

Abstract
Plasma membrane monoamine transporter (PMAT or ENT4) is a newly cloned transporter assigned to the equilibrative nucleoside transporter (ENT) family (SLC29). Unlike ENT1–3, PMAT mainly functions as a polyspecific organic cation transporter. In this study, we investigated the molecular mechanisms underlying the unique substrate selectivity of PMAT. By constructing chimeras between human PMAT and ENT1, we showed that a chimera consisting of transmembrane domains (TM) 1–6 of PMAT and TM7–11 of hENT1 behaved like PMAT, transporting 1-methyl-4-phenylpyridinium (MPP+, an organic cation) but not uridine (a nucleoside), suggesting that TM1–6 contains critical domains responsible for substrate recognition. To identify residues important for the cation selectivity of PMAT, 10 negatively charged residues were chosen and substituted with alanine. Five of the alanine mutants retained PMAT activity, and four were non-functional due to impaired targeting to the plasma membrane. However, alanine substitution at Glu206 in TM5 abolished PMAT activity without affecting cell surface expression. Eliminating the charge at Glu206 (E206Q) resulted in loss of organic cation transport activity, whereas conserving the negative charge (E206D) restored transporter function. Interestingly, mutant E206Q, which possesses the equivalent residue in ENT1, gained uridine transport activity. Thr220, another residue in TM5, also showed an effect on PMAT activity. Helical wheel analysis of TM5 revealed a distinct amphipathic pattern with Glu206 and Thr220 clustered in the center of the hydrophilic face. In summary, our results suggest that Glu206 functions as a critical charge sensor for cationic substrates and TM5 forms part of the substrate permeation pathway in PMAT.
Membrane transporters play pivotal roles in sustaining the normal life of cells (1, 2). The solute carrier (SLC)4 proteins constitute a large series of membrane transporters currently consisting of 360 members in 46 gene families in humans (1). Transporters from the same SLC gene family share at least 20–25% amino acid sequence identity and are thought to be evolved from a common ancestor (1, 2). Whereas it is generally anticipated that transporters from the same gene family possess similar functional properties, several SLC families appear to consist of members with great functional diversity (2, 3). For example, the Na+-glucose transporter family, SLC5, comprises not only Na+-coupled glucose transporters, but also transporters for iodide, choline, vitamins, and short-chain fatty acids (3).
We recently discovered an interesting functional divergence in the equilibrative nucleoside transporter (ENT) family, SLC29. The human and rodent genomes encode four SLC29 isoforms, SLC29A1–4. SLC29A1–3, known as ENT1–3, are nucleoside transporters that specifically transport nucleosides (e.g. uridine, adenosine, etc.) and their structural analogs (4, 5). ENT1 and ENT2 play important roles in nucleoside salvage pathways, regulation of adenosine signaling, and cellular disposition of anticancer and antiviral nucleoside analogs (4, 5). ENT3 is an intracellular nucleoside transporter, which may play a role in lysosomal transport of nucleosides (6). The fourth member, SLC29A4, was originally identified as ENT4 and was expected to function as a nucleoside transporter (7). However, our recent cloning and characterization work clearly showed that SLC29A4 functions as a polyspecific organic cation transporter that transports prototype organic cations (e.g. 1-methyl-4-phenylpyridinium (MPP+), tetraethylammonium, and biogenic amines) but minimally interacts with nucleosides or nucleoside analogs (8, 9). Because its typical endogenous substrates are monoamine neurotransmitters (e.g. dopamine, serotonin), we have proposed a functional name, plasma membrane monoamine transporter (PMAT), for SLC29A4 (9). The substrate selectivity of PMAT is strikingly similar to that of the organic cation transporters (OCTs) in the SLC22 family (10–13). In humans, PMAT may play a role in regulating central nervous system homeostasis of monoamine neurotransmitters (9, 14), as well as in tissue-specific transport of organic cations (8, 15).
