acid were tabulated for the unmodified amino acid and all distinct modified forms of that amino acid (if any). The modification abundance estimate was calculated as the ratio of the spectral count of a particular modified amino acid form to the total spectral count for that amino acid (all modified and unmodified forms). The spectral counts of the eleven major types of modifications at the 155 in vivo crystallin sites were averaged across the soluble and insoluble fractions of the three aged lenses for each type of modification, and are listed along with data from the 3-day old control lens in Table 1. The data in Table 1 are average modification abundances over the amino acids that had detectible modifications, and are not the overall modification abundances in the samples. Protein N-terminal acetylation (a co-translational modification), oxidation, and cysteine methylation were abundant, but deamidation was by far the major PTM present in aged human lens crystallins.
Table 1.
Averaged human lens PTM spectral counts and PTM rates (for peptides containing detected modifications) for aged water-insoluble and water-soluble crystallins.
| Average Aged Human Lens Spectral Counts (N=3)a,b | 3-Day Old Spectral Countsc | |||||||
|---|---|---|---|---|---|---|---|---|
| Overall | Water-Insoluble | Water-Soluble | Water-Soluble | |||||
| Modification | aa, ΔM | Relative Rankd | Modified Count | Ratee (%) |
Modified Count | Ratee (%) |
Modified Count | Ratee (%) |
| deamidation | N,Q +1 | 93 | 1,739 | 18.52 | 796 | 8.77 | 176 | 2.30 |
| S-methylation | C -43f | 28 | 390 | 28.80 | 374 | 20.30 | 6 | 0.45 |
| acetylationg | nt +42 | 24 | 300 | 84.89 | 355 | 83.78 | 420 | 80.15 |
| oxidation | M,W +16 | 11 | 143 | 2.76 | 169 | 3.57 | 153 | 4.27 |
| carboxymethyl lysine | K +58 | 8 | 99 | 4.19 | 107 | 3.95 | 0 | 0.00 |
| phosphorylation | S,T +80 | 7 | 80 | 3.83 | 113 | 5.46 | 20 | 1.12 |
| carbamylation | K +43 | 3 | 33 | 4.28 | 39 | 4.52 | 5 | 0.68 |
| carboxyethyl lysine | K +72 | 2 | 33 | 2.71 | 33 | 2.84 | 1 | 0.12 |
| R +55 | R +55 | 2 | 34 | 3.01 | 25 | 2.60 | 0 | 0.00 |
| H-methylation | H +14 | 2 | 33 | 5.01 | 21 | 3.33 | 0 | 0.00 |
| S,H +28 | S,H +28 | 1 | 14 | 0.69 | 14 | 0.72 | 14 | 1.00 |
Aged lenses were 70-year normal, 70-year mild cataract, and 93-year moderate cataract.
Overall spectral counts for the 11 major in vivo human lens crystallin modifications at 155 PTM sites.
Similar quantities for water-soluble protein from a 3-day old lens are shown as a control.
Relative to least abundant modification. Ranked by total modified spectral count.
Modification rate was calculated as the number of spectra assigned to a given amino acid modification divided by total number of spectra that contained those amino acids.
Net methylation mass shift after cysteine +57 Da carbamidomethylation.
Protein co-translational N-terminal acetylation.
The fraction of water-insoluble protein in human lens increases with age, and eventually becomes more abundant than the water-soluble protein.56 A reasonable hypothesis for this is the accumulated damaging effect of PTMs on crystallin structure and stability. Starting with the 491 PTM sites listed in Supplemental Table 3, we tested for sites that were more abundant in aged lenses and also more abundant in the water-insoluble fraction. A chi-square test with one degree of freedom was used to identify sites more abundant (p>0.05) in aged lenses than the 3-day old control, and sites more abundant (p>0.05) in the water-insoluble fractions than in the water-soluble fractions. One carbamylated lysine, five methylated cysteine sites and 36 deamidation sites from lens proteins passed the tests. No other sites passed the filtering. Figure 7 shows the abundances for the 31 crystallin deamidation sites passing the above criteria that were also greater than 2-fold more abundant in the water-insoluble fractions compared to the water-soluble fractions.
