

Abstract
Cancer results from the concerted performance of malignant cells and stromal cells. Cell types populating the microenvironment are enlisted by the tumor to secrete a host of growth-promoting cues, thus upholding tumor initiation and progression. Platelet-Derived Growth Factors (PDGFs) support the formation of a prominent tumor stromal compartment by as of yet unidentified molecular effectors. While PDGF-CC induces fibroblast reactivity and fibrosis in a range of tissues, little is known about the function of PDGF-CC in shaping the tumor-stroma interplay. Herein, we present evidence for a paracrine signaling network involving PDGF-CC and PDGFR-α in malignant melanoma. Expression of PDGF-C in a mouse model accelerated tumor growth through recruitment and activation of different subsets of cancer-associated fibroblasts. In seeking the molecular identity of the supporting factors provided by cancer-associated fibroblasts we made use of antibody arrays and an in vivo co-injection model to identify osteopontin as the effector of the augmented tumor growth induced by PDGF-CC. In conclusion, we establish paracrine signaling by PDGF-CC as a potential drug target to reduce stromal support in malignant melanoma.
Keywords: Cancer-associated fibroblast, Malignant melanoma, Osteopontin, Platelet Derived Growth Factor
Introduction
Cancer has traditionally been studied from the viewpoint of the tumor cell. However, the notion of the tumor as an organ in its own right is gaining acceptance. Recent studies have highlighted a carefully orchestrated and intricate interplay between transformed and genetically unstable tumor cells with local or bone-marrow derived ostensibly normal cells (1). Indeed, certain cancers, such as breast carcinomas and pancreatic adenocarcinomas, display prominent stromal compartments comprising a large fraction of the tumor volume (2, 3). The reactive tumor stroma is characterized by expansion and activation of the fibroblast population, excessive production of extra-cellular matrix, and persistent inflammation (4-6). The functional importance of cell types in the tumor stroma is highlighted by studies demonstrating a higher growth rate of the same tumor transplanted into mice deficient for Trp53 compared to wildtype mice (7). Gross phenotypic changes in terms of gene expression, and even genetic alterations, have been described in tumor stromal cells compared to their normal counterparts (8-10). In addition, an expanded and fibrotic tumor stroma presents a problem in terms of drug penetration, and thereby efficacy. In light of these findings, the tumor stromal compartment is an exciting and largely unchartered territory for exploring novel strategies to treat cancer.
A number of studies demonstrate the importance of platelet-derived growth factors (PDGFs) for the recruitment and phenotypic character of cancer-associated fibroblasts (CAFs). Indeed, ectopic expression of PDGF-B in immortalized, but non-tumorigenic keratinocytes, is sufficient to confer tumorigenic potential through the formation of a rich stroma characterized by mesenchymal cell proliferation and angiogenesis (11). In addition, PDGF-BB induces the formation of a prominent and growth-promoting stroma in xenograft studies of melanoma (12). Furthermore, paracrine signaling by tumor cell-derived PDGF-AA was identified as a major driving force in the recruitment of CAFs in xenograft studies of breast and lung carcinoma (13, 14). Lastly, tumor cells which were experimentally deprived of their ability to produce vascular endothelial growth factor-A (VEGF-A) were found to compensate by secreting PDGF-AA to attract stromal fibroblasts, which in turn provided VEGF-A to stimulate angiogenesis (15). The functional importance of tumor cell expression of the PDGF ligands PDGF-C and -D is, however, less well known. Ectopic expression of PDGF-D enhances tumor growth both through direct growth stimulatory effects, as well as through promotion of angiogenesis and pericyte recruitment (16, 17). Also, transgenic expression of PDGF-C in the mouse liver results in tissue fibrosis and subsequent development of hepatocellular carcinoma (18).
Despite the fact that many human tumors secrete PDGF ligands, and display expression of PDGF receptors on various stromal cell populations such as CAFs and pericytes (19), the molecular details of the tumor growth-promoting effects of paracrine PDGF signaling are not well understood. Prompted by findings of prominent expression of PDGF-C by many common skin cancers, including melanoma, basal cell carcinoma and squamous cell carcinoma, in contrast to the corresponding normal skin, we set out to investigate the phenotypic significance of PDGF-C expression and to molecularly define the role for PDGF-CC in maintaining a growth-promoting tumor microenvironment.
Materials and Methods
Skin tumor tissue array
The human skin tumor and normal tissue array (Tissue Array Network, BC21011) was immunostained for PDGF-CC as described (20) and for PDGFR-α (1:40, Cell Signaling) as described below.
Generation of cell lines stably expressing PDGF-C
Full-length human PDGF-C cDNA was cloned into the pcDNA3.1 vector to generate the PDGF-C expression plasmid. For generation of stably expressing cell lines, B16/F10 cells were transfected with the PDGF-C plasmid (B16/PDGF-C), or empty vector (B16/mock), and 72 h post transfection 500 μg/ml Zeocin (Invitrogen) was added to the cultures for selection of resistant mass cultures. For propagation, these cell lines were kept under continuous selection (250 μg/ml). The mass culture was tested for PDGF-CC expression by immunostaining using a monoclonal antibody generated in-house.
Receptor stimulation and immunoprecipitation
B16/mock, B16/PDGF-C, and porcine aortic endothelial cells stably expressing PDGFR-α (α-PAE) were stimulated with 100 ng/ml core PDGF-CC for 1 h on ice. Immunoprecipitation was performed with PDGFR-α rabbit antiserum (LICR, Uppsala branch), an antibody against osteopontin (R&D Systems) or a pool of antibodies against FGF-2 (Chemicon, Advanced Targeting Systems, and Santa Cruz Biotechnology) and western blot analysis was performed using 1 μg/ml antibody against phospho-tyrosine (Santa Cruz Biotechnology), osteopontin, PDGF-CC or FGF-2, respectively.
In vitro proliferation assay
Fifteen thousand B16/mock or B16/PDGF-C cells were seeded in triplicates in 12-well plates. Cells were subsequently counted using a Beckman Coulter Z2 particle counter 24, 48 and 72 h after seeding.