The unique substrate selectivity of PMAT in the SLC29 family has brought up intriguing questions: why does PMAT behave so differently from the ENTs (i.e. ENT1–3) and is PMAT structurally related to the ENTs or to the OCTs? At the protein level, human and rodent PMATs exhibit a low overall sequence identity (~20%) to the ENTs (9) (Table 1), which is on the borderline of classifying a transporter into a specific gene family. However, in the TM regions, sequence identity between PMAT and the ENTs significantly increases (up to 35–40%), and PMAT does not show significant sequence homology to other known gene families (9). Its sequence identity to the OCTs is only 11–14% (Table 1). Moreover, PMAT and ENTs both possess an 11-transmembrane topology (9, 16). Finally, Barnes et al. (17) recently showed that PMAT also transports adenosine at acidic pH. These data suggest an intrinsic relationship in protein structure between PMAT and the ENTs. In this study, we investigated the molecular determinants and structural elements underlying the unique substrate selectivity of PMAT in the SLC29 family.
TABLE 1.
Sequence identity and similarity of PMAT with human ENTs and OCTs
|
hENT1 (SLC29A1) |
hENT2 (SLC29A2) |
hENT3 (SLC29A3) |
PMAT (SLC29A4) |
||
| 46 (60)* | 29 (48) | 19 (32) |
hENT1 (456 a.a) |
||
|
hOCT3 (556 a.a) |
11 (24) | 28 (44) | 20 (33) |
hENT2 (456 a.a) |
|
|
hOCT2 (555 a.a) |
12 (23) | 49 (67) | 20 (35) |
hENT3 (475 a.a) |
|
|
hOCT1 (554 a.a) |
14 (27) | 48 (65) | 70 (80) | ||
|
PMAT (530 a.a) |
hOCT3 (SLC22A3) |
hOCT2 (SLC22A2) |
hOCT1 (SLC22A1) |
Values are expressed in “identity (similarity).”
EXPERIMENTAL PROCEDURES
Sequence Alignment and Analysis
Sequences of mammalian ENTs and human OCTs were obtained from Gen-Bank™ with the following accession numbers: hENT1 (AF_079117), rENT1 (NM_031684), mENT1 (AF_218255), hENT2 (AF_034102), rENT2 (NM_031738), mENT2 (AF_183397), hENT3 (AF_326987), mENT3 (AF_326986), PMAT (AY_485959), rPMAT (XP_001072979), mPMAT (NM_146257), hOCT1 (X_98332), rOCT1 (NP_036829), mOCT1 (NP_033228), hOCT2 (X_98333), rOCT2 (NP_113772), mOCT2 (NP_038695), hOCT3 (AJ_001417), rOCT3 (NP_062103), and mOCT3 (NP_035525). ClustalW and Vector NTI Suite 8 software were used for sequence analysis and multiple alignments.
Generation of Chimeric and Mutant Constructs
To facilitate the determination of membrane localization, chimeras and mutants were constructed using yellow fluorescence protein (YFP)-tagged wild type (WT) PMAT and hENT1 as templates. The WT PMAT and hENT1 cDNAs (9) were subcloned into the YFP vector pEYFP-C1 (Clontech, Palo Alto, CA) using the BglII/XbaI and BglII/KpnI sites, respectively. To construct chimeras T1–6 and T7–11, an internal EcoRI site of hENT1 in the loop region between TM6 and 7 was used. An engineered EcoRI site was introduced into WT PMAT cDNA by site-directed mutagenesis, and chimeras were generated using a method described by Wang and Giacomini (18). To generate additional chimeras with smaller replacement, an engineered site was introduced into both WT human PMAT and hENT1. Junctions in all chimeras were made within homologous regions. No foreign amino acid was introduced to any of the chimera constructs. For site-directed mutagenesis, mutants were generated using the QuikChange kit (Stratagene) as described previously (19). The sequence of each chimera and mutant was confirmed by DNA sequencing in the Department of Biochemistry at the University of Washington.
