

Abstract
Nuclear hormone receptors are hormone-regulated transcription factors that bind to specific sites on DNA and modulate the expression of adjacent target genes. Many nuclear hormone receptors display bimodal transcriptional properties; thyroid hormone receptors, for example, typically repress target gene expression in the absence of hormone, but activate target gene expression in the presence of hormone. The ability to repress is closely linked to the ability of the apo-receptor to physically bind to auxiliary corepressor proteins denoted SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) and N-CoR (nuclear receptor corepressor), which, in turn, help mediate the actual molecular events involved in transcriptional silencing. We report here that repression by thyroid hormone receptors can be regulated not only by cognate hormone, but also by certain tyrosine kinase signal transduction pathways, such as that represented by the epidermal growth factor-receptor. Activation of tyrosine kinase signaling leads to inhibition of T3R-mediated repression with relatively little effect on activation. These effects appear to be mediated by a kinase-initiated disruption of the ability of T3R to interact with SMRT corepressor. Intriguingly, tyrosine kinase signaling similarly disrupted the interactions of SMRT with v-Erb A, with retinoic acid receptors, and with PLZF, a nonreceptor transcriptional repressor. We conclude that tyrosine kinase signaling exerts potentially important regulatory effects on transcriptional silencing mediated by a variety of transcription factors that operate through the SMRT corepressor complex.
INTRODUCTION
Many crucial processes of vertebrate homeostasis, reproduction, and differentiation are regulated by the actions of small, lipophilic hormones (reviewed in Refs. 1-7). These lipophilic hormones include the steroids, retinoids, and thyroid hormones are sensed by nuclear hormone receptors that bind to cognate hormone and function as ligand-regulated transcription factors (1-7). Each nuclear hormone receptor binds to specific sites on the DNA genome and modulates the expression of specific sets of target genes. Intriguingly, nuclear hormone receptors can exert both positive and negative effects on transcription. These bimodal transcriptional properties are manifested through the ability of these receptors to associate with auxiliary regulator proteins, denoted corepressors and coactivators, that mediate the actual transcriptional response (8). For example, thyroid hormone receptors (T3Rs) and retinoic acid receptors (RARs) can function as transcriptional silencers in the absence of hormone, a context in which these receptors bind to a class of corepressor proteins denoted SMRT (silencing mediator of retinoic acid and thyroid hormone receptor)/N-CoR (nuclear receptor corepressor) (also known as TRAC and RIP13) (9-21). Binding of hormone converts T3Rs and RARs into strong transcriptional activators, a process that is accomplished by the release of the SMRT/N-CoR corepressors and the subsequent association of the receptors with a new set of proteins that function as transcriptional coactivators (9-12, 14, 18, 19, 22).
Thus, repression by nuclear hormone receptors involves a physical recruitment of SMRT/N-CoR corepressor proteins to the target promoter. The SMRT/N-CoR proteins can recruit, in turn, an ensemble of additional polypeptides that includes mSin3A/B, histone deacetylases (HDAC-1 and 2), retinoblastoma protein-associated proteins 46 and 48, and several other polypeptides of as-yet-unknown function (reviewed in Refs. 23 and 24). It is this larger corepressor complex that is thought to be the actual effector of transcriptional repression. Repression of gene expression probably involves multiple mechanisms, including both modifications of the chromatin template and inhibitory interactions with the general transcriptional machinery itself (17, 23-24b). Notably, although SMRT and N-CoR were initially identified as corepressors for the nuclear hormone receptors, the different components of the SMRT/N-CoR/Sin3/HDAC complex also appear to play a key role in transcriptional silencing by a wide variety of nonreceptor transcription factors, including Mad/Max, YY-1, PLZF, BCL-6, the retinoblastoma gene product, and several yeast transcriptional regulators (23-28).
Nuclear hormone receptors appear to serve as an important molecular nexus at which a variety of hormonal and nonhormonal signals converge to generate a combinatorial regulation of target gene expression. The actual transcriptional response in vivo is determined not only by the hormone status, but also by the nature of the target promoter, and by the actions of nonligand signal transduction pathways in the cell (29-34). Particularly interesting is the ability of certain protein kinases to modulate, both negatively and positively, nuclear hormone receptor function (reviewed in Refs. 31-34). For example, certain nonligand signal transduction pathways can induce target gene activation, even in the absence of ligand, or can further enhance the activation observed in the presence of hormone ligand (e.g. Refs. 32, 35-45). In some cases these effects may be mediated through posttranslational modifications of the receptor itself (32, 35, 41, 44-46). In other contexts, however, the mechanism by which a signal transduction pathway alters the regulatory properties of the nuclear hormone receptor does not appear to involve a detectable alteration in the receptor itself (36, 43, 47).
We wished to investigate whether the SMRT corepressor might represent a previously unrecognized target for these nonligand signal transduction pathways. We report here that activation of a cell surface tyrosine kinase, the epidermal growth factor-receptor [EGF-R (48)] or its constitutively activated allele, the v-Erb B oncogene (49), can interfere with T3R-mediated repression with little or no effect on T3R-mediated gene activation. This relief of repression by tyrosine kinase signaling is manifested as a severe inhibition of the interaction between T3R and SMRT corepressor. Notably, the interactions between SMRT and RARs, or between SMRT and PLZF (a nonreceptor transcriptional repressor), are similarly strongly inhibited by activation of the EGF-R/v-Erb B tyrosine kinase-signaling pathway. We conclude that certain cell surface tyrosine kinases can mediate effects on transcription by nuclear hormone receptors, and by POZ domain proteins such as PLZF, by interfering with ability of the SMRT corepressor complex to tether to these transcription factors.
RESULTS
Activation of a Tyrosine Kinase Signal Transduction Pathway Interferes with T3R-Mediated Transcriptional Repression in CV-1 Cells
We assayed the transcriptional properties of T3R by transient transfections of CV-1 cells. The reporter gene was a thymidine kinase promoter-luciferase (tk-luc) construct bearing T3R-binding sites (i.e. thyroid hormone response elements, or TREs). CV-1 cells possess very low levels of endogenous T3Rs, and the basal expression of the TRE-tk-luc reporter exhibited little or no response to thyroid hormone if no exogenous receptor was introduced (Fig. 1). Consistent with previous work (19), cointroduction of T3Rα into the CV-1 cells resulted in a repression of this reporter gene in the absence of T3 hormone and in a stimulation of luciferase expression in the presence of T3 hormone (Fig. 1A). This bimodal hormone-dependent activity required the presence of TRE sites in the reporter and was not observed for a β-galactosidase reporter, lacking TREs, employed as an internal negative control (data not shown).
Fig. 1. Tyrosine Kinase Inhibition of Transcriptional Repression by T3Rα and by v-Erb A.