Animal care and tumor establishment
All animal experiments were approved by the local committee for animal experiments. To establish B16/mock and B16/PDGF-C tumors, 2*106 cells were injected subcutaneously in the dorsal left flank into young C57Bl/6J mice. For co-injection studies, C57Bl/6J SCID mice were injected with 5*105 B16 cells, wt mouse embryonic fibroblasts (MEFs) or MEFs from osteopontin deficient mice (21), alone or in combination at a 1:1 ratio. Tumors were considered as established when larger than 1×1 mm. Tumor volume was calculated as π/6×length×width2.
Tissue preparation, histology and immunostaining
Mice were perfused with PBS followed by 2% PFA. For paraffin-embedding, tumors were postfixed in 2% paraformaldehyde at 4°C overnight. For cryo-preservation, tumors were kept in 30% sucrose at 4°C overnight, followed by embedding in cryosectioning medium.
Frozen sections were air dried and fixed in cold acetone for 10 min. Paraffin embedded sections were deparaffinized followed by quenching of endogenous peroxidase activity using 3% H2O2 for 10 min at room temperature. Antigen retrieval was performed using 0.25 mg/ml trypsin diluted in PBS at 37°C for 30 min (TUNEL), 20 min (PDGFR-α), or using 0.5 mg/ml trypsin for 5 min (CD31, NG2), followed by boiling in 10 mM citric acid pH 6.0 for 20 min (BrdU, PDGFRα, FSP-1, F4/80, osteopontin). Antibodies and dilutions used were: TUNEL according to manufacturer’s instructions (Roche), PDGFR-α (2 μg/ml, R&D Systems), CD31 (0.16 μg/ml, BD Biosciences), BrdU according to manufacturer’s instructions (Labeling and detection kit II, Roche), NG2 (1:200, Chemicon), FSP-1 (1:300), F4/80 (1:50, BD Biosciences) and osteopontin (1:100, R&D Systems). Appropriate HRP-conjugated secondary antibodies were applied followed by amplification using the Vectastain ABC system (Vector Laboratories). For immunofluorescence, fluorochrome-conjugated secondary antibodies (Invitrogen) were used and sections mounted using media containing DAPI (Vector Laboratories), where appropriate.
Picrosirius red staining was performed according to standard protocol using 1% Direct Red 80 diluted in picric acid (Fluka).
Antibody array
Antibody arrays (RayBio Mouse Cytokine Antibody Array C series 1000.1) were used according to provided instructions. A total of 500 μg of pooled protein lysate from three different B16/mock or B16/PDGF-C tumors was used. Quantification was digitally performed using the Image Reader LAS-1000 Pro software (Fujifilm). Signals were normalized against the internal standard of each membrane. Final signal intensity was taken as the average of two separate experiments.
PCGF-CC stimulation of NIH/3T3 cells
NIH/3T3 cells were stimulated with 50 ng/ml PDGF-CC for 6 h at 37°C. Analysis of transcript expression was performed using quantitative RT-PCR in relation to L19.
| Primers: | ||
| L19 | 5’-GGT GAC CTG GAT GAG AAG GA -3’ | forward |
| 5’-TTC AGC TTG TGG ATG TGC TC -3’ | reverse | |
| FGF-2 | 5’-GGC TGC TGG CTT CTA AGT GT -3’ | forward |
| 5’-CCG TTT TGG ATC CGA GTT TA -3’ | reverse | |
| osteopontin | 5’-TGC ACC CAG ATC CTA TAG CC-3’ | forward |
| 5’-CTC CAT CGT CAT CAT CAT CG-3’ | reverse |
Statistical analysis
All measurements are depicted as average ± standard deviation. All analyses utilized the Student’s double-sided, unpaired t-test with statistical significance defined as p<0.05.
Results
Expression of PDGF-C by tumor cells and PDGFR-α by stromal cells is an apparent feature of human skin tumors
To assess the expression pattern of PDGF-C in a variety of skin cancers, we made use of tissue arrays comprised of normal skin and a large selection of samples from a variety of skin cancers, including melanoma, basal cell carcinoma, and squamous cell carcinoma. Immunohistochemical staining for PDGF-CC revealed prominent expression by the tumor cell compartment in malignant specimens, whereas PDGF-CC was exclusively expressed in hair follicles of corresponding normal skin (Fig. 1a). In particular, production of PDGF-CC by tumor cells was noted in malignant melanomas (Fig. 1a), prompting us to further study the role of PDGF-CC in the development of this disease. To localize cell types expressing the receptor for PDGF-CC, i.e. PDGFR-α, in human melanomas, we performed immunostaining of identical tissue arrays. As detailed in Fig. 1a, normal skin exhibited only very weak staining for PDGFR-α. In contrast, we found abundant expression of PDGFR-α in cells histologically and morphologically identified as fibroblasts populating the stromal compartment of malignant melanoma (Fig. 1a). Thus, we have identified PDGF ligand and receptor expression as an apparent feature of a subset of human malignant melanoma.
Figure 1. PDGF-CC and PDGFR-α are expressed in human malignant melanoma.

(a) Representative pictures of immunostaining for PDGF-CC and PDGFR-α in normal human skin and in malignant melanoma (400x magnification). (b) Immunostaining for PDGF-CC in B16 cells transfected with either human PDGF-C (B16/PDGF-C) or empty vector (B16/mock) (630x magnification). (c) Immunoprecipitation (IP) of PDGFR-α followed by western blot (WB) for phosphorylated proteins from a receptor stimulation assay using PDGF-CC to stimulate B16/PDGF-C and B16/mock cells. Porcine aortic endothelial cells stably transfected with PDGFR-α (α-PAE) were used as a positive control. (d) Left panel, assessment of in vitro growth rate of B16/mock and B16/PDGF-C cells in medium supplemented with serum. Similar results were obtained using serum-free conditions (data not shown). Right panel, in vivo growth rate of B16/mock and B16/PDGF-C cells injected subcutaneously into syngeneic C57Bl/6J mice (n=6). *p<0.05, Student’s t-test.