Stable Expression in MDCK Cells
YFP-tagged chimera and mutant constructs were transfected into MDCK cells using a Lipofectamine (Invitrogen) method as previously described (9, 15). Stably transfected cell lines were obtained by culturing cells in minimal essential medium containing 10% fetal bovine serum and G418 (1000 μg/ml). Empty pEYFP-C1 vector was transfected into MDCK cells to obtain the control cell line. After 2–3 weeks selection, fluorescence-positive cells were purified by a FACS Vantage SE sorter (BD Biosciences) at the Cell Analysis Center at the University of Washington Health Science Center. The sorted cells were cultured and maintained in minimal essential medium containing G418 (200 μg/ml).
Confocal Fluorescence Microscopy
To determine the cellular localization of YFP-tagged chimeric and mutant transporters, 106 cells were grown in two-well Lab-Tek borosilicated coverglass chambers (Nalge Nunc International Corp.) and visualized with a Leica Spectral confocal microscope equipped with a krypton/argon laser as the light source. Images were captured by excitation at 488 nm and emission at 610–640 nm.
Functional Characterization in MDCK Cells
Stably transfected MDCK cells were plated in 24-well plates and allowed to grow for 3 days. Transport assays were performed at 37 °C in Krebs-Ringer-Henseleit buffer (5.6 mM glucose, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM CaCl2, 1.2 mM MgSO4, 25 mM HEPES, pH 7.4) containing a 3H-labeled ligand. [3H]5-HT (5-hydroxy-[1,2-3H]tryptamine creatinine sulfate, 27.1 Ci/mmol) and [3H]dopamine (3,4-dihydroxy-[2,5,6-3H]phenyl-ethylamine, 59.7 Ci/mmol) were from PerkinElmer Life Sciences, Inc. [3H]MPP+ (39.3 Ci/mmol) and [3H]uridine ([5,6-3H]uridine, 30.0 Ci/mmol) were from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Uptake was terminated by washing the cells three times with ice-cold Krebs-Ringer-Henseleit buffer. Cells were then solubilized and the radioactivity was quantified by liquid scintillation counting. Protein content in each uptake well was measured using a BCA protein assay kit (Pierce) and the uptake in each well was normalized to its protein content. In all studies, cells transfected with an empty vector were served as a background control. Transporter-specific uptake was calculated by subtracting the background uptake in vector-transfected cells.
Isolation of Plasma Membrane Proteins by Cell Surface Biotinylation
Confluent cells were washed twice with 3 ml of ice-cold phosphate-buffered saline/CM (pH 8.0). Biotinylation was carried out on ice by incubation with 1 ml of ice-cold phosphate-buffered saline/CM containing a membrane-impermeable biotinylation reagent NHS-SS-biotin (0.5 mg/ml). After two successive 20-min incubations on ice with freshly prepared NHS-SS-biotin and gentle shaking, cells were briefly rinsed with 3 ml of phosphate-buffered saline/CM containing 100 mM glycine. Cells were then incubated on ice with the same solution for 20 min to ensure complete quenching of the unreacted NHS-SS-biotin. Cells were then solubilized on ice by incubating in 400 μl of lysis buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, protease inhibitors phenylmethyl-sulfonyl fluoride, 200 μg/ml, leupeptin, 3 μg/ml, pH 7.4) for 1 h. Fifty microliters of streptavidin-agarose beads was then added to the supernatant to isolate cell membrane protein, which are subjected to Western blot using a mouse monoclonal anti-yellow fluorescent protein antibody (JL-8) (BD Biosciences) with 1:1000 dilution, followed by horseradish peroxidase-conjugated goat anti-mouse IgG (1:20,000 dilution).
Helical Wheel Analysis
The helical wheel was generated using the Wisconsin Package version 10.3-UNIX as described before (19). The transmembrane domain is assumed to be a standard α-helix (3.6 residues/helical turn). Each residue in TM is plotted every 100° around the center of a circle. The projection of the positions of the residues was shown on a plane perpendicular to the helical axis. Hydrophobicity and hydrophilicity are assigned according to the consensus scale of Eisenberg et al. (20).