Figure 7.

A comparison of the averaged relative deamidation abundances between water-soluble and water-insoluble proteins from the three aged lenses. Abundances were estimated from spectral counts and only the sites having a 2-fold or greater abundance difference are shown. The data shows not only increased deamidation present in the water-insoluble lens proteins, but also widely different extents of deamidation depending on the site.
Discussion
In this study, complex digests of lens proteins were analyzed using 2D-LC and blind PTM identification techniques used to globally map human lens protein PTMs. An automatic validation method was employed to identify 155 age-related crystallin PTM sites, including 78 previously unreported sites. Modification abundances at specific sites were estimated from spectral counts and three aged lenses compared to a young lens to identify age-related PTMs. Our most significant finding was that large numbers of asparagine and glutamine sites were found to be significantly more deamidated in the water-insoluble proteins of aged lenses.
PTM abundance estimates from spectral counting
There are few quantitative data reported for lens modification abundances making it difficult to access the accuracy of the spectral counting abundance estimates. In Lapko, et al.,8 the overall extent of methylation was reported for γD (56%) and γC (12%) in a similar aged lens (72-year old). These overall lens average values were calculated using the reported weights of each dissected region and taking a weighted averaging of the two abundance values. In comparison, our observed rates were 66% for C110 in γD, and 16% (C22) and 14% (C79) for two sites in γC, in good agreement with the above published values. In an earlier report,24 a value of 31% was reported for γS, which is predominantly methylated at C26, and we observed a methylation rate of 43% at this site.
We were able to integrate peptide ion currents for 14 unmodified peptides and their deamidated forms taken from the sites shown in Figure 7. These peptides had a sufficiently intense 1+ ion which improved the accuracy of the extracted ion chromatograms. A comparison between the average deamidation abundances determined from the integrated ion currents and the abundances based on spectral counting is shown in Figure 8 for the water-insoluble fractions. A positive correlation (correlation coefficient of 0.83) indicated good agreement between the two methods to estimate deamidation abundances. We also checked the method using the (protein) N-terminal acetylated peptides in βB2, αA, and αB where the co-translational modifications were expected to have relative abundance estimates of 100%. We measured values of 92%, 95%, and 96%, respectively. We consider the spectral counts to be tentative abundance estimates until additional comparisons can be made with relative peptide ion currents and stable isotope ratio measurements that are planned.
Figure 8.
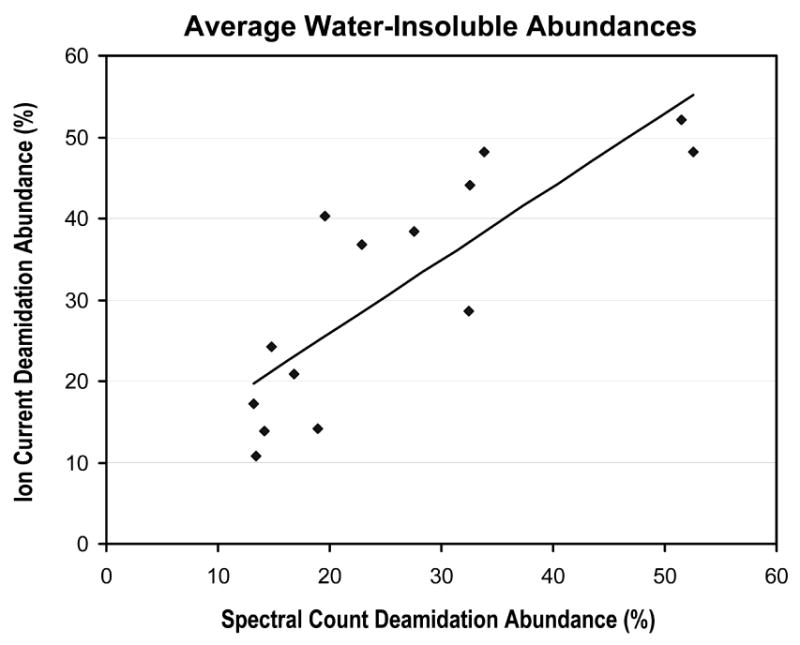
Correlation between average deamidation abundances determined from integrated ion currents and from spectral counting for the three aged lens water-insoluble fractions at 14 different crystallin deamidation sites.