Expression of PDGF-C by tumor cells accelerates tumor growth in vivo
To study the functional implications of paracrine PDGF-CC/PDGFR-α signaling, we made use of the B16 mouse model of melanoma. B16 tumors display a thin fibrous capsule surrounding the tumor parenchyma, but a relative lack of infiltrating stroma (data not shown), making it a suitable model with high sensitivity to determine the effects of paracrine PDGF-CC signaling. To investigate the functional significance of expression of PDGF-C by tumor cells, we therefore transfected B16 cells with a vector containing the cDNA for full-length human PDGF-C, or with empty vector (B16/PDGF-C and B16/mock, respectively). RT-PCR analysis and immunostaining confirmed expression of PDGF-C transcript and PDGF-CC protein by B16/PDGF-C cells, but not by B16/mock cells (Fig. 1b and data not shown). While exceedingly low expression levels of PDGFR-α were detected by quantitative RT-PCR (data not shown), we were unable to detect activated PDGFR-α following stimulation of either B16/mock or B16/PDGF-C cells with PDGF-CC (Fig. 1c). Importantly, B16/PDGF-C cells did not display an altered growth rate in vitro compared to B16/mock cells (Fig. 1d, left panel), indicating that B16 tumors are not intrinsically dependent on the PDGF/PDGFR system for autocrine growth stimulation, consistent with earlier reports (17). In contrast, when injected into syngeneic C57Bl/6J mice, B16/PDGF-C tumors exhibited a dramatically accelerated growth rate compared to B16/mock tumors (Fig. 1d, right panel). At the completion of the study, mice with B16/PDGF-C tumors presented with an average tumor volume of 410±109 mm3, whereas mice with B16/mock tumors displayed a significantly smaller average tumor volume of 122±36 mm3 (p<0.05, Student’s t-test). The growth-promoting effects of PDGF-CC on B16 tumors were further confirmed using an independently established mass culture of B16/PDGF-C cells (data not shown).
Next, we sought to assess the general effects of PDGF-C expression on tumor growth characteristics. Quantitation of tumor cell proliferation by incorporation of BrdU revealed that B16/PDGF-C tumor cells exhibited a 1.6-fold higher rate of proliferation compared to B16/mock cells (Fig. 2a; p<0.001, Student’s t-test). Moreover, analysis of tumor cell apoptotic rate using TUNEL staining demonstrated that B16/PDGF-C tumor cells were 1.9-fold less prone to undergo apoptosis compared to B16/mock cells (Fig. 2b; p<0.05, Student’s t-test). Since various PDGF isoforms are known to stimulate angiogenesis, either by direct or indirect mechanisms, we investigated the angiogenic phenotype in B16/PDGF-C and B16/mock tumors. B16/PDGF-C tumors displayed a significantly higher blood vessel density compared to B16/mock tumors, as assessed by immunostaining for the endothelial cell marker CD31 (Fig. 2c; p<0.05, Student’s t-test). However, as judged by pericyte coverage and by morphological criteria, no difference was found in the degree of maturation or in the gross anatomy of the vascular tree in the two different cohorts of B16 tumors (Fig. 2d and data not shown). We conclude that B16/PDGF-C tumors exhibited a faster growth rate due to an increased rate of proliferation and a decreased rate of apoptosis, most likely consequential to an enhanced angiogenic response.
Figure 2. B16/PDGF-C tumors display an increased proliferation, decreased apoptosis and enhanced angiogenic response.

(a) Proportion of BrdU positive proliferating cells (upper panel) and representative pictures of BrdU immunostaining of B16/mock and B16/PDGF-C tumor sections (lower panel; 400x magnification). (b) Proportion of TUNEL positive apoptotic cells (upper panel) and representative pictures of apoptotic labeling of B16/mock and B16/PDGF-C tumor sections (lower panel; 400x magnification). Arrows denote apoptotic cells. (c) Number of CD31+ blood vessels per field (upper panel) and representative pictures of CD31 immunostaining of B16/mock and B16/PDGF-C tumor sections (lower panel; 400x magnification). (d) Pericyte coverage assessed as NG2+ vessels in relation to number of CD31+ vessels (upper panel). Representative pictures of NG2 immunostaining of B16/mock and B16/PDGF-C tumor sections (lower panel; 400x magnification). All quantifications were made on at least 4 tumors of each type using at least 10 randomly selected fields of vision per tumor. *p <0.05; *** p<0.001, Student’s t-test.
PDGF-CC induces the recruitment of cancer-associated fibroblasts into tumors
Since B16 cells did not express significant levels of PDGFR-α, or exhibit growth stimulation consequential to PDGF-C expression (Fig. 1c and d), we analyzed B16/PDGF-C tumors for possible paracrine interactions involving tumor cell-derived PDGF-CC and the stromal compartment. First, we assessed the presence of cells expressing PDGFR-α by immunostaining. The fibrous capsule surrounding B16 tumors consisted predominantly of cells morphologically identified as fibroblasts with prominent expression of PDGFR-α (Fig. 3a, top panel). Whereas the fibrous capsule was thin and sharply defined in B16/mock tumors, B16/PDGF-C tumors displayed a thicker capsule with a diffuse border with the tumor parenchyma (Fig. 3a, top panel). To enable easier visualization of cells expressing PDGFR-α, we made use of knock-in mice in which a nuclearly targeted green fluorescent protein (GFP) construct was substituted for one of the PDGFR-α alleles (PDGFR-α/GFP; (22)). B16/PDGF-C and B16/mock cells were injected into PDGFR-α/GFP mice and allowed to form tumors, which were excised and analyzed for their content of cells expressing GFP. As shown in Fig. 3a (bottom panel), whereas B16/mock tumors and the center of B16/PDGF-C tumors were devoid of cells expressing PDGFR-α, the edge of B16/PDGF-C tumors displayed a substantial infiltration of PDGFR-α/GFP+ cells. To establish the identity of the PDGFR-α+ cells infiltrating the B16/PDGF-C tumors, we performed immunostaining for the fibroblast marker fibroblast-specific protein-1 (FSP-1; S100A4). In contrast to B16/mock tumors, which were devoid of fibroblasts, cells expressing FSP-1 were prevalent throughout B16/PDGF-C tumors (Fig. 3b, top panel). Interestingly, the recruitment of FSP-1+ cells into B16 tumors was dependent on activation of PDGFR-α, since analysis of tumors derived from B16 cells transfected with PDGF-B (17), a PDGFR-β ligand, revealed a comparable lack of infiltrating stromal fibroblasts expressing FSP-1 (Fig. 3b, top panel). Consistent with the notion that signaling by PDGFR-α is crucial for the recruitment and phenotype of fibroblasts, co-expression analysis of PDGFR-α and FSP-1 revealed three different populations of infiltrating stromal fibroblasts (Fig. 3b, bottom panel). On the edge of B16/PDGF-C tumors, a population of cells with high expression of PDGFR-α, as assessed by expression of GFP, but lacking expression of FSP-1, was apparent (designated PDGFR-αhigh; Fig. 3b, bottom panel, arrows). Secondly, a significant proportion of cells expressing PDGFR-α albeit at lower levels, in the surrounding fibrous capsule and on the edge of tumors, also produced FSP-1 (designated PDGFR-αlow/FSP-1; Fig. 3b, bottom panel, arrowheads). Finally, a third population of stromal fibroblasts exclusively expressing FSP-1, but not PDGFR-α resided in the center of B16/PDGF-C tumors, but not in B16/mock tumors (designated FSP-1; Fig. 3b, bottom panel, stars). In addition, all three fibroblast populations expressed the myofibroblast marker α-smooth muscle actin to various degrees, indicating that they are indeed CAFs (data not shown). To further substantiate an increased presence of stromal fibroblasts in B16/PDGF-C tumors compared to B16/mock tumors, we performed picrosirius red staining for the presence of interstitial collagen, as fibroblasts are the main producers of collagen-I. Whereas B16/mock tumors mainly exhibited deposits of collagen in the surrounding capsule, B16/PDGF-C tumors displayed large streaks of collagen deposits invading into the tumor mass (Fig. 3c), indicative of an increased fibroblast presence within the tumor. Additionally, interstitial collagen between the tumor cells was found to be moderately increased in B16/PDGFC tumors (data not shown).