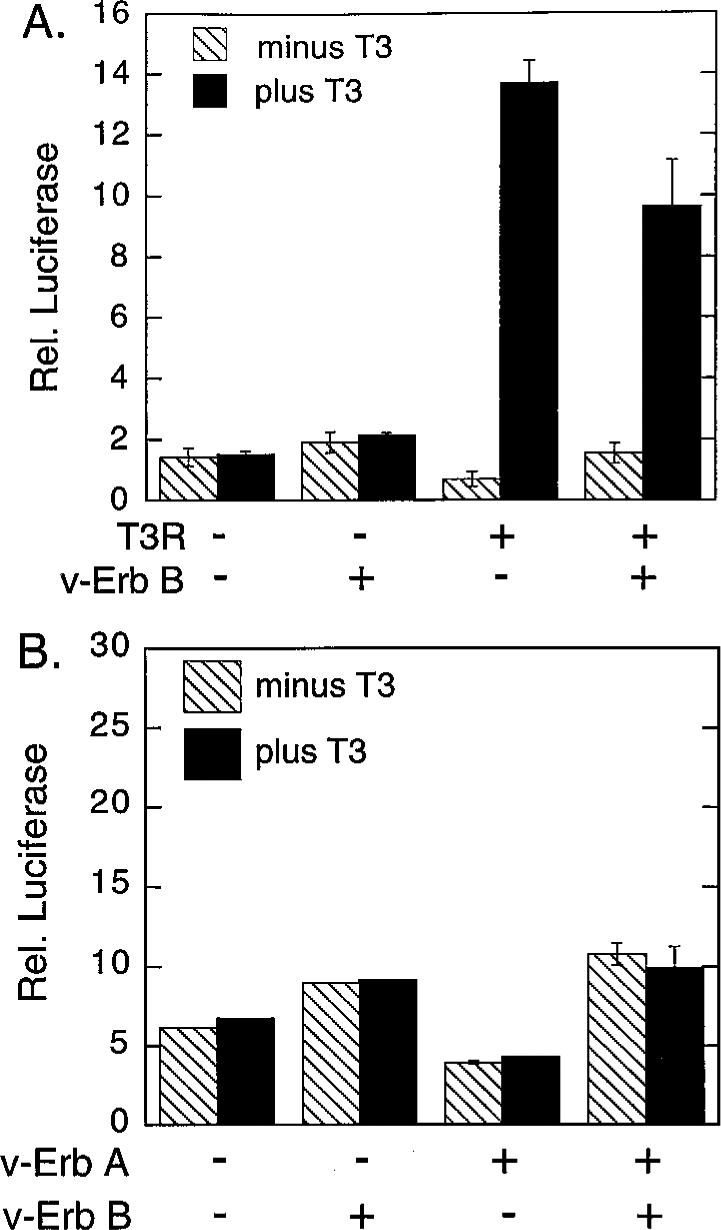
A, Inhibition of T3Rα-mediated repression. CV-1 cells were transfected with either an empty pSG5 vector (−) or a pSG5-T3Rα vector (+) in the absence (−) or presence (+) of a v-Erb B expression vector, as indicated below the panel. All transfections also included a tk-luciferase reporter containing a TRE (DR-4) and a pCMV-lac Z reporter employed as an internal transfection standard. The cells were subsequently incubated in the presence (solid bars) or absence (cross-hatched bars) of 1 μm T3-thyronine and harvested, and the relative luciferase activity (Rel. luciferase) was determined relative to that of the β-galactosidase control. The data represent the average and range of duplicate experiments. B, Inhibition of v-Erb A-mediated repression. The same experimental protocol was employed as in panel A, but with a pSG5-v-Erb A expression vector substituted in place of the pSG5-T3Rα vector.
We next repeated these experiments, but with the cointroduction of v-Erb B, a constitutively-activated derivative of the host cell EGF-R; v-Erb B is a membrane-associated tyrosine kinase that functions independently of EGF ligand and was employed to uniformly and strongly activate the EGF-R signal transduction pathway in the transfected cells (48, 49). Intriguingly, the introduction of v-Erb B significantly impaired T3R-mediated repression in the absence of T3 hormone, but had only a modest effect on T3R-mediated activation in the presence of T3 hormone (Fig. 1). In contrast, introduction of v-Erb B in the absence of T3R had little or no effect on the basal level of expression of the TRE-tk-luc reporter, indicating that the primary effect of v-Erb B was to counteract T3R repression, rather than to stimulate reporter gene expression independent of the actions of T3R.
A retrovirus-transduced, mutant version of a normal cellular T3R, denoted v-Erb A, is in fact, encoded by the same retrovirus as is v-Erb B, and the two oncoproteins operate together in leukemogenesis (49). V-Erb A has lost the ability to bind to T3 hormone and functions in many contexts as a hormone-independent transcriptional repressor (50, 51). Notably, cointroduction of v-Erb B interfered with v-Erb A-mediated repression in a fashion similar to the effects of v-Erb B on repression mediated by the wild-type T3R (Fig. 1B; the very modest change in reporter expression observed on introduction of v-Erb B in the absence of v-Erb A likely reflects small differences in the overall physiology of the transfected cells in the presence and absence of the tyrosine kinase). We conclude that the ability of v-Erb B to reverse repression by T3R derivatives is independent of the ability of the nuclear receptor itself to bind, or to respond directly, to cognate T3 hormone.
Activation of the Tyrosine Kinase-Signaling Pathway Is Manifested as Inhibition of the Interaction between T3R and SMRT Corepressor
The abrogation of T3R-mediated repression by v-Erb B suggested that the EGF-R signal transduction pathway might be capable of altering the interaction between T3R and SMRT corepressor. We first tested this possibility by use of a mammalian two-hybrid interaction assay. For this assay, the receptor-interaction domains of SMRT were fused to the DNA-binding domain (DBD) of GAL4 (GAL4-DBD) and inserted into a mammalian expression vector, pSG5. In parallel, relevant portions of the nuclear hormone receptors were fused to the activation domain of GAL4 (GAL4-AD) and placed into the same pSG5 vector. In this manner, interaction between SMRT and receptor should lead to a functional reconstitution of the GAL4 transcriptional activator, assayed as stimulation of a GAL4(17-mer)-luciferase reporter, when all three constructs are cointroduced into mammalian CV-1 cells (26).
Introduction of the GAL4DBD-SMRT fusion together with an “empty” pGAL4-AD construct, or an empty GAL4DBD construct together with the GAL4-AD-T3Rα fusion, had little or no effect on expression of the GAL4(17-mer)-luciferase reporter (Fig. 2A). In contrast, cointroduction of both constructs led to a strong stimulation of luciferase expression, consistent with the known ability of SMRT and T3R to strongly interact (Fig. 2A). This SMRT/T3R interaction assayed by the two-hybrid protocol displayed all of the properties expected from prior in vitro and in vivo studies: 1) it was abolished by addition of thyroid hormone; 2) mutations of T3R that are unable to interact with SMRT in vitro (such as the P156R substitution) fail to function in the mammalian two-hybrid assay; 3) deletions of SMRT that are defective for receptor binding in vitro (such as the SMRT ΔRID construct) fail to function in the mammalian two-hybrid assay; 4) T3R mutants, such as v-Erb A, that exhibit a hormone-resistant association with SMRT in vitro display a hormone-resistant function in the two-hybrid assay; and 5) nuclear hormone receptors that fail to interact with SMRT in vitro (such as vitamin D3 receptor) fail to function in the two-hybrid assay in vivo (Fig. 2A).
Fig. 2. Tyrosine Kinase-Mediated Inhibition of the Interaction between T3Rα and SMRT, Determined by a Mammalian Two-Hybrid Assay.