Age-related changes in human lens
The small number of lenses processed so far provides a preliminary picture of the aging changes occurring in the human lens. PTM abundance estimates were averaged over all three aged lenses to minimize the unknown uncertainty associated with the spectral counting estimates, and comparisons between the water-soluble and water-insoluble fractions were made. Deamidation was found to be the major age-related PTM and was significantly more abundant in the water-insoluble fractions. Deamidation introduces a negative charge into the protein and may alter the tertiary structure.57 Accumulated deamidations may lead to partial protein denaturation and disrupt protein-protein interactions22, 58 necessary for the highly ordered structure of the lens. It is clear from Figure 7 that there is a striking increase in deamidation rates associated with water-insolubility. Asparagine sites are expected to deamidate faster than glutamine sites,59, 60 and it is not surprising that 74% of the sites are asparagine, and the 12 most extensively deamidated sites were at asparagine residues.
Since deamidation causes only a +1 Da mass shift, it was possible that ion trap mass inaccuracy contributed to incorrect assignments of some deamidation sites. Several precautions were taken to reduce this effect. Reverse phase chromatography conditions were employed so that unmodified and deamidated peptides did not co-elute. Mass corrections using y- and b-ion pairing were done to reduce parent ion mass errors that could result in unmodified peptides being incorrectly interpreted as deamidated forms. Lastly, deamidation abundances in the aged lenses were compared to the 3-day old control lens, and only those sites where deamidation was increased relative to the young lens were reported (see Results section). Our criteria for accepting identified deamidation sites was extremely conservative since 24 of the 53 rejected sites were, in fact, previously reported sites of deamidation.
Another significant finding was the far greater extent of S-methylation than was previously known in lens. The estimated abundances for S-methylation in αA, βB1, and βB2 were very low compared to previously identified sites in the γ crystallins and βA3. The mechanism of S-methylation in the lens remains unknown, but the wide variety of methylated sites suggests an enzyme able to recognize cysteine sites independent of primary sequence or three-dimensional structure. There was no methylation in the 3-day old lens and additional lenses at intermediate ages are needed to determine when methylation first occurs in the lens.
Carboxymethyl lysine (CML) and carboxyethyl lysine (CEL) are advanced glycation end products and have been suggested as general markers of advanced age and oxidation.61, 62 This study is the first to identify specific CML and CEL sites in lens crystallins, however, there did not appear to be any correlation between CML/CEL abundances and protein insolubility (data not shown), and these modifications were far less abundant than deamidation or methylation (see Table 1). Methionine and tryptophan oxidation (+16) can occur during sample processing and we did not observe a significant increase in +16 oxidation rate between the 3-day old control lens and aged lenses.
The novel R+55 modification is similar in mass to previously reported63 advanced glycation end-product modifications of arginine in lens, but higher mass accuracy measurements are necessary to determine the chemical structure of the modification. We were able to confirm serine and histidine +28 modifications3 at very low levels in the lens, and determining the identity of these modifications will also require further experiments.
Blind PTM search results
Blind searches of large data sets containing high frequencies of modified peptides, such as lens, produce rather extensive lists of putative modifications. Modifications on closely located amino acids often produce similar theoretical spectra, thus making it difficult to identify the exact position of modification. These can be difficult to assign reliably if the mass accuracy of the instrument is limited or the spectrum is of poor quality. Multiple modifications per peptide are even more complicated, and, unfortunately, occur frequently in aged lens samples. It is challenging to distinguish between a sum of known modifications and a potentially novel single-site PTM, a problem that grows when larger numbers of potential modifications are considered. Also, if partially tryptic peptides are considered, addition or subtraction of amino acids can be difficult to distinguish from larger PTM mass shifts.