Figure 3. PDGF-C expression promotes recruitment of CAFs.

(a) Top panel, immunostaining for PDGFR-α in B16/mock and B16/PDGF-C tumor sections (400x magnification). Bars represent the thickness of the fibrous capsule surrounding the tumor. Bottom panel, visualization of infiltrating PDGFR-α+ cells in PDGFR-α/GFP mice. Representative pictures from B16/mock and B16/PDGF-C tumors (100x magnification). Dotted line denotes boundary between skin and tumor. (b) Top panel, immunostaining for FSP-1 in B16/mock, B16/PDGF-C and B16/PDGF-B tumor sections (400x magnification). Bottom panel, immunostaining for FSP-1 (blue) of tumor sections from B16/PDGF-C tumors grown in PDGFR-α/GFP mice (PDGFR-α, green) at the tumor edge and tumor center (400x magnification). Arrows denote CAFs of the PDGFR-αhigh subclass, arrowheads denote the PDGFR-αlow/FSP-1 subclass of CAFs, and stars denote the FSP-1 subclass of CAFs. (c) Picrosirius red staining for collagen in B16/mock and B16/PDGF-C tumor sections (200x magnification). (d) Immunostaining for the macrophage marker F4/80 of B16/mock and B16/PDGF-C tumor sections (200x magnification).
To investigate whether PDGFR-α designated a population of macrophages, a cell type reported to express PDGFRs upon activation, we performed immunostaining for the macrophage marker F4/80. As seen in Fig. 3d, macrophages were prevalent to a similar degree in necrotic areas of both B16/mock and B16/PDGF-C tumors, indicating that macrophages are not recruited into tumors by secretion of PDGF-CC by B16 cells.
PDGF-CC induces production of tumor growth-promoting factors
Having documented that ectopic expression of PDGF-C in B16 melanoma cells accelerated tumor growth and recruited distinct populations of stromal fibroblasts into tumors, we sought to identify the molecular identity of the tumor growth-promoting factors supplied by the CAFs. We made use of antibody arrays to simultaneously and quantitatively probe the abundance of 96 different secreted growth factors, cytokines, chemokines and transmembrane receptors in pools of lysates from B16/PDGF-C tumors compared to B16/mock tumors. In agreement with our recent studies, paracrine signaling by PDGF was found to stimulate the secretion of fibroblast growth factor-2 (FGF-2; bFGF), a prototypical angiogenic factor (23). Moreover, B16/PDGF-C tumor lysates contained a significantly higher concentration of osteopontin (SPP1), a soluble extracellular matrix protein with known effector functions in tumor growth, angiogenesis and metastasis. As assessed by densitometric quantitation of two independent hybridizations of the antibody arrays, FGF-2 and osteopontin were present at 1.4-fold and 2-fold higher abundance, respectively, in lysates from B16/PDGF-C tumors compared with B16/mock tumors (Fig. 4a). The increased presence of PDGF-CC, FGF-2 and osteopontin in B16/PDGF-C tumor lysates was further validated by an immunoprecipitation and western blot strategy using materials from three independent B16/PDGF-C and B16/mock tumors. The results corroborated an increased expression of PDGF-CC, as expected, and a dramatically increased presence of both FGF-2 and osteopontin in lysates from B16/PDGF-C compared to B16/mock tumors (Fig. 4b). To exclude the possibility that intrinsic differences in B16/PDGF-C and B16/mock cells accounted for the augmented secretion of FGF-2 and osteopontin, we performed quantitative RT-PCR analyses of RNA isolated from B16/PDGF-C and B16/mock cells. As shown in Fig. 4c, expression of neither FGF-2, nor osteopontin by B16 cells was increased following transfection of PDGF-C, indicating that the higher production of FGF-2 and osteopontin in whole tumor lysates was the result of paracrine signaling events induced by PDGF-CC.
Figure 4. B16/PDGF-C tumors show an increased abundance of FGF-2 and osteopontin.