Data Analysis
All uptake experiments were performed in triplicate and repeated three times. Data were expressed as mean ± S.D. Statistical significance was determined by Student’s t test, and the kinetic parameters were determined by nonlinear least squares regression fitting as described previously (8).
RESULTS
Function and Substrate Selectivity of PMAT and hENT1 Chimeric Transporters
Among all known human genes, PMAT only shows a low but significant sequence homology to ENTs (9) (Table 1). Multiple computer algorithms also predicted that PMAT possesses a similar membrane topology to the ENTs with 11 putative transmembrane helices, a cytoplasmic N terminus, and an extracellular C terminus (Fig. 1a). Based on these data, we hypothesized that despite the difference in substrate selectivity, PMAT and ENTs may share a similar tertiary structure and a similar arrangement of functional domains for substrate recognition and translocation. Earlier studies have suggested that the N-terminal half (TM1–6) of ENT1 and ENT2 contains critical domains for substrate/inhibitor recognition and translocation (21, 22). To test our hypothesis, we first constructed two chimeric transporters, chimeras T1–6 and T7–11, between human PMAT and the prototype human equilibrative nucleoside transporter, hENT1 (Fig. 1). Chimera T1–6 consists of the N-terminal half (TM1–6) of PMAT and the C-terminal half (TM7–11) of hENT1, whereas chimera T7–11 was constructed in a reciprocal manner (Fig. 1a). To monitor membrane trafficking of chimeric proteins, YFP was tagged to the N termini of both chimeric and WT transporters. After being stably transfected into MDCK cells, the substrate specificity of the chimeras were determined by uptake assays using MPP+ and uridine for the PMAT and hENT1 phenotype, respectively. YFP-tagged WT PMAT and hENT1 were expressed predominantly on the plasma membranes in MDCK cells. Tagging of YFP had little effect on substrate selectivity and kinetic behaviors of WT transporters (data not shown) (23). As expected, cells expressing WT PMAT showed uptake activity for MPP+ but not for uridine (Fig. 1b). In contrast, cells expressing WT hENT1 transported uridine but not MPP+. Chimera T1–6 showed plasma membrane localization (data not shown) and behaved like PMAT, transporting MPP+ but not uridine (Fig. 1b). The PMAT-like substrate selectivity of chimera T1–6 strongly suggests that TM1–6 contains the key domains for substrate recognition and translocation in PMAT. The reciprocal chimera T7–11, however, was not functional due to intra-cellular trapping in MDCK cells as revealed by confocal microscopy (data not shown). Further attempts to make chimeras with smaller replacements were not successful, as all resultant chimeras were not functional when expressed in MDCK cells (data not shown).
FIGURE 1.

a, secondary structures of wild type PMAT, hENT1, and chimeric transporters. Chimera T1– 6 contains residues 1–309 of PMAT (530 amino acids) and 232 to 456 of hENT1 (456 amino acids); and chimera T7–11 consists of residues 1–231 of hENT1 and 310 –530 of PMAT. b, uptake of MPP+ and uridine by wild type PMAT, hENT1, and chimera T1– 6 stably expressed in MDCK cells. Each value represents mean ± S.D. (n = 3).
Selection of Candidate Amino Acid Residues for Site-directed Mutagenesis
To further identify regions and residues important for the substrate selectivity of PMAT, we employed site-directed mutagenesis based on multiple sequence alignments. Because PMAT has been shown to transport a variety of organic cations (8, 9), we proposed that negatively charged residues are important for its cation selectivity, particularly those conserved in PMAT orthologs but substituted by a non-charged or positively charged residues in the ENTs. Of the 33 glutamate (Glu) and aspartate (Asp) residues conserved in human and rodent PMATs, 13 residues were not taken into consideration because they are located in the very hydrophilic N or C terminus or the large intracellular loop linking TM6 and -7. Among the remaining 20 negatively charged residues, 10 showed charge differences from mammalian ENTs and were thus selected for mutagenesis. Fig. 2a shows an example of two selected residues in TM3 and TM5. In addition, we also performed forced sequence alignments of PMATs with human and rodent OCT1–3. Despite the low sequence homology between PMATs and the OCTs, there are 23 conserved residues. Among these residues, Thr226 in the OCTs was previously suggested to participate in the binding of organic cations (24). We thus included the equivalent residue in PMAT, Thr220, for mutational analysis. Together, 11 residues, Asp91, Asp107, Glu128, Asp154, Asp163, Glu206, Thr220, Glu227, Glu242, Asp336, and Glu375 were chosen as candidate residues. As shown in Fig. 2b, 9 of the 11 residues are located in the N-terminal half (TM1–6) and only two are located in the C-terminal half in TM7.