Requiring PTM site agreement from multiple analysis programs was one way to address these issues, and was used in place of time-consuming manual validation. Since it was possible that the high sensitivity of the MS-Alignment/InsPecT analysis could result in less accuracy because of lower quality spectra, we chose two additional programs having different approaches to PTM detection. OpenSea tries to match PTMs to de novo sequencing results and only the higher quality spectra will be considered. X! Tandem uses a more traditional database searching method, and a restricted number of modifications and consideration of only tryptic peptides (due to performance limitations) may increase scoring accuracy. However, consensus is more likely for shorter peptides or peptide having only one or two possible modification sites (fewer ambiguous site possibilities), for modifications of a reasonable delta mass value (larger than +1 Da), and for higher quality spectra. This automated validation scheme likely had a high false negative rate, but was necessary to generate reliable annotations for the large number of modified MS/MS spectra in this study. Although many spectra identified by MS-Alignment/InsPecT did not pass our strict validation protocol, this does not imply that all rejected MS-Alignment/InsPecT interpretations were incorrect. There are undoubtedly many lens PTM sites that we rejected in this initial study, and future experiments will likely identify additional novel lens modifications.
The previously reported methylated arginine, methylated lysine, oxidized tyrosine, and phosphorylated tyrosine modifications by MacCoss et al.6 could not be confirmed. That study analyzed a 4-year old congenital cataractous lens using 2-DLC MS, and used different proteases to create overlapping peptides. Since a normal young lens was not processed as a control, it is difficult to determine if the unique modifications were associated with the congenital cataract. It is also possible that the modifications they observed were below the sensitivity of our experiment. However, there have been several analyses of aged lens since MacCoss et al. published their results, and none of those modifications have been reported in any aged lenses (see Supplemental Table 1). Unfortunately, deamidation was not considered in their analysis.
There were many PTMs identified in our data that are not shown in Figures 2 through 4. Many peptides were observed where loss of water or ammonia had occurred. There did not seem to be any correlation between S-18 sites and any of the known phosphorylation sites, so -18 and -17 PTMs were classified as in vitro artifacts in our analysis. We observed a large number of N-terminally carbamylated peptides, probably as a result of using urea as a denaturant. We distinguished in vivo carbamylated lysines from artifactual modifications by comparing carbamylation rates between the aged lenses and the 3-day old control lens processed under identical conditions, and their appearance at missed cleavage sites. There were several tryptic peptides that had glutamines located at the N-termini, and the most abundant forms of those peptides were often the pyroglutamic acid forms (a -17 Da N-terminal modification). Supplemental Table 4 is a partially annotated PTM frequency table generated from the complete MS-Alignment/InsPecT results and gives a sense of the PTM complexity in aged lens proteins.
There were many modifications detected at sites in non-crystallin lens proteins such as filensin and phakinin that passed “SameSite” consensus validation, but were not summarized here (see Supplemental Table 3). Also, some lower frequency modifications were not included in the X! Tandem searches, but may have been proper in vivo modifications (W+4 is a likely conversion of tryptophan to kynurinine, for example). In Supplemental Table 5, we list the putative sites of six additional modifications that were present in multiple crystallins and in peptides containing single modifications. Verification of these modifications will be the subject of future studies.
Conclusions
We employed methods for blind PTM identification and automated validation that allowed the majority of the abundant modifications in aged human lens data sets to be reliably identified. This greatly extended our previous analysis3 of PTMs in human lens crystallins, and further increased the number of modifications that could be detected from a single sample. This is the first study to use spectral counting to estimate modification abundances, and it was found that the major lens PTM that was more abundant in the water-insoluble lens fractions was deamidation. Since cataract can be thought of, in very simple terms, as a crystallin insolubility disease, modifications exclusively associated with water-insoluble proteins may have implications in cataractogenesis.
These experiments generated a large dataset of automatically validated annotated spectra of modified peptides that are publicly available at http://bioinfo2.ucsd.edu. While there exist a variety of publicly available annotated MS/MS samples of unmodified peptides,64-66 this is the first publicly available dataset of MS/MS spectra of modified peptides with a broad spectrum of PTMs. These data should be useful to test new PTM-specific scoring functions for MS/MS database search programs.