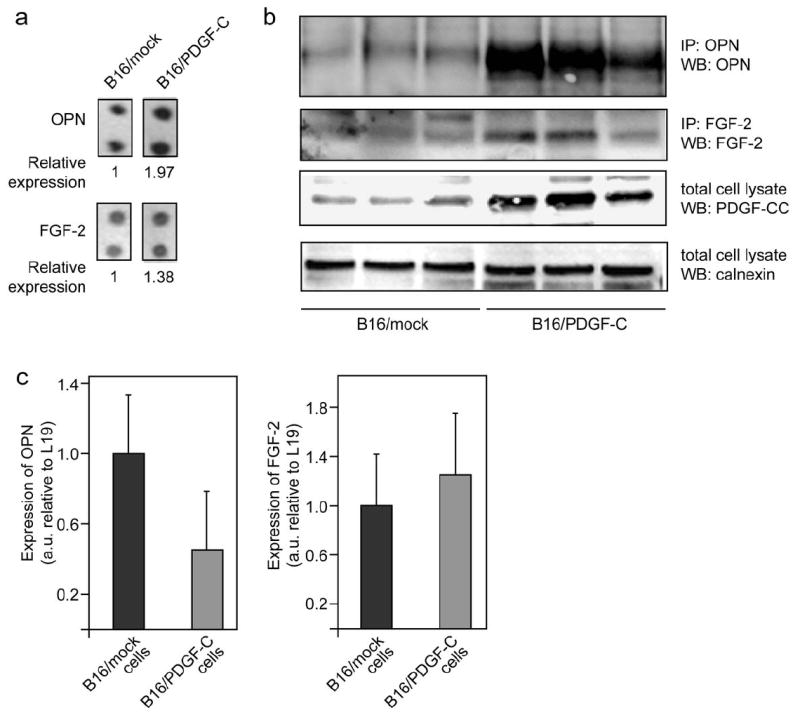
(a) Pooled protein lysates from 3 separate B16/mock and B16/PDGF-C tumors were applied to antibody arrays. Densitometry was used to quantify the intensity of each spot following normalization to internal standards. (b) Validation of increased osteopontin (OPN) and FGF-2 levels in B16/PDGF-C tumors using immunoprecipitation (IP) and western blot (WB) on separate protein lysates from 3 tumors of each type. WB for PDGF-C verifying increased levels. Calnexin was used as an independent loading control. (c) Relative expression of transcripts for osteopontin and FGF-2, respectively, as measured by quantitative RT-PCR of RNA derived from B16/mock and B16/PDGF-C cell lines.
Osteopontin is produced by cancer-associated fibroblasts in B16/PDGF-C tumors and confers growth-stimulatory functions
We decided to focus our efforts on the regulation, expression and functional significance of osteopontin by stromal fibroblasts in B16/PDGF-C tumors. To determine which cell type within B16 tumors was responsible for the expression of osteopontin, we performed immunostainings. Osteopontin was found to be expressed by cells populating the fibrous capsule surrounding the tumor, and in single cells infiltrating B16/PDGF-C tumors (Fig. 5a). Co-immunostaining analysis revealed that most cells producing osteopontin in the tumor capsule were of the PDGFR-αlow/FSP-1 class of CAFs, as they expressed all three proteins (Fig. 5b, arrowheads), although a subset of fibroblasts resident in the tumor capsule of the FSP-1 class also produced osteopontin (Fig. 5b, stars). The majority of osteopontin-producing cells within the tumor parenchyma were of the FSP-1 class, as they did not express PDGFR-α (Fig. 5b, stars). Thus, we conclude that osteopontin is predominantly expressed by CAFs in B16/PDGF-C tumors. Furthermore, we stimulated cultured fibroblasts with PDGF-CC and assessed expression of osteopontin. As detailed in Fig. 5c, fibroblasts expressed high amounts of osteopontin, and stimulation with PDGF-CC for 6 h increased the production even further, by 2.4-fold compared to unstimulated fibroblasts, demonstrating that the expression of osteopontin is directly regulated by signaling induced by PDGF-CC in fibroblasts.
Figure 5. Osteopontin is expressed by CAFs, and is functionally important for tumor growth.

(a) Immunostaining for osteopontin (OPN) in B16/PDGF-C tumor sections (400x magnification). (b) Immunostaining for FSP-1 (blue), osteopontin (red) in tumor sections from B16/PDGF-C tumors grown in PDGFR-α/GFP mice (PDGFRα, green) (400x magnification). Arrowheads denote the PDGFR-αlow/FSP-1 subclass of CAFs; stars denote the FSP-1 subclass of CAFs. (c) Relative expression of osteopontin transcript, as measured by quantitative RT-PCR, following stimulation of NIH/3T3 fibroblasts with PDGF-CC. (d) Left panel, latency of B16 tumors injected alone (n=10), or in combination with a 1:1 ratio of wt (n=10) or osteopontin deficient (OPN KO) (n=9) mouse embryonic fibroblasts (MEFs). Right panel, growth of tumors established from B16 cells co-injected with a 1:1 ratio of wt or osteopontin deficient MEFs, compared to injection of each cell type separately (MEFs alone, n=3)). *p<0.05; **p<0.01; ***p<0.001, Student’s t-test.
Finally, we sought to assess the functional significance of the expression of osteopontin by CAFs. To this end, we established a co-injection model of B16 melanoma cells with mouse embryonic fibroblasts (MEFs) in C57Bl/6J SCID mice. MEFs isolated from wildtype mice (wt MEFs; (21)) were co-injected with B16 melanoma cells at a ratio of 1:1. As shown in Fig. 5d, wt MEFs reduced the latency of B16 tumors from 10±1.5 days to 8.2±1.2 days (p<0.01, Student’s t-test). Moreover, wt MEFs conferred a significant growth advantage to established B16 tumors (Fig. 5d; p<0.05, Student’s t-test). In contrast, MEFs isolated from osteopontin-deficient mice (21) and co-injected with B16 melanoma cells neither affected the latency of tumors (9.8±1.8 days), nor accelerated the growth of established tumors (Fig. 5d), indicating that osteopontin supplied by CAFs is functionally important for the growth of B16 tumors. Thus, we conclude that PDGF-CC enhances the growth of B16 tumors by recruitment of CAFs that secrete osteopontin.
Discussion
Herein we present evidence for a paracrine signaling circuitry involving tumor cell-derived PDGF-CC signaling to PDGFR-α expressed by stromal fibroblasts in human skin malignancies. We further demonstrate a functional significance of paracrine PDGF-CC signaling in the recruitment of CAFs into experimental mouse melanoma, thereby augmenting both tumor growth and angiogenesis (Fig. 6). The molecular effectors of the promotion of tumorigenesis were identified as the prototypical angiogenic factor FGF-2 and the diffusible extracellular matrix protein osteopontin (Fig. 6). Accordingly, fibroblast-supported tumor growth was dependent on expression of osteopontin in co-injection studies. Hence, we have revealed a paracrine signaling network involving PDGF-CC that serves to regulate the recruitment and phenotype of CAFs into tumors and which is readily available for therapeutic intervention (Fig. 6).