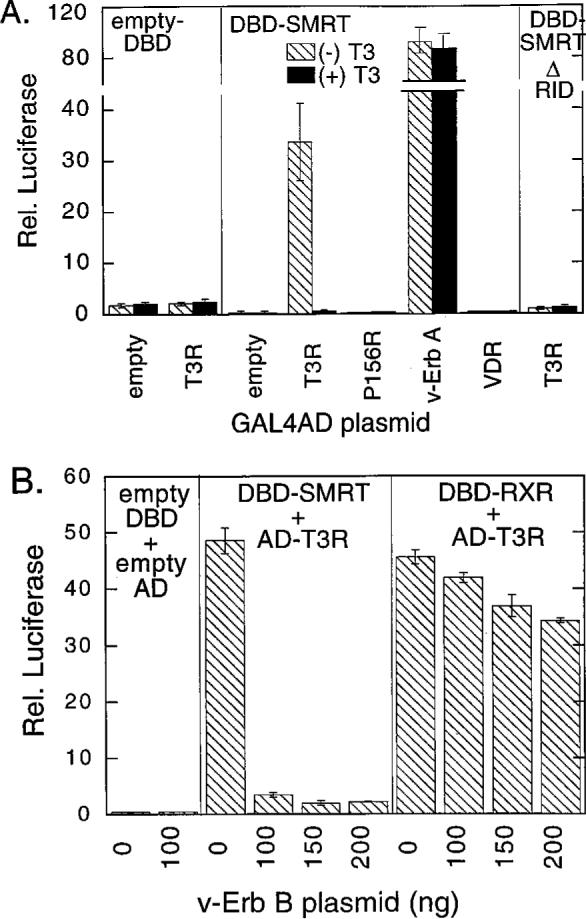
A, Mammalian cell-based two-hybrid assay of the interaction between SMRT and T3Rα. A pSG5 vector expressing either a GAL4DBD alone (empty DBD), a GAL4DBD fused with the receptor interaction domain of SMRT (amino acids 751−1495; DBD-SMRT), or a GAL4DBD fused with a subdomain of SMRT lacking the receptor interaction domain (amino acids 751−1074; DBD-SMRT ΔRID), as indicated within each panel, were introduced into CV-1 cells by calcium phosphate coprecipitation together with a GAL4 (17-mer) luciferase reporter and a pSG5-GAL4-AD construct. As indicated below the panels, the GAL4-AD constructs contained either a GAL4-AD domain alone (empty), the T3Rα hormone-binding domain (T3R), the same T3Rα construct with a mutation that disrupts SMRT-association (P156R), the analogous region of v-Erb A (v-Erb A), or the vitamin D3 receptor hormone-binding domain (VDR). The cells were subsequently incubated in the absence (cross-hatched bars) or presence (solid bars) of 1 μm cognate hormone, the cells were harvested, and the relative luciferase activity was determined normalized to that of a pCH110-lac Z plasmid introduced as an internal control. The data represent the average and range of duplicate experiments. B, V-Erb B inhibition of the two-hybrid interaction between T3Rα and SMRT. A pSG5 vector expressing either an GAL4DBD alone (empty DBD), a GAL4DBD fused with the receptor interaction domain of SMRT (amino acids 751−1495, DBD-SMRT), or a GAL4DBD fused with the hormone- binding domain of RXRα were introduced into CV-1 cells by lipofection, together with either the empty pSG5-GAL4-AD vector or the pSG5-GAL4-AD fused to the hormone-binding domain of T3Rα. In addition, each transfection included the GAL4 (17-mer) luciferase reporter, a pCMV-lac Z internal standard, and the amount of the v-Erb B expression plasmid indicated below the panel (0 to 200 ng). The cells were subsequently incubated and harvested, and the luciferase activity was determined and normalized to β-galactosidase activity. The SMRT/T3Rα interactions were assayed in the absence of T3.
We next tested the effect of v-Erb B on this SMRT/T3R interaction. The introduction of v-Erb B virtually abolished the two-hybrid interaction between SMRT and T3R, resulting in a greater than 20-fold inhibition of luciferase expression (Fig. 2B). This inhibition by v-Erb B appeared to be operative on the SMRT/T3R interaction itself: v-Erb B had little or no effect on the basal level of reporter expression if either (or both) of the GAL4DBD-SMRT or GAL4-AD-T3R fusions were omitted from the transfection (Fig. 2B). Similarly, v-Erb B had little or no effect on expression of a β-galactosidase reporter, lacking GAL4-binding sites, employed in the same experiments as an internal control (data not shown). We conclude that the effects of v-Erb Bin the two-hybrid assay require the presence of the T3R and SMRT constructs and are therefore unlikely to be mediated by a nonspecific inhibition of the activity of the reporter promoter itself, or by a disruption of the stability or enzymatic activity of the encoded luciferase protein.
We also wished to exclude the possibility that v-Erb B was acting by inhibiting the expression or function of either the GAL4-DBD or GAL4-AD moieties, rather than by interfering with the SMRT/T3R interaction itself. We therefore assayed the effects of v-Erb B on the two-hybrid interaction between T3R and retinoid × receptor (RXR); RXRs are heterodimer partners for T3Rs, and the two receptor classes can physically associate in vitro and in vivo (6). T3R exhibited a strong interaction with RXR in our mammalian two-hybrid system that was relatively unaltered by cointroduction of v-Erb B; only a very modest inhibition of the T3R/RXR interaction was observed at high levels of v-Erb B, perhaps reflecting very small differences in the overall physiology of these cells in the presence or absence of the tyrosine kinase. This relative lack of an effect of v-Erb B on the T3R/RXR interaction was in notable contrast to the dramatic v-Erb B-mediated inhibition of the T3R/SMRT interaction (Fig. 2B). These results indicate that the interaction between SMRT and T3R is specifically inhibited by cointroduction of an activated EGF-R/v-Erb B allele.
We also repeated these experiments using the v-Erb A oncoprotein derivative of T3R. In common with wild-type T3R, the v-Erb A protein exhibited a strong two-hybrid interaction with SMRT (Fig. 3). This two-hybrid interaction was again severely impaired by the cointroduction of v-Erb B. In contrast, substitution of the pGAL4-AD-v-Erb A construct with one containing a v-Erb A-mutant defective for SMRT binding (v-Erb A P144R) abolished, as expected, both the two-hybrid interaction and the effects of v-Erb B (Fig. 3). We conclude that v-Erb B signaling disrupts the ability of SMRT to interact with both hormone-dependent and hormone-independent forms of the T3R.
Fig. 3. Tyrosine Kinase Inhibition of the Mammalian Two-Hybrid Interaction between SMRT and the v-Erb A Derivative of T3Rα.

The same protocol as in Fig. 2B was repeated, but utilizing either an empty pSG5-GAL4-AD construct (empty), a pSG5-GAL4-AD-v-Erb A fusion (v-Erb A), or the same v-Erb A fusion bearing a mutation disrupting the SMRT association domain (P144R). The transfections were performed in the absence (−) or presence (+) of 100 ng of the v-Erb B expression construct, as indicated below the panel.