The InsPecT software, which implements the MS-Alignment algorithm, is available for download at http://peptide.ucsd.edu and program instructions are the subject of a recent published protocol.67 Availability of OpenSea is discussed in Searle, et al.3
Supplementary Material
Acknowledgments
This project was supported by National Institutes of Health grants NEI EY007755 (OHSU), NEI EY10572 (OHSU), and NIGMS 1-R01-RR16522 (UCSD). This research was also supported in part by the UCSD FWGrid Project, NSF Research Infrastructure Grant EIA-0303622. SD acknowledges support from NIH grant U19ES11384. We thank Brain Searle for facilitating the initial phase of this collaboration.
References
- 1.Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL. Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes the identification of the major proteins in young human lens. J Biol Chem. 1997;272(4):2268–75. doi: 10.1074/jbc.272.4.2268. [DOI] [PubMed] [Google Scholar]
- 2.Searle BC, Dasari S, Turner M, Reddy AP, Choi D, Wilmarth PA, McCormack AL, David LL, Nagalla SR. High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal Chem. 2004;76(8):2220–30. doi: 10.1021/ac035258x. [DOI] [PubMed] [Google Scholar]
- 3.Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, Nagalla SR. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J Proteome Res. 2005;4(2):546–54. doi: 10.1021/pr049781j. [DOI] [PubMed] [Google Scholar]
- 4.Lapko VN, Smith DL, Smith JB. In vivo carbamylation and acetylation of water-soluble human lens alphaB-crystallin lysine 92. Protein Sci. 2001;10(6):1130–6. doi: 10.1110/ps.40901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin PP, Barry RC, Smith DL, Smith JB. In vivo acetylation identified at lysine 70 of human lens alphaA-crystallin. Protein Sci. 1998;7(6):1451–7. doi: 10.1002/pro.5560070622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR., 3rd Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A. 2002;99(12):7900–5. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lapko VN, Smith DL, Smith JB. Expression of betaA2-crystallin in human lenses. Exp Eye Res. 2003;77(3):383–5. doi: 10.1016/s0014-4835(03)00156-8. [DOI] [PubMed] [Google Scholar]
- 8.Lapko VN, Smith DL, Smith JB. Methylation and carbamylation of human gamma-crystallins. Protein Sci. 2003;12(8):1762–74. doi: 10.1110/ps.0305403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kramps JA, de Jong WW, Wollensak J, Hoenders HJ. The polypeptide chains of alpha-crystallin from old human eye lenses. Biochim Biophys Acta. 1978;533(2):487–95. doi: 10.1016/0005-2795(78)90394-x. [DOI] [PubMed] [Google Scholar]
- 10.Miesbauer LR, Zhou X, Yang Z, Yang Z, Sun Y, Smith DL, Smith JB. Post-translational modifications of water-soluble human lens crystallins from young adults. J Biol Chem. 1994;269(17):12494–502. [PubMed] [Google Scholar]
- 11.Lund AL, Smith JB, Smith DL. Modifications of the water-insoluble human lens alpha-crystallins. Exp Eye Res. 1996;63(6):661–72. doi: 10.1006/exer.1996.0160. [DOI] [PubMed] [Google Scholar]
- 12.Takemoto LJ. Quantitation of asparagine-101 deamidation from alpha-A crystallin during aging of the human lens. Curr Eye Res. 1998;17(3):247–50. doi: 10.1076/ceyr.17.3.247.5218. [DOI] [PubMed] [Google Scholar]
- 13.Takemoto L. Increased deamidation of asparagine-101 from alpha-A crystallin in the high molecular weight aggregate of the normal human lens. Exp Eye Res. 1999;68(5):641–5. doi: 10.1006/exer.1999.0668. [DOI] [PubMed] [Google Scholar]
- 14.Takemoto L, Boyle D. Deamidation of alpha-A crystallin from nuclei of cataractous and normal human lenses. Mol Vis. 1999;5:2. [PubMed] [Google Scholar]