Figure 6. Schematic illustration of the effect of PDGF-C expression by tumor cells.
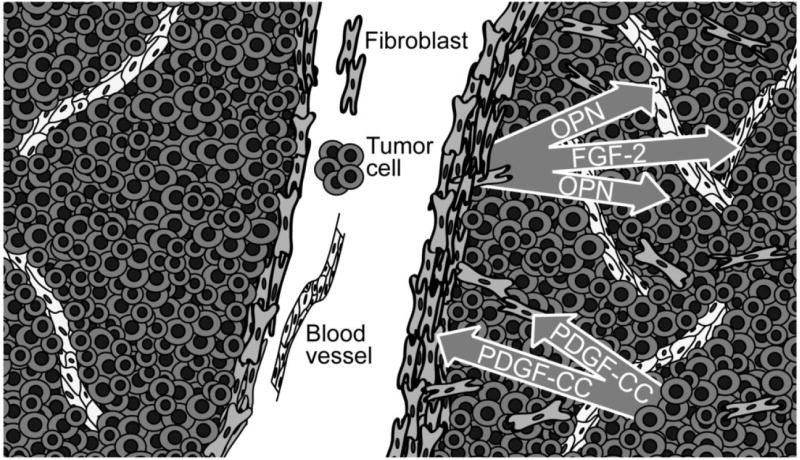
Malignant cells (dark grey) express PDGF-C that signals in a paracrine manner to recruit fibroblasts (light grey), thereby promoting formation of a thicker fibrous capsule surrounding the tumor and migration of fibroblasts into the tumor mass. Cancer-associated fibroblasts produce osteopontin (OPN) and fibroblast growth factor-2 (FGF-2), which in turn exert tumor growth-stimulatory and pro-angiogenic effects (endothelial cells, white).
Recruitment of cancer-associated fibroblasts into tumors
CAFs are known to modulate tumorigenic properties of neoplastic cells, including proliferation, apoptosis and angiogenesis (6). Experiments involving co-injection of fibroblasts of different origin and tumor cells demonstrate that, in contrast to normal fibroblasts, CAFs promote tumor growth (5). Moreover, recent studies have revealed extensive changes in the phenotype, and even the genotype, of CAFs, compared to their normal counterparts (9, 10). However, the source for recruitment of stromal fibroblasts into tumors has not been clearly defined yet. While local, resident fibroblasts are in many cases abundant and the most likely pool for recruitment, there is also evidence suggesting that bone-marrow derived mesenchymal stem cells are recruited into experimental tumors (24). There is abundant evidence that PDGF-CC induces fibrosis in a range of tissues, including liver, kidney and heart (18, 25, 26). Our studies reveal that expression of PDGF-C by tumor cells serves to engage fibroblasts also in the malignant process. Interestingly, the recruited fibroblasts were of different subtypes, as defined by their expression of PDGFR-α, FSP-1 and α-SMA. Paradoxically, not all of the CAF subtypes displayed PDGFR-α at readily detectable levels, expression of which would have been assumed to be required for recruitment by PDGF-CC. Also, a majority of the fibroblasts within the tumor parenchyma that expressed the growth-stimulatory protein osteopontin expressed FSP-1, but not PDGFR-α. We speculate that a progenitor population of stromal fibroblasts, which express high levels of PDGFR-α, are recruited and expanded by tumor-derived PDGF-CC, and subsequently educated by the tumor microenvironment into mature and tumor growth-promoting CAFs, denoted by the activated fibroblast markers FSP-1 and α-SMA. Interestingly, a similar role has been described for PDGFR-β in determining pericyte fate, i.e. PDGFR-β denotes a progenitor population of pericytes which is recruited to tumor blood vessels and subsequently differentiated into fully mature pericytes distinguished by expression of NG2, desmin and α-SMA, while loosing expression of PDGFR-β (27). Intriguingly, PDGFR-α is a marker for mesenchymal stem cells (28), and PDGF-CC has been identified as an autocrine growth factor for Ewing’s sarcoma, a cancer thought to originate from mesenchymal stem cells (29). The notion that growth-promoting mesenchymal stem cells are recruited into tumors by PDGF-CC warrants further examination.
Cancer-associated fibroblast effector functions aid in tumor growth and progression
Stromal fibroblasts are rich sources for growth factors, survival factors and pro-angiogenic factors during wound healing (30), as well as in tumorigenesis (4, 6, 31, 32). Nevertheless, the molecular identity of factors of direct functional importance for the tumor-growth promoting effects or for the maintenance of the CAF phenotype is still not understood in detail. A survey of angiogenic inducers in a xenograft study showed that all factors, apart from VEGF-A, were produced in higher quantities by stromal cells, as compared to the overt tumor cells (33). Moreover, a recent study employing laser capture microdissection of normal and prostate cancer-derived stroma identified an autocrine loop within CAFs involving the chemokine CXCL14. Here, CXCL14 was found to be an effector of the stimulatory support provided to the tumor in terms of regulation of angiogenesis (Augsten M, Östman A et al, personal communication). Another chemokine, fibroblast-derived CXCL12/SDF-1, serves to recruit endothelial progenitor cells into the neo-vasculature of tumors, thereby promoting angiogenesis and tumor growth (34). Finally, using mice expressing GFP under the VEGF promoter, Fukumura et al. demonstrated a dramatic induction of VEGF promoter activity in stromal cells in spontaneous mammary tumors or upon implantation of solid tumors (35). Indeed, consistent with this, an inability of tumor cells to produce VEGF-A mandates recruitment of VEGF-secreting stromal fibroblasts in a PDGFR-α dependent manner (15).