Stimulation of Endogenous EGF-R Can Substitute for Transfection of v-Erb B
The experiments described above relied on introduction of v-Erb B as a means of strongly activating the EGF-R signal transduction pathway. CV-1 cells express modest levels of endogenous EGF-R, and we therefore extended our experiments to test the ability of transforming growth factor-α (TGF-α), a physiological agonist for EGF-R (52), to substitute for the transfection of v-Erb B. Incubation of CV-1 cells with TGF resulted in a reproducible 2-fold inhibition of the T3R/SMRT interaction (Fig. 4A). A similar effect could be observed with EGF (data not shown). Although the effects of TGF-α and EGF incubation were much more modest than that observed with transfection of v-Erb B, this differential is not unanticipated. When transfected, v-Erb B is probably expressed at much higher levels than is endogenous EGF-R and is likely to be a much stronger inducer of downstream signal transduction than is EGF-R ligand. In addition, v-Erb B is refractory to many of the negative feedback mechanisms that constrain the actions of the wild-type EGF-R; v-Erb B-induced signaling, therefore, is likely to be more sustained than that of EGF or TGF ligand. We conclude that stimulation of the EGF-R-signaling pathway, by either EGF-R itself or by a ligand-independent form of EGF-R, results in inhibition of the T3R/SMRT interaction, although the effect is more dramatic with the latter.
Fig. 4. Inhibition of the Mammalian Two-Hybrid Interaction between SMRT and v-Erb A by Various Signal Effectors/Transducers.

The same protocol as in Fig. 2B was repeated, but testing the ability of different signal effectors/transducers to replace v-Erb B. A, Effect of transforming growth factor (TGF)α on the mammalian two-hybrid interaction between SMRT and v-Erb A. pSG5 vectors expressing the GAL4-DBD-SMRT fusion were introduced into CV-1 cells together with the GAL4 (17-mer) luciferase reporter and either an empty pSG5-GAL4-AD or the pSG5-GAL4-AD-v-Erb A fusion, as indicated below the panel. The cells were then treated (solid bars) or not (cross-hatched bars) with 50 μm TGFα and harvested, and the relative luciferase activity, normalized to β-galactosidase activity, was determined. The data represent the average and range of duplicate experiments. B, Effect of different signal transducers on the mammalian two-hybrid interaction between SMRT and v-Erb A. The CV-1 cells were transfected as described in panel A, and either not manipulated further (None), treated with the concentrations of 8-bromo-cAMP indicated (cyclic AMP), or transfected with expression plasmids for v-Erb B, v-Ras, v-Raf, or the p110 catalytic subunit of phosphatidylinositol-3-kinase (PI3-K), as indicated below the panel. The cells were subsequently harvested, and the relative luciferase activity, normalized to β-galactosidase activity, was determined. The data represent the average and range of duplicate experiments.
We also performed a preliminary dissection of the downstream signaling pathways that may be operating in this EGF-R/v-Erb B signaling pathway. Ras has been implicated as one (of several) downstream effectors involved in EGF-R signaling (48): notably, substitution of v-Erb B with v-Ras also inhibited the two-hybrid interaction between T3R and SMRT, although not as strongly as did v-Erb B itself (Fig. 4B). cAMP also exerted a modest inhibition, but only at relatively high concentrations that also lead to detectable cell toxicity (Fig. 4B). In contrast, introduction of a number of other potential signal transducers, such as v-Raf and the catalytic subunit of phosphatidyl-inositol 3-kinase, failed to inhibit the T3R/SMRT interaction (Fig. 4B). We conclude that c-Ras may be one downstream mediator of the v-Erb B effects on corepressor function, but that other pathways are also likely to contribute to the effects observed here.
Activation of the EGF-R Signaling Pathway Inhibits the Ability of SMRT to Interact with RARs, and with the PLZF Transcriptional Repressor
RARs share the ability to bind to the SMRT corepressor complex and to repress target gene transcription in vivo. Nonetheless, RARs diverge from T3Rs in primary amino acid sequence, hormone specificity, and target gene specificity (7). We therefore tested the effects of the EGF-R signaling pathway on the SMRT/RAR interaction. In the absence of v-Erb B, a strong two-hybrid interaction was observed between RARα and SMRT that could be inhibited, as expected, by cognate hormone (Fig. 5). Introduction of increasing amounts of v-Erb B virtually abolished this interaction, resulting in a more than 10-fold reduction in GAL4(17-mer) luciferase reporter activity (Fig. 5). As a negative control we examined the effects of v-Erb B on the two-hybrid interaction between RAR and RXR [RXRs are heterodimer partners for RARs as well as for T3Rs (6)]; consistent with the results we observed for T3R, the two-hybrid interaction between RAR and RXR was relatively unaffected, or even slightly stimulated, by coexpression of v-Erb B (Fig. 5).
Fig. 5. Tyrosine Kinase Inhibition of the Mammalian Two-Hybrid Interaction between RARα and SMRT.
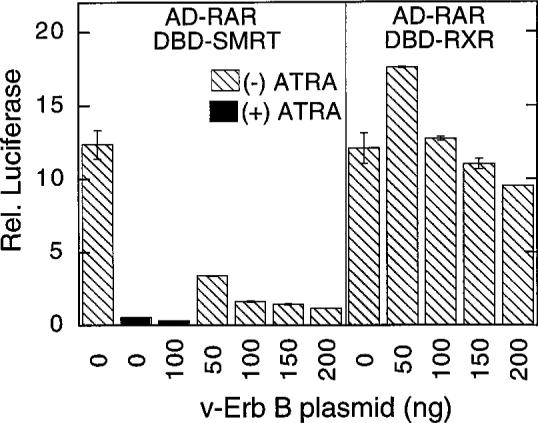
The same protocol as in Fig. 2B was repeated, but utilizing a pSG5-GAL4-AD-RARα fusion. Two samples were also treated with all-trans-retinoic acid (ATRA; indicated by solid bars) to demonstrate the hormone-labile nature of the SMRT/RAR interaction.
In addition to nuclear hormone receptors, SMRT has also been implicated in transcriptional repression mediated by many nonreceptor transcription factors, such as the PLZF protein (26-28). PLZF possesses little or no detectable amino acid relatedness to the nuclear hormone receptors, and the interactions between PLZF and SMRT appear to be mediated by protein determinants distinct from those implicated in the nuclear hormone receptor interaction (26). Once again, however, the two-hybrid interaction between PLZF and SMRT observed in CV-1 cells in the absence of v-Erb B was strongly inhibited in the presence of v-Erb B (Fig. 6B). Consistent with this v-Erb B-mediated inhibition of the interaction between PLZF and SMRT, cointroduction of v-Erb B counteracted PLZF-mediated transcriptional repression when tested using a suitable PLZF-fusion/reporter construct system (Fig. 6A). We conclude that the effects of tyrosine kinase signaling on SMRT function are manifested on a variety of transcriptional repressors.
Fig. 6. Tyrosine Kinase-Mediated Inhibition of Transcriptional Repression by PLZF and of the Two-Hybrid Interaction between PLZF and SMRT.

A, Inhibition by v-Erb B of transcriptional repression by PLZF. The GAL4 (17-mer) luciferase reporter was introduced by lipofection into CV-1 cells, together with the concentrations of a pSG5-GAL4-DBD fusion of PLZF (amino acids 1−456) indicated below the panel. The transfections were performed in the absence (cross-hatched bars) or presence (solid bars) of a v-Erb B expression plasmid; the cells were incubated and harvested, and the luciferase activity was determined relative to that of the pCMV-lac Z reporter employed as an internal transfection standard. Fold repression was calculated relative to the levels of reporter gene expression detected in the absence of the PLZF plasmid. B, Tyrosine kinase inhibition of the mammalian two-hybrid interaction between SMRT and PLZF. The same protocol as in Fig. 2B was repeated, but utilizing either an empty pSG5-GAL4-AD plasmid (“empty”) or a pSG5-GAL4-AD-PLZF fusion (“v-Erb A”), together with the pSG5-GAL4DBD-SMRT construct. The transfections were performed in the absence (cross-hatched bars) or presence (solid bars) of the v-Erb B expression plasmid.