- 15.Takemoto L, Boyle D. Increased deamidation of asparagine during human senile cataractogenesis. Mol Vis. 2000;6:164–8. [PubMed] [Google Scholar]
- 16.Hanson SR, Hasan A, Smith DL, Smith JB. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp Eye Res. 2000;71(2):195–207. doi: 10.1006/exer.2000.0868. [DOI] [PubMed] [Google Scholar]
- 17.Takemoto L. Deamidation of Asn-143 of gamma S crystallin from protein aggregates of the human lens. Curr Eye Res. 2001;22(2):148–53. doi: 10.1076/ceyr.22.2.148.5524. [DOI] [PubMed] [Google Scholar]
- 18.Lapko VN, Purkiss AG, Smith DL, Smith JB. Deamidation in human gamma S-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry. 2002;41(27):8638–48. doi: 10.1021/bi015924t. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, David LL, Smith DL, Smith JB. Resistance of human betaB2-crystallin to in vivo modification. Exp Eye Res. 2001;73(2):203–11. doi: 10.1006/exer.2001.1023. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava OP, Srivastava K. Existence of deamidated alphaB-crystallin fragments in normal and cataractous human lenses. Mol Vis. 2003;9:110–8. [PubMed] [Google Scholar]
- 21.Zhang Z, Smith DL, Smith JB. Human beta-crystallins modified by backbone cleavage, deamidation and oxidation are prone to associate. Exp Eye Res. 2003;77(3):259–72. doi: 10.1016/s0014-4835(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 22.Harms MJ, Wilmarth PA, Kapfer DM, Steel EA, David LL, Bachinger HP, Lampi KJ. Laser light-scattering evidence for an altered association of beta B1-crystallin deamidated in the connecting peptide. Protein Sci. 2004;13(3):678–86. doi: 10.1110/ps.03427504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrington V, McCall S, Huynh S, Srivastava K, Srivastava OP. Crystallins in water soluble-high molecular weight protein fractions and water insoluble protein fractions in aging and cataractous human lenses. Mol Vis. 2004;10:476–89. [PubMed] [Google Scholar]
- 24.Lapko VN, Smith DL, Smith JB. S-methylated cysteines in human lens gamma S-crystallins. Biochemistry. 2002;41(50):14645–51. doi: 10.1021/bi0267700. [DOI] [PubMed] [Google Scholar]
- 25.Lapko VN, Cerny RL, Smith DL, Smith JB. Modifications of human betaA1/betaA3-crystallins include S-methylation, glutathiolation, and truncation. Protein Sci. 2005;14(1):45–54. doi: 10.1110/ps.04738505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takemoto L, Horwitz J, Emmons T. Oxidation of the N-terminal methionine of lens alpha-A crystallin. Curr Eye Res. 1992;11(7):651–5. doi: 10.3109/02713689209000738. [DOI] [PubMed] [Google Scholar]
- 27.Schey KL, Finley EL. Identification of peptide oxidation by tandem mass spectrometry. Acc Chem Res. 2000;33(5):299–306. doi: 10.1021/ar9800744. [DOI] [PubMed] [Google Scholar]
- 28.Kamei A, Hamaguchi T, Matsuura N, Iwase H, Masuda K. Post-translational modification of alphaB-crystallin of normal human lens. Biol Pharm Bull. 2000;23(2):226–30. doi: 10.1248/bpb.23.226. [DOI] [PubMed] [Google Scholar]
- 29.Kamei A, Hamaguchi T, Matsuura N, Masuda K. Does post-translational modification influence chaperone-like activity of alpha-crystallin? I. Study on phosphorylation. Biol Pharm Bull. 2001;24(1):96–9. doi: 10.1248/bpb.24.96. [DOI] [PubMed] [Google Scholar]
- 30.Ma Z, Hanson SR, Lampi KJ, David LL, Smith DL, Smith JB. Age-related changes in human lens crystallins identified by HPLC and mass spectrometry. Exp Eye Res. 1998;67(1):21–30. doi: 10.1006/exer.1998.0482. [DOI] [PubMed] [Google Scholar]
- 31.Takemoto L. Increased cleavage of the c-terminal serine from alpha-A crystallin present in the high molecular weight aggregate fraction from human and bovine lenses. Curr Eye Res. 1999;19(5):450–5. doi: 10.1076/ceyr.19.5.450.5295. [DOI] [PubMed] [Google Scholar]