FIGURE 2.

a, examples of multiple sequence alignment of PMATs (ENT4s) with mammalian ENTs in TM3 and TM5 regions. The residues Asp154 and Glu206 are conserved in PMATs (red) but are substituted with uncharged residues in the ENTs (yellow). b, positions of candidate residues selected for alanine substitution.
Analysis of Candidate Residues by Alanine Substitution
Alanine substitution was used to test the functional significance of the 11 candidate residues. YFP-tagged PMAT mutants (D91A, D107A, E128A, D154A, D163A, E206A, T220A, E227A, E242A, D336A, and E375A) were constructed and stably expressed in MDCK cells. The function of these alanine mutants was analyzed by uptake studies using PMAT substrates MPP+, serotonin, and dopamine. As shown in Fig. 3, mutation of Asp91 and Glu128 to alanine did not result in a significant change in transport activity. Mutants E227A and E375A were also functional and had a slightly increased (~1.5–2-fold) transport activity. Mutants E242A and T220A exhibited reduced transport activity thatwasabout30%oftheWTPMAT. In contrast, mutants D107A, D154A, D163A, E206A, and D336A completely lost transport activity toward all three tested substrates.
FIGURE 3. Uptake of organic cations by various PMAT alanine mutants in stably transfected MDCK cells.

The uptake of 10 μM 3H-labeled MPP+, dopamine, and serotonin was measured at 37 °C for 1 min. Substrate uptake was corrected by subtracting nonspecific uptake in vector-transfected cells. Values are expressed as percentage of uptake in cells expressing wild type YFP-PMAT measured in parallel. Each value represents the mean ± S.D. (n = 3).
Cellular Localization of Alanine Mutants
The observed change in the uptake activity of an alanine mutant could be due to a change in intrinsic transport activity or an alteration of cell surface expression levels of the functional transporter. To distinguish these two circumstances, confocal microscopy was used to visualize the cellular localization of all tested alanine mutants (Fig. 4). For mutants D91A, E128A, T220A, E227A, E242A, and E375A, which retained full or partial transport activity, a predominant plasma membrane localization pattern was observed (Fig. 4). Mutants D107A, D154A, D163A, and D336A, which are not functional in MDCK cells, displayed altered cellular localization. D154A and D336A were localized intracellularly to perinuclear membrane network, indicating that substitutions with alanine affected PMAT trafficking to the plasma membrane. The other two mutants, D107A and D163A, showed diffused fluorescence throughout the cytoplasm, which was similar to the distribution of YFP. Western blot analysis revealed degraded protein products in cells expressing D107A and D163A (data not shown), suggesting that the alanine substitutions at these two positions may have affected protein stability. Most importantly, mutant E206A, which showed complete loss of transporter activity (Fig. 3), exhibited normal plasma membrane expression (Fig. 4). These data suggest that unlike D107A, D154A, D163A, and D336A, the loss of function in the E206A mutant is not caused by protein trafficking or stability problems, but rather is due to a defect in transport activity.
FIGURE 4. Confocal imaging of cellular localization of wild type PMAT, YFP, and various PMAT alanine mutants in stably transfected MDCK cells.