There are two distinct domains of SMRT that mediate the interaction of corepressor with T3R and with v-Erb A in vitro and in vivo; these are denoted SMRT receptor interaction domains (RID)-1 and RID-2 (12, 14-16). We therefore next tested whether the inhibitory effects of v-Erb B were specific to one or the other SMRT domain. In the absence of v-Erb B, both RID-1 (SMRT amino acids 1055−1291) and RID-2 (SMRT amino acids 1291−1495) independently conferred a two-hybrid interaction with v-Erb A, with the RID-2/v-Erb A interaction somewhat the stronger of the two (Fig. 7A). No interaction was observed with a region of SMRT mapping upstream of the RID domains (SMRT amino acids 751−1074). Notably, both the RID-1 and RID-2 interactions with v-Erb A were clearly inhibited by cointroduction of v-Erb B, although the magnitude of this inhibition was somewhat greater for the RID-1 interaction. In contrast to the ability of v-Erb A and T3Rs to contact both RID-1 and RID-2 regions of SMRT, RARs contact primarily the RID-1 domain (SMRT amino acids 1055−1291; Ref. 14). Consistent with our results for v-Erb A, the two-hybrid interaction between RAR and the SMRT RID-1 domain was also strongly inhibited by the introduction of v-Erb B (Fig. 7B). We conclude that tyrosine kinase signaling exerts an overall inhibition of the interaction between SMRT and nuclear hormone receptors, with this effect particularly pronounced for the interactions manifested by the SMRT RID-1 domain.
Fig. 7. Mapping of the SMRT/Receptor Interactions That Are Inhibited by v-Erb B.

A, Tyrosine kinase inhibition of the two-hybrid interactions between SMRT and v-Erb A. The same procedure as in Fig. 3 was repeated using the pSG5-GAL4-AD-v-Erb A construct and a variety of pSG5-GAL4DBD fusions, each representing a distinct subdomain of SMRT as indicated below the panel. The transfections were performed in the absence (cross-hatched bars) or presence (solid bars) of the v-Erb B expression plasmid. B, Tyrosine kinase inhibition of the interactions between SMRT and RARα. The same procedure as in Fig. 3 was repeated, but using a pSG5-GAL4-AD-RARα construct and a variety of pSG5-GAL4DBD fusions, each representing a distinct subdomain of SMRT as indicated below the panel.
The Tyrosine Kinase-Mediated Inhibition of the RAR/SMRT Interaction Observed in Vivo Is Also Observed in Vitro
We next exploited a protein-protein binding assay to determine whether the tyrosine-kinase signaling effects in vivo could be mimicked in an in vitro system. In this assay, radiolabeled RARα, synthesized by in vitro transcription and translation, was tested for the ability to bind to an immobilized glutathione-S-transferase (GST)-SMRT construct, synthesized in and purified from Escherichia coli. RARα bound to the GST-SMRT was subsequently eluted and analyzed by SDS-PAGE. As previously shown, RAR strongly bound to the GST-SMRT fusion under these conditions, but not to nonrecombinant GST (Fig. 8). Addition of lysates of control COS-1 cells, transfected with an empty pSG5 plasmid, had little or no effect on the GST-SMRT/RAR interaction. In contrast, addition of lysates of COS-1 cells transfected by v-Erb B inhibited the ability of radiolabeled RAR to bind to the GST-SMRT matrix in vitro (Fig. 8). We conclude that activation of the EGF-R/v-Erb B signaling pathway in CV-1 cells results in the production of an inhibitory activity that can subsequently function to interfere with the physical association between RAR and SMRT in vitro.
Fig. 8. Inhibition by v-Erb B of the in Vitro Interaction between SMRT and RARα.

Nonrecombinant GST, or a GST-SMRT (amino acids 1055−1291) fusion protein, were synthesized in Escherichia coli and were immobilized independently on glutathione agarose. The immobilized proteins were then incubated with radiolabeled RARα in the presence of lysates of COS cells transfected by an empty pSG5 vector, or by a v-Erb B expression vector, as indicated. The agarose matrices were extensively washed, and radiolabeled RARα remaining bound to the immobilized GST proteins was eluted with solubilized glutathione, and was resolved by SDS-PAGE. The amount of bound RARα was quantified by PhosphorImager analysis. The data represent the normalized average and range of duplicate experiments.
DISCUSSION
Transcriptional Repression by Nuclear Hormone Receptors Is Opposed by Tyrosine Kinase-Mediated Signaling
Although originally believed to be regulated exclusively by cognate hormone, many nuclear hormone receptors are actually subject to control by an assortment of nonligand signals. For example, the nature of the DNA-binding site, dimerization with other nuclear hormone receptors, protein-protein interactions with other nonreceptor proteins, and covalent modifications, such as receptor phosphorylation, can all alter nuclear hormone receptor function (11, 29, 30-47). These extrahormonal signals can exert both positive and negative effects on receptor activity in the absence of hormone and can either enhance or preclude the effects of hormone. In this manuscript, we report that tyrosine kinase signaling, such as that mediated by EGF-R/v-Erb B, can specifically abrogate transcriptional repression by the T3R, with little or no effect on transcriptional activation. Thus, the transcriptional properties of T3R are subject to regulation both by T3 hormone and by a distinct set of signals arising from tyrosine kinase/growth factor receptors.
What is the nature of this tyrosine kinase-initiated inhibition of T3R-mediated repression? As demonstrated here, activation of the EGF-R/v-Erb B signaling pathway virtually abolished the ability of SMRT to interact with T3R in a mammalian two-hybrid assay. Although the two-hybrid assay is inherently an indirect measure of protein-protein interaction, our experiments strongly indicate that the effect of tyrosine kinase signaling is to inhibit the actual interaction between T3R and SMRT corepressor, rather than to interfere with an incidental or extraneous aspect of the two-hybrid assay itself. For example, tyrosine kinase signaling had little or no effect on the two-hybrid interaction between T3Rα and its heterodimer partner RXR (or between RARα and RXR) in an assay dependent on virtually the same reagents and subject to the same constraints as the T3R/SMRT two-hybrid assay. Additional controls demonstrated that EGF-R/v-Erb B had little or no effect on basal reporter gene function, on the enzymatic activity of the luciferase itself, or on the DNA binding or transactivation properties of the GAL4DBD or GAL4-AD fusions. Notably, the inhibitory effects of EGF-R/v-Erb B signaling are also demonstrable in vitro, at a reduced intensity, using extracts of transfected cells in a protein-protein binding protocol. These results further support our hypothesis that the tyrosine kinase-signaling pathways act to disrupt the physical interaction between unliganded nuclear hormone receptor and the SMRT corepressor.