- 32.Thampi P, Hassan A, Smith JB, Abraham EC. Enhanced C-terminal truncation of alphaA- and alphaB-crystallins in diabetic lenses. Invest Ophthalmol Vis Sci. 2002;43(10):3265–72. [PubMed] [Google Scholar]
- 33.Srivastava OP, Srivastava K. BetaB2-crystallin undergoes extensive truncation during aging in human lenses. Biochem Biophys Res Commun. 2003;301(1):44–9. doi: 10.1016/s0006-291x(02)02975-3. [DOI] [PubMed] [Google Scholar]
- 34.Ball LE, Little M, Nowak MW, Garland DL, Crouch RK, Schey KL. Water permeability of C-terminally truncated aquaporin 0 (AQP0 1-243) observed in the aging human lens. Invest Ophthalmol Vis Sci. 2003;44(11):4820–8. doi: 10.1167/iovs.02-1317. [DOI] [PubMed] [Google Scholar]
- 35.Ball LE, Garland DL, Crouch RK, Schey KL. Post-translational modifications of aquaporin 0 (AQP0) in the normal human lens: spatial and temporal occurrence. Biochemistry. 2004;43(30):9856–65. doi: 10.1021/bi0496034. [DOI] [PubMed] [Google Scholar]
- 36.Aquilina JA, Truscott RJ. Cysteine is the initial site of modification of alpha-crystallin by kynurenine. Biochem Biophys Res Commun. 2000;276(1):216–23. doi: 10.1006/bbrc.2000.3461. [DOI] [PubMed] [Google Scholar]
- 37.Aquilina JA, Truscott RJ. Kynurenine binds to the peptide binding region of the chaperone alphaB-crystallin. Biochem Biophys Res Commun. 2001;285(5):1107–13. doi: 10.1006/bbrc.2001.5288. [DOI] [PubMed] [Google Scholar]
- 38.Aquilina JA, Truscott RJ. Identifying sites of attachment of UV filters to proteins in older human lenses. Biochim Biophys Acta. 2002;1596(1):6–15. doi: 10.1016/s0167-4838(01)00313-2. [DOI] [PubMed] [Google Scholar]
- 39.Eng JK, McCormack AL, Yates JR., III An Approach to Correlate Tandem Mass Sectral Data of Peptides with Amino Acid Sequences in a Protein Database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 40.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Yates JR, 3rd, Eng JK, McCormack AL. Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal Chem. 1995;67(18):3202–10. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- 42.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 43.Creasy DM, Cottrell JS. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics. 2002;2(10):1426–34. doi: 10.1002/1615-9861(200210)2:10<1426::AID-PROT1426>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 44.Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun Mass Spectrom. 2003;17(20):2310–6. doi: 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- 45.Pevzner PA, Dancik V, Tang CL. Mutation-tolerant protein identification by mass spectrometry. J Comput Biol. 2000;7(6):777–87. doi: 10.1089/10665270050514927. [DOI] [PubMed] [Google Scholar]
- 46.Pevzner PA, Mulyukov Z, Dancik V, Tang CL. Efficiency of database search for identification of mutated and modified proteins via mass spectrometry. Genome Res. 2001;11(2):290–9. doi: 10.1101/gr.154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han Y, Ma B, Zhang K. SPIDER: Software for Protein Identification from Sequence Tags with De Novo Sequencing Error. Proceedings of the 2004 IEEE Computational Systems Bioinformatics Conference; 2004. [DOI] [PubMed] [Google Scholar]
- 48.Hansen BT, Davey SW, Ham AJ, Liebler DC. P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J Proteome Res. 2005;4(2):358–68. doi: 10.1021/pr0498234. [DOI] [PubMed] [Google Scholar]
- 49.Tang WH, Halpern BR, Shilov IV, Seymour SL, Keating SP, Loboda A, Patel AA, Schaeffer DA, Nuwaysir LM. Discovering known and unanticipated protein modifications using MS/MS database searching. Anal Chem. 2005;77(13):3931–46. doi: 10.1021/ac0481046. [DOI] [PubMed] [Google Scholar]