We have recently identified FGF-2 derived from stromal fibroblasts as a crucial mediator of the angiogenic response in cervical squamous cell carcinoma (23). The production of FGF-2 was attenuated by administration of PDGFR inhibitors, yielding therapeutic efficacy in a stringent genetically engineered mouse model of cervical cancer (23). Consistent with our earlier study, we here found that FGF-2 is present in higher abundance in tumors expressing PDGF-C. In addition, we found that stromal fibroblasts within B16/PDGF-C tumors produce high levels of osteopontin. Intriguingly, FGF-2 and osteopontin are known to act in concert to promote tumor angiogenesis (36). Osteopontin is a soluble extra-cellular matrix protein harboring an RGD-site with a known function in suppression of apoptosis, promotion of angiogenesis and induction of metastasis (37). While many cell-surface proteins are known to bind osteopontin, including CD44 and αv, α5, β1 and β3 integrins, the exact signaling receptor(s) for the various functions of osteopontin is not known. In agreement with our finding of osteopontin as a growth-stimulatory factor for mouse melanoma, osteopontin was identified by molecular profiling as a progression marker for human melanomas (38). Furthermore, in a recent study, osteopontin was part of a 26-gene expression profile with potent predictive power for good or bad prognosis of breast cancer patients (39). High expression of osteopontin corresponded to a bad prognosis for recurrence of metastatic breast cancer, suggestive of functional importance also in other cancers than melanoma.
Further evidence for a role for CAF-derived osteopontin in tumor biology comes from a study in which PDGFR-α+ CAFs were isolated from a transgenic mouse model of squamous cell carcinoma of the skin. In contrast to fibroblasts isolated from non-transgenic littermates, PDGFR-α+ CAFs from K14-HPV16 mice expressed abundant levels of osteopontin (Erez N, Hanahan D et al., personal communication). Also, in agreement with our studies, and with the notion that the tumor stroma is in many aspects similar to a wound that does not heal (40), an analogous situation was recently described for skin wound healing, during which macrophage-derived PDGF(s) induced production of osteopontin by reactive fibroblasts (41). In this context, osteopontin hindered wound healing and induced excessive fibrosis. Taken together with our data, osteopontin appears to be of particular importance for various aspects of skin pathology, begging the question whether fibroblast-derived osteopontin constitutes a general therapeutic target for skin malignancies; a question that deserves further studies. Osteopontin was recently also identified as a factor necessary for systemic instigation of indolent tumors by activating and recruiting bone marrow-derived cells (42). Whether osteopontin in addition serves to directly regulate the recruitment and phenotype of CAFs remains to be studied. However, mice deficient for osteopontin display fewer interstitial fibroblasts in a mouse model of renal fibrosis (43).
Therapeutic targeting of cancer-associated fibroblasts
The notion of targeting CAFs to inhibit tumor growth and progression is attractive due to the increasing identification of stimulatory factors derived from the tumor stroma. Targeting of stromal fibroblast effector functions by drugs inhibiting the action of osteopontin, FGF-2, CXCL12, CXCL14, and many more, singularly or in combination, holds promise of producing therapeutic efficacy. In addition, approaches to prevent the recruitment and phenotypic conversion of CAFs are highly warranted. Based on our studies, as well as earlier work (14, 15, 23), we propose antagonism of paracrine signaling by PDGF-CC and PDGFR-α as one such strategy. Recent genetic evidence supports a functional role for epithelial-mesenchymal crosstalk directed by PDGFR-α during the embryonal development of the palate (44). We speculate that pharmacological disruptance of such signaling may be fruitful also in treating various types of cancers. Tyrosine kinase inhibitors incorporating action against the PDGFRs, such as imatinib, sunitinib and sorafenib, are already in clinical use with demonstrable efficacy (45). Consistent with this idea, treatment with imatinib attenuates fibroblast support and tumor growth in mouse models of cervical carcinoma and PDGF-CC induced hepatocellular carcinoma (23, 46). However, in a small Phase II-trial where 16 evaluable patients with metastatic melanoma had received treatment with imatinib, no objective clinical efficacy was observed (47). There may be several reasons for this, including the low number of patients included in the trial and the unknown expression status of PDGF-C, its activating enzyme tissue plasminogen activator (20), and stromal PDGFR-α by these tumors. Moreover, experience from the past years of clinical trials using small molecule tyrosine kinase inhibitors points to that clinical efficacy is more likely if the target is genetically altered by mutation. In contrast, inhibition by neutralizing antibodies has displayed demonstrable efficacy on stromal targets, as in the case of inhibition of VEGF-A signaling by bevacizumab. Stromally expressed PDGFR-α would likely appear in a non-mutated form, making inhibition of PDGF ligands by neutralizing antibodies a more attractive therapeutic alternative from an empirical point of view.
Nevertheless, it is likely that therapeutic modalities targeting the tumor stroma will prove to be most efficacious in combination with simultaneous targeting of other tumor compartments. Rational drug combinations would conceivably weaken the stromal pro-angiogenic, pro-growth and anti-apoptotic support, thereby sensitizing the tumor to conventional chemotherapeutic drugs or anti-angiogenic treatments. It is imperative that the substances used for inhibition are specific for their targets in order to avoid excessive side effects from combining several drugs. In fact, several early Phase I-studies of the combination of imatinib with chemotherapeutic drugs have been terminated due to disproportionate toxicity (48, 49). Hence, efforts to effectively target the tumor stroma should be helped by the development of more specific inhibitors, such as neutralizing PDGF-CC antibodies. In conclusion, mounting evidence points to paracrine PDGF signaling as being crucial for the recruitment and phenotypic conversion of CAFs, and provides further rationale for clinical testing of the concept of therapeutic targeting of stromal fibroblasts in cancer.
Acknowledgments
We would like to gratefully acknowledge David Denhardt, Rutgers, NJ for the wt and osteopontin deficient MEFs; Janna Paulsson and Arne Östman, Karolinska Institutet for sections from B16/PDGF-B tumors; Eric Neilson, Vanderbilt University, TN for the FSP-1 antibody. This study was supported by the Swedish Research Council’s Linnaeus grant to the STARGET consortium (KP, CB and UE), the Swedish Cancer Society (KP and UE), Karolinska Institutet’s Cancer Strategic grant (KP), and Karolinska Institutet (CA and UE).