The Inhibitory Effects of Tyrosine Kinase Signaling Extend to SMRT Interactions with v-Erb A, with RARs, and with PLZF
The SMRT corepressor complex has been implicated in transcriptional silencing by T3Rs, by RARs, and by an assortment of nonreceptor transcriptional repressors, such as BCL-6 and PLZF (9, 13-18, 20, 25-28). Paralleling the inhibition of the SMRT/T3R interaction, the capacity of SMRT to interact with RARs and with PLZF was also dramatically inhibited by activation of the EGF-R/v-Erb B-signaling pathway. Thus, the SMRT corepressor may represent a common regulatory link through which gene silencing by an assortment of transcription factors can be controlled by tyrosine kinase signaling.
Although RARs and T3Rs possess distinct hormone and DNA recognition properties, they share clusters of related amino acid sequence and interact with overlapping, if somewhat distinct, portions of SMRT. In contrast, PLZF possesses no detectable amino acid relatedness with the nuclear hormone receptors and appears to interact with SMRT through a POZ protein motif not present in the nuclear hormone receptors (26-28). We do not yet know the precise nature of the tyrosine kinase-initiated inhibitory effect, although our results imply that whatever the molecular basis of this phenomenon, the components and mechanism must be able to survive cell lysis to function in vitro. Given the near-identical effects of tyrosine kinase signaling on T3R-, RAR-, and PLZF-mediated silencing, it is tempting to conclude that it is the shared SMRT corepressor, not these divergent transcription factors, that represents the actual target of this inhibition. Plausible mechanisms may therefore include the covalent modification of SMRT or of another component of the corepressor complex or, alternatively, the altered expression or altered function of a modulator of SMRT function. Of course, we cannot fully exclude that the transcription factors are themselves the targets of the actions of v-Erb B, possibly through the actions of an as-yet-unidentified, but shared modulator.
Our preliminary use of known components of the signal transduction pathway that lie downstream of EGF-R/v-Erb B suggests that the effects of v-Erb Bon SMRT function can be partially mimicked by v-Ras, but not by v-Raf, protein kinase C, or moderate levels of cAMP. It is somewhat intriguing that v-Raf does not mimic the actions of v-Ras in the inhibition of SMRT function; presumably the actions of v-Ras in this context are not mediated through c-Raf, or perhaps v-Raf lacks some functions present in the c-Raf protein. It is also important to note that v-Ras appears capable of only partial substitution for v-Erb B in this phenotype; further work is in progress to better define the signal transduction pathway involved and to identify any additional intermediates and effectors.
Tyrosine Kinases and Transcriptional Repression: A Consistent Theme
A multiplicity of circumstantial linkages have been documented between the actions of the nuclear hormone receptors and the signal transduction events initiated by tyrosine kinases. For example, as already described, the two oncogenes encoded by the avian erythroblastosis virus, v-erb A and v-erb B, are aberrant versions of the normal cellular genes for T3Rs and EGF-R, and these two oncogenes function synergistically in erythroleukemogenesis. V-Erb A has been proposed to function in oncogenesis as a transcriptional repressor (49-51). This may appear at first to be contradictory to our own observations that v-Erb B counteracts repression by v-Erb A in CV-1 cells. However, the precise actions of v-Erb A in the erythroleukemic cell remain incompletely understood and may extend beyond simple transcriptional repression, perhaps involving functions that are enhanced by v-Erb B. Alternatively, the v-Erb B signal transduction pathway in avian erythroleukemic cells may be different from that in the primate-derived CV-1 kidney cells employed here and may not result in an antagonism of v-Erb A repression in the former.
A variety of other tantalizing links have been established between the actions of tyrosine kinases and those of nuclear hormone receptors. EGF treatment, for example, has been reported to activate the estrogen receptor, even in the absence of estrogen agonist, possibly through phosphorylation of mitogen-activated protein kinase sites in the nuclear receptor and/or by phosphorylation of tyrosine 537 in its ligand-binding domain (e.g. Refs. 32, 38, 41, 53, and 54). This observation is likely to have important medical implications: mammary carcinomas that display aberrant estrogen receptor function often also exhibit aberrant expression of EGF-R family members, such as the closely related HER-2 tyrosine kinase (55, 56). More broadly, interactions between tyrosine kinases and transcriptional repressors occur in a variety of eukaryotic systems. In Drosophila embryonic development, for example, activation of torso, a membrane-associated tyrosine kinase, inhibits transcriptional repression by dorsal, an ortholog of mammalian NF-κB (57). These interactions between tyrosine kinases and nuclear hormone receptors can operate in both directions: for example, the promoter for human EGF-R contains a binding site for T3R, and endogenous EGF-R expression is repressed by T3R in at least certain cell types (58).
Notably, the SMRT/N-CoR corepressor has also been implicated in mediating the inhibitory effects of certain ligand antagonists on the progesterone and estrogen receptors (47, 59, 60). When this manuscript was in the final stages of preparation, McDonnell and colleagues (47) reported that cAMP treatment can counteract the effect of antagonists on progesterone receptor, apparently by interfering with corepressor function (47). Although we observe a much more modest effect of cAMP on the receptors and transcription factors tested here, it appears reasonable that the progesterone receptor studies, and our own, reflect a common phenomenon by which transcriptional repression is made subservient to a variety of cellular signal transduction pathways. These observations may also prove relevant to the actions of chicken ovalbumin upstream promoter transcription factor (COUP-TF), a member of the orphan class of nuclear hormone receptors (61). COUP-TF lacks a known hormone ligand and functions in most contexts as a strong transcriptional repressor, apparently through the ability of COUP-TF to bind to the SMRT/N-CoR corepressor complex (61). Intriguingly, COUP-TF repression is strongly counteracted by dopamine, by cAMP, and by okadaic acid, even though none of these reagents bind directly to the COUP-TF protein (62). Given the results presented here and by McDonnell and co-workers, it will be very interesting to determine whether the actions of these nonligand signals in COUP-TF-mediated regulation are mediated through a similar signal transduction pathway that leads to disruption of the ability of SMRT to interact with COUP-TF.
In conclusion, we have elucidated a previously unrecognized mechanism through which the functions of the nuclear hormone receptors (and of nonreceptor transcription factors such as PLZF) can be regulated. This mechanism involves tyrosine kinase signal transduction pathways that alter the interactions of these transcription factors with essential cofactors, such as the SMRT corepressor complex. These alterations to corepressor function are likely to represent an important component of signal convergence and integration in eukaryotic gene regulation.
MATERIALS AND METHODS
Transient Transfections
Transfections of CV-1 cells were performed by a lipofectin-mediated method (19). Approximately 4 × 105 cells were transfected with 50 ng of pSG5-T3Rα or pSG5-v-erb A plasmid, 100 ng of pCMV-lac Z as an internal control, and 100 ng of ptk-luc-TRE with or without 200 ng of a pSG5-v-erb B expression plasmid. Cells were harvested at 48 h post transfection. Relative luciferase activity was measured and normalized to β-galactosidase activity (19).