- 50.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol. 2005;23(12):1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 51.Tanner S, Shu H, Frank A, Wang LC, Zandi E, Mumby M, Pevzner PA, Bafna V. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77(14):4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 52.Savitski MM, Nielsen ML, Zubarev RA. ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol Cell Proteomics. 2006;5(5):935–48. doi: 10.1074/mcp.T500034-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 54.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4(10):1487–502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Zybailov B, Coleman MK, Florens L, Washburn MP. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem. 2005;77(19):6218–24. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]
- 56.Spector A. The search for a solution to senile cataracts. Proctor lecture. Invest Ophthalmol Vis Sci. 1984;25(2):130–46. [PubMed] [Google Scholar]
- 57.Kim YH, Kapfer DM, Boekhorst J, Lubsen NH, Bachinger HP, Shearer TR, David LL, Feix JB, Lampi KJ. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, betaB1-crystallin. Biochemistry. 2002;41(47):14076–84. doi: 10.1021/bi026288h. [DOI] [PubMed] [Google Scholar]
- 58.Lampi KJ, Oxford JT, Bachinger HP, Shearer TR, David LL, Kapfer DM. Deamidation of human beta B1 alters the elongated structure of the dimer. Exp Eye Res. 2001;72(3):279–88. doi: 10.1006/exer.2000.0950. [DOI] [PubMed] [Google Scholar]
- 59.Robinson NE, Robinson AB. Prediction of primary structure deamidation rates of asparaginyl and glutaminyl peptides through steric and catalytic effects. J Pept Res. 2004;63(5):437–48. doi: 10.1111/j.1399-3011.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 60.Robinson NE, Robinson ZW, Robinson BR, Robinson AL, Robinson JA, Robinson ML, Robinson AB. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J Pept Res. 2004;63(5):426–36. doi: 10.1111/j.1399-3011.2004.00151.x. [DOI] [PubMed] [Google Scholar]
- 61.Frye EB, Degenhardt TP, Thorpe SR, Baynes JW. Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem. 1998;273(30):18714–9. doi: 10.1074/jbc.273.30.18714. [DOI] [PubMed] [Google Scholar]
- 62.Zarina S, Zhao HR, Abraham EC. Advanced glycation end products in human senile and diabetic cataractous lenses. Mol Cell Biochem. 2000;210(12):29–34. doi: 10.1023/a:1007015416572. [DOI] [PubMed] [Google Scholar]
- 63.Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, Haik GM. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest Ophthalmol Vis Sci. 2003;44(12):5287–92. doi: 10.1167/iovs.03-0573. [DOI] [PubMed] [Google Scholar]
- 64.Keller A, Purvine S, Nesvizhskii AI, Stolyar S, Goodlett DR, Kolker E. Experimental protein mixture for validating tandem mass spectral analysis. Omics. 2002;6(2):207–12. doi: 10.1089/153623102760092805. [DOI] [PubMed] [Google Scholar]
- 65.Deutsch EW, Eng JK, Zhang H, King NL, Nesvizhskii AI, Lin B, Lee H, Yi EC, Ossola R, Aebersold R. Human Plasma PeptideAtlas. Proteomics. 2005;5(13):3497–500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 66.Prince JT, Carlson MW, Wang R, Lu P, Marcotte EM. The need for a public proteomics repository. Nat Biotechnol. 2004;22(4):471–2. doi: 10.1038/nbt0404-471. [DOI] [PubMed] [Google Scholar]
- 67.Tanner S, Pevzner PA, Bafna V. Unrestrictive identification of post-translational modifications through peptide mass spectrometry. Nature Protocols. 2006;1(1):X. doi: 10.1038/nprot.2006.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.