References
- 1.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001;3:143–5. doi: 10.1186/bcr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–97. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 4.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001;166:2472–83. [PubMed] [Google Scholar]
- 6.Micke P, Ostman A. Exploring the tumour environment: cancer-associated fibroblasts as targets in cancer therapy. Expert Opin Ther Targets. 2005;9:1217–33. doi: 10.1517/14728222.9.6.1217. [DOI] [PubMed] [Google Scholar]
- 7.Kiaris H, Chatzistamou I, Trimis G, Frangou-Plemmenou M, Pafiti-Kondi A, Kalofoutis A. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 2005;65:1627–30. doi: 10.1158/0008-5472.CAN-04-3791. [DOI] [PubMed] [Google Scholar]
- 8.Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11:539–54. doi: 10.1016/j.ccr.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allinen M, Beroukhim R, Cai L, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–6. [PubMed] [Google Scholar]
- 11.Skobe M, Fusenig NE. Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc Natl Acad Sci USA. 1998;95:1050–5. doi: 10.1073/pnas.95.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsberg K, Valyi-Nagy I, Heldin C-H, Herlyn M, Westermark B. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc Natl Acad Sci USA. 1993;90:393–7. doi: 10.1073/pnas.90.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shao Z-M, Nguyen M, Barsky SH. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 2000;19:4337–45. doi: 10.1038/sj.onc.1203785. [DOI] [PubMed] [Google Scholar]
- 14.Tejada ML, Yu L, Dong J, et al. Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin Cancer Res. 2006;12:2676–88. doi: 10.1158/1078-0432.CCR-05-1770. [DOI] [PubMed] [Google Scholar]
- 15.Dong J, Grunstein J, Tejada M, et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23:2800–10. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Fredriksson L, Li X, Eriksson U. PDGF-D is a potent transforming and angiogenic growth factor. Oncogene. 2003;22:1501–10. doi: 10.1038/sj.onc.1206223. [DOI] [PubMed] [Google Scholar]
- 17.Furuhashi M, Sjoblom T, Abramsson A, et al. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004;64:2725–33. doi: 10.1158/0008-5472.can-03-1489. [DOI] [PubMed] [Google Scholar]
- 18.Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–94. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 20.Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Denhardt DT, Rittling SR. Osteopontin is required for full expression of the transformed phenotype by the ras oncogene. Br J Cancer. 2000;83:156–63. doi: 10.1054/bjoc.2000.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003;23:4013–25. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. Int J Hematol. 2007;86:8–16. doi: 10.1532/IJH97.06230. [DOI] [PubMed] [Google Scholar]
- 25.Eitner F, Bucher E, van Roeyen C, et al. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19:281–9. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponten A, Li X, Thoren P, et al. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am J Pathol. 2003;163:673–82. doi: 10.1016/S0002-9440(10)63694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–9. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ball SG, Shuttleworth CA, Kielty CM. Platelet-derived growth factor receptor-alpha is a key determinant of smooth muscle alpha-actin filaments in bone marrow-derived mesenchymal stem cells. Int J Biochem Cell Biol. 2007;39:379–91. doi: 10.1016/j.biocel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11:421–9. doi: 10.1016/j.ccr.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 30.Grazul-Bilska AT, Johnson ML, Bilski JJ, et al. Wound healing: the role of growth factors. Drugs Today (Barc) 2003;39:787–800. doi: 10.1358/dot.2003.39.10.799472. [DOI] [PubMed] [Google Scholar]
- 31.Desmouliere A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–17. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 32.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 33.Thijssen VL, Brandwijk RJ, Dings RP, Griffioen AW. Angiogenesis gene expression profiling in xenograft models to study cellular interactions. Exp Cell Res. 2004;299:286–93. doi: 10.1016/j.yexcr.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 35.Fukumura D, Xavier R, Sugiura T, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–25. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 36.Leali D, Dell’Era P, Stabile H, et al. Osteopontin (Eta-1) and fibroblast growth factor-2 cross-talk in angiogenesis. J Immunol. 2003;171:1085–93. doi: 10.4049/jimmunol.171.2.1085. [DOI] [PubMed] [Google Scholar]
- 37.Jain S, Chakraborty G, Bulbule A, Kaur R, Kundu GC. Osteopontin: an emerging therapeutic target for anticancer therapy. Expert Opin Ther Targets. 2007;11:81–90. doi: 10.1517/14728222.11.1.81. [DOI] [PubMed] [Google Scholar]
- 38.Jaeger J, Koczan D, Thiesen HJ, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007;13:806–15. doi: 10.1158/1078-0432.CCR-06-1820. [DOI] [PubMed] [Google Scholar]
- 39.Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 40.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 41.Mori R, Shaw TJ, Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205:43–51. doi: 10.1084/jem.20071412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAllister SS, Gifford AM, Greiner AL, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo KH, Thornhill BA, Forbes MS, et al. Osteopontin regulates renal apoptosis and interstitial fibrosis in neonatal chronic unilateral ureteral obstruction. Kidney Int. 2006;70:1735–41. doi: 10.1038/sj.ki.5000357. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, Bringas P, Jr, Soriano P, Chai Y. PDGFR-alpha signaling is critical for tooth cusp and palate morphogenesis. Dev Dyn. 2005;232:75–84. doi: 10.1002/dvdy.20197. [DOI] [PubMed] [Google Scholar]
- 45.Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): a target for anticancer therapeutics. Drug Resist Updat. 2005;8:75–83. doi: 10.1016/j.drup.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Campbell JS, Johnson MM, Bauer RL, et al. Targeting stromal cells for the treatment of platelet-derived growth factor C-induced hepatocellular carcinogenesis. Differentiation. 2007;75:843–52. doi: 10.1111/j.1432-0436.2007.00235.x. [DOI] [PubMed] [Google Scholar]
- 47.Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92:1398–405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson FM, Krug LM, Tran HT, et al. Phase I studies of imatinib mesylate combined with cisplatin and irinotecan in patients with small cell lung carcinoma. Cancer. 2006;106:366–74. doi: 10.1002/cncr.21640. [DOI] [PubMed] [Google Scholar]
- 49.Lin AM, Rini BI, Derynck MK, et al. A phase I trial of docetaxel/estramustine/imatinib in patients with hormone-refractory prostate cancer. Clin Genitourin Cancer. 2007;5:323–8. doi: 10.3816/CGC.2007.n.011. [DOI] [PubMed] [Google Scholar]