Mammalian Two-Hybrid Assays
Construction of the pSG5 GAL4 DNA DBD-SMRT derivatives and GAL4-AD-T3Rα vectors were previously described (26). The pSG5 GAL4-AD-RARα and RXRα vectors were constructed by inserting the appropriate PCR-generated DNA fragments, bearing terminal EcoRV and XhoI sites, into the pSG5 GAL4-AD vector (26). If a calcium phosphate coprecipitation protocol was employed, the transfections were performed in 60-mm plates containing approximately 2.5 × 105 CV-1 cells. Each plate was transfected with 500 ng each of the pSG5 GAL4DBD vector, the pSG5 GAL4-AD vector, the pGL2-GAL4(17-mer)-luciferase reporter, and a pCH110-lac Z vector (employed as an internal control). If the lipofectin protocol was employed, the transfections were performed in 12-well culture plates, each well containing 7 × 104 CV-1 cells; each well was transfected with 25 ng of the pSG5 GAL4DBD vector, 100 ng of the pSG5 GAL4-AD vector, 100 ng of pGL2-GAL4(17-mer)-luciferase reporter, and 100 ng of a pCMV-lac Z as an internal control, with either 200 ng of an empty pSG5 vector or 200 ng of the pSG5 v-erb B plasmid. Luciferase and β-galactosidase assays were performed as previously described (19).
In Vitro Receptor/Corepressor Binding Assays
The nonrecombinant GST and the GST-SMRT (codons 1055−1291) constructions were created in a pGEX-KG vector background (14, 18, 19, 26). GST-fusion proteins were expressed in E. coli, were purified, and were immobilized on glutathione agarose as previously described (14, 18, 19, 26). The immobilized GST fusion proteins were then incubated at 37 C for 30 min with lysates of COS-1 cells, prepared as described below, and then incubated for 1 h at 4 C with 35S-labeled RARα, prepared in a coupled in vitro transcription and translation system (Promega TnT procedure, Promega, Madison, WI). The agarose matrix was extensively washed, and the bound proteins were eluted with free glutathione and were analyzed by SDS-PAGE (18, 19, 26). The electrophoretograms were visualized by autoradiography and were quantified by PhosphorImager analysis (Molecular Dynamics STORM system, Molecular Dynamics, Sunnyvale, CA).
Cell extracts were prepared as follows: COS-1 cells, maintained in 60-mm tissue culture dishes, were transiently transfected with 1 μg per dish of either an empty pSG5 vector or a pSG5 v-Erb B expression vector, using the lipofectin-mediated method. At 60 h post transfection, the cells were harvested, and then lysed by sonication in WCE buffer (25 mm HEPES, pH 7.7, 0.3 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.1% Triton X-100, 0.5 mm dithiothreitol, 20 μm β-glycerol-phosphate, 0.1 mm Na3VO4, 2 μg/ml leupeptin, and 100 μg/ml phenylmethylsulfonyl fluoride).
Acknowledgments
We thank H.-J. Kung, B. Vennstrom, and A. Dejean for generously providing molecular clones employed in this research.
This work was supported by Public Health Service/NIH Grants R37 CA-53394 and R01 DK-53528.
REFERENCES
- 1.Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14:184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro RC, Apriletti JW, West BL, Wagner RL, Fletterick RJ, Schaufele F, Baxter JD. The molecular biology of thyroid hormone action. Ann NY Acad Sci. 1993;758:366–389. doi: 10.1111/j.1749-6632.1995.tb24843.x. [DOI] [PubMed] [Google Scholar]
- 3.Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid hormone receptor superfamily members. Annu Rev Biochem. 1994;63:451–483. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 4.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–858. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 5.Kastner P, Mark M, Chambon P. Nonsteroidal nuclear receptors: what are genetic studies telling us about their role in real life? Cell. 1995;83:859–870. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 6.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Overview: the nuclear receptor superfamily: the second decade. Cell. 1995;83:835–840. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear hormone receptor coactivators and corepressors. Mol Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 9.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 10.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamel Y, Soderstrom M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 11.Kurokawa RM, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld MG, Glass CK. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature. 1995;377:451–454. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen JD, Umesono K, Evans RM. SMRT isoforms mediate repression and anti-repression of nuclear receptor heterodimers. Proc Natl Acad Sci USA. 1996;93:7567–7571. doi: 10.1073/pnas.93.15.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Downes M, Burke LJ, Bailey PJ, Muscat GE. Two receptor interaction domains in the corepressor, N-CoR/RIP13, are required for an efficient interaction with ReverbA alpha and RVR: physical association is dependent on the E region of the orphan receptors. Nucleic Acids Res. 1996;2:4379–4386. doi: 10.1093/nar/24.22.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sande S, Privalsky ML. Identification of TRACs (T3 Receptor-associating cofactors) a family of cofactors that associate with, and modulate the activity of, nuclear hormone receptors. Mol Endocrinol. 1996;10:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 15.Seol W, Mahon MJ, Lee YK, Moore DD. Two receptor interacting domains in the nuclear hormone receptor corepressor RIP13/N-CoR. Mol Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- 16.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld M, Lazar MA. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Leo C, Schroen DJ, Chen JD. Characterization of receptor interaction and transcriptional repression by the corepressor SMRT. Mol Endocrinol. 1997;11:2025–2037. doi: 10.1210/mend.11.13.0028. [DOI] [PubMed] [Google Scholar]
- 18.Lin BC, Hong S-H, Krig S, Yoh SM, Privalsky ML. A conformational switch in nuclear hormone receptors is involved in coupling hormone binding to corepressor release. Mol Cell Biol. 1997;17:6131–6138. doi: 10.1128/mcb.17.10.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoh SM, Chatterjee VKK, Privalsky ML. Thyroid hormone resistance syndrome manifests as an aberrant interaction between mutant T3 receptors and transcriptional corepressors. Mol Endocrinol. 1997;11:470–480. doi: 10.1210/mend.11.4.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamir I, Zhang J, Lazar MA. Stoichiometric and steric principles governing repression by nuclear hormone receptors. Genes Dev. 1997;11:835–846. doi: 10.1101/gad.11.7.835. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Zamir I, Lazar MA. Differential recognition of liganded and unliganded thyroid hormone receptor by retinoid X receptor regulated transcriptional repression. Mol Cell Biol. 1997;17:6887–6897. doi: 10.1128/mcb.17.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulman IG, Juguilon H, Evans RM. Activation and repression by nuclear hormone receptors: hormone modulates an equilibrium between active and repressive states. Mol Cell Biol. 1996;16:3807–3813. doi: 10.1128/mcb.16.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pazin MJ, Kadonaga JT. What's up and down with histone deacetylation and transcription? Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 24.Wolffe AP. Sinful repression. Nature. 1997;387:16–17. doi: 10.1038/387016a0. [DOI] [PubMed] [Google Scholar]
- 24a.Wong C-W, Privalsky ML. Transcriptional repression by the SMRT/mSin3 corepressor: multiple interactions, multiple mechanisms, and a potential role for TFIIβ. Mol Cell Biol. 1998 doi: 10.1128/mcb.18.9.5500. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b.Muscat GEO, Burke LJ, Downes M. The corepressor N-CoR and its variants RIP13a and RIP13Δ1 directly interact with the basal transcription factors, TFIIβ, TAFII32 and TAFII70. Nucleic Acids Res. 1998;26:2899–2907. doi: 10.1093/nar/26.12.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhordain PO, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A, Kerckaert JP, Evans RM, Leprince D. Corepressor SMRT binds the BTB/POZ repressing domain of the LAZ3/BCL-6 oncoprotein. Proc Natl Acad Sci USA. 1997;94:10762–10767. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong S-H, David G, Wong C-W, Dejean A, Privalsky ML. SMRT corepressor interacts with PLZF, and with the PML-RARα and PLZF-RARα oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci USA. 1997;94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 28.Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukemia. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 29.Burnstein KL, Cidlowski JA. Multiple mechanisms for regulation of steroid hormone action. J Cell Biochem. 1993;51:130–134. doi: 10.1002/jcb.240510203. [DOI] [PubMed] [Google Scholar]
- 30.Saatcioglu F, Claret FX, Karin M. Negative transcriptional regulation by nuclear receptors. Semin Cancer Biol. 1994;5:347–359. [PubMed] [Google Scholar]
- 31.Blok LJ, P de Ruiter PE, Brinkmann AO. Androgen receptor phosphorylation. Endocr Res. 1996;22:197–219. doi: 10.3109/07435809609030508. [DOI] [PubMed] [Google Scholar]
- 32.Katzenellenbogen BS. Estrogen receptors: bioactivities and interactions with cell signaling pathways. Biol Reprod. 1966;54:287–293. doi: 10.1095/biolreprod54.2.287. [DOI] [PubMed] [Google Scholar]
- 33.McEwan IJ, Wright APH, Gustafsson J-A. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions. Bioessays. 1997;19:153–160. doi: 10.1002/bies.950190210. [DOI] [PubMed] [Google Scholar]
- 34.Meier CA. Regulation of gene expression by nuclear hormone receptors. J Recept Signal Transduction Res. 1997;17:319–335. doi: 10.3109/10799899709036612. [DOI] [PubMed] [Google Scholar]
- 35.Huggenvik JI, Collard MW, Kim YW, Sharma RP. Modification of the retinoic acid signaling pathway by the catalytic subunit of protein kinase-A. Mol Endocrinol. 1993;7:543–550. doi: 10.1210/mend.7.4.8388997. [DOI] [PubMed] [Google Scholar]
- 36.Sartorius CA, Tung L, Takimoto GS, Horwitz KB. Antagonist-occupied human progesterone receptors bound to DNA are functionally switched to transcriptional agonists by cAMP. J Biol Chem. 1993;268:9262–9266. [PubMed] [Google Scholar]
- 37.Tahayato A, Lefebvre P, Formstecher P, Dautrevaux M. A protein kinase C-dependent activity modulates retinoic acid-induced transcription. Mol Endocrinol. 1993;7:1642–1653. doi: 10.1210/mend.7.12.8145770. [DOI] [PubMed] [Google Scholar]
- 38.Arnold SF, Vorojeikina DP, Notides AC. Phosphorylation of tyrosine 537 on the human estrogen receptor is required for binding to an estrogen response element. J Biol Chem. 1995;270:30205–3012. doi: 10.1074/jbc.270.50.30205. [DOI] [PubMed] [Google Scholar]
- 39.Matkovits T, Christakos S. Ligand occupancy is not required for vitamin D receptor and retinoid receptor-mediated transcriptional activation. Mol Endocrinol. 1995;9:232–242. doi: 10.1210/mend.9.2.7776973. [DOI] [PubMed] [Google Scholar]
- 40.O'Malley BW, Schrader WT, Mani S, Smith C, Weigel NL, Conneely OM, Clark JH. An alternative ligand-independent pathway for activation of steroid receptors. Recent Prog Horm Res. 1995;50:333–347. doi: 10.1016/b978-0-12-571150-0.50020-2. [DOI] [PubMed] [Google Scholar]
- 41.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 42.Leitman DC, Costa CH, Graf H, Baxter JD, Ribeiro RC. Thyroid hormone activation of transcription is potentiated by activators of cAMP-dependent protein kinase. J Biol Chem. 1996;271:21950–21955. doi: 10.1074/jbc.271.36.21950. [DOI] [PubMed] [Google Scholar]
- 43.Bai W, Rowan BG, Allgood VE, O'Malley BW, Weigel NL. Differential phosphorylation of chicken progesterone receptor in hormone-dependent and ligand-independent activation. J Biol Chem. 1997;272:10457–10463. doi: 10.1074/jbc.272.16.10457. [DOI] [PubMed] [Google Scholar]
- 44.Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3947–3954. doi: 10.1128/mcb.17.7.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol. 1997;11:353–365. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 46.Weigel NL. Steroid hormone receptors and their regulation by phosphorylation. Biochem J. 1996;319:657–667. doi: 10.1042/bj3190657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner BL, Norris JD, Knotts TA, Weigel NL, McDonnell DP. The nuclear corepressors NCoR and SMRT are key regulators of both ligand and 8-bromo-cyclic AMP-dependent transcriptional activity of the human progesterone receptor. Mol Cell Biol. 1998;18:1369–1378. doi: 10.1128/mcb.18.3.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 49.Graf T, Beug H. Role of the v-erb A and v-erb B oncogenes of avian erythroblastosis virus in erythroid cell transformation. Cell. 1983;34:7–9. doi: 10.1016/0092-8674(83)90130-7. [DOI] [PubMed] [Google Scholar]
- 50.Damm K, Thompson CC, Evans RM. Protein encoded by v-Erb A functions as a thyroid hormone receptor antagonist. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- 51.Sap J, Munoz A, Schmitt H, Stunnenberg H, Vennstrom B. Repression of transcription mediated by a thyroid hormone response element by the v-Erb A oncogene product. Nature. 1989;340:242–244. doi: 10.1038/340242a0. [DOI] [PubMed] [Google Scholar]
- 52.Kumar V, Bustin SA, McKay IA. Transforming growth factor alpha. Cell Biol Int Rep. 1995;19:373–388. doi: 10.1006/cbir.1995.1083. [DOI] [PubMed] [Google Scholar]
- 53.Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, Korach KS. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci USA. 1996;93:12626–12630. doi: 10.1073/pnas.93.22.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–1398. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 55.Lupu R, Cardillo M, Harris L, Hijazi M, Rosenberg K. Interaction between erb B-receptors and heregulin in breast cancer tumor progression and disease. Semin Cancer Biol. 1995;6:135–145. doi: 10.1006/scbi.1995.0016. [DOI] [PubMed] [Google Scholar]
- 56.Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- 57.Rusch J, Levine M. Regulation of the dorsal morphogen by the Toll and torso signalling pathway: a receptor tyrosine kinase selectively masks transcriptional repression. Genes Dev. 1994;8:1247–1257. doi: 10.1101/gad.8.11.1247. [DOI] [PubMed] [Google Scholar]
- 58.Thompson KL, Santon JB, Shephard LB, Walton GM, Gill GN. A nuclear protein is required for thyroid hormone receptor binding to an inhibitory half-site in the epidermal growth factor receptor promoter. Mol Endocrinol. 1992;6:627–635. doi: 10.1210/mend.6.4.1584225. [DOI] [PubMed] [Google Scholar]
- 59.Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol. 1997;11:693–705. doi: 10.1210/mend.11.6.0004. [DOI] [PubMed] [Google Scholar]
- 60.Smith CL, Nawaz Z, O'Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogn, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 61.Shibata H, Nawaz Z, Tsai SY, O'Malley BW. Gene silencing by COUP-TF is mediated by transcriptional corepressors, N-CoR and SMRT. Mol Endocrinol. 1997;11:714–724. doi: 10.1210/mend.11.6.0002. [DOI] [PubMed] [Google Scholar]
- 62.Power RF, Lydon JP, Conneely OM, O'Malley BW. Dopamine activation of an orphan of the steroid receptor superfamily. Science. 1991;252:1546–1548. doi: 10.1126/science.2047861. [DOI] [PubMed] [Google Scholar]