Role of Glu206 in the Interactions with MPP+ and Monoamine Neurotransmitters
To further investigate the role of Glu206 in PMAT interactions with cationic substrates, three additional mutants, E206Q, E206D, and E206R, were constructed and stably expressed in MDCK cells. All mutants exhibited plasma membrane expression patterns similar to that of the WT PMAT (Fig. 5a). Cell surface biotinylation followed by Western analysis with anti-yellow fluorescent protein antibody also revealed comparable membrane expression of the 75-kDa YFP-tagged fusion protein in all Glu206 mutant cell lines (Fig. 5b). Functional analysis showed that elimination of the negative charge (E206Q) or switching to a positively charged residue (E206R) resulted in complete loss of transporter activity (Fig. 6). In contrast, substitution of Glu206 with a charge-conserved residue (E206D) generated a fully functional transporter (Fig. 6) with similar transport efficiency toward various cationic substrates (Table 2). These data strongly suggest that the negative charge at position 206 is essential for the transport function of PMAT, and organic cation binding to PMAT may involve electrostatic interaction with Glu206.
FIGURE 5. Plasma membrane expression of Glu206 and Thr220 mutants as revealed by confocal fluorescence microscopy (a) or biotinylation followed by Western blot with an anti-yellow fluorescent protein monoclonal antibody (b).
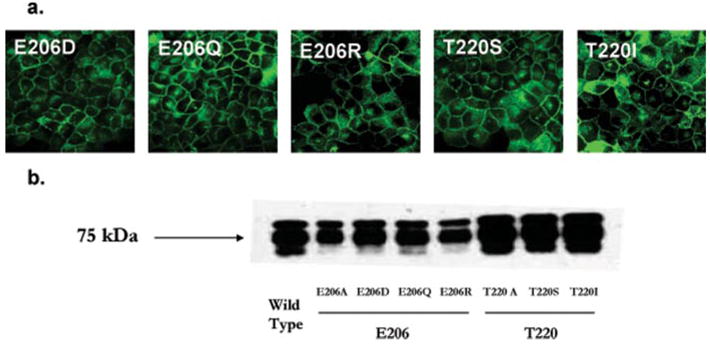
FIGURE 6. Uptake of organic cations by Glu206 and Thr220 mutants in stably transfected MDCK cells.

The uptake of 10 μM 3H-labeled MPP+, dopamine, and serotonin was measured at 37 °C for 1 min. Substrate uptake was corrected by subtracting nonspecific uptake in vector-transfected cells. Values are expressed as percentage of uptake in cells expressing wild type YFP-PMAT measured in parallel. Each value represents the mean ± S.D. (n = 3).
TABLE 2.
Kinetic parameters of PMAT mutants
| MPP+ |
Dopamine |
Serotonin |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Km | Vmax | Vmax/Km | Km | Vmax | Vmax/Km | Km | Vmax | Vmax/Km | |
| Wild type | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| E206D | 2.19 | 1.98 | 0.91 | 1.57 | 1.09 | 0.70 | 0.86 | 0.66 | 0.76 |
| T220A | 0.79 | 0.23 | 0.29 | 0.58 | 0.21 | 0.35 | 0.69 | 0.20 | 0.29 |
| T220S | 0.86 | 0.24 | 0.26 | 0.58 | 0.19 | 0.31 | 0.64 | 0.18 | 0.27 |
All experiments were performed in triplicate and repeated three times. Values were expressed as relative ratio to the wild type transporter by setting parameters of wild type PMAT = 1.
Uridine Uptake by PMAT Mutants
At the equivalent position of Glu206 in PMAT, hENT1 processes a non-charged residue glutamine (Gln180), which is conserved among mammalian ENT1–2. We thus tested whether Glu206 mutants of PMAT showed increased activity toward the ENT substrate uridine, an uncharged molecule. Interestingly, whereas mutants E206Q and E206A completely lost activity toward organic cations (Figs. 3 and 6), they consistently exhibited ~2-fold increase in uridine uptake as compared with WT PMAT (Fig. 7a). In marked contrast, the E206D mutant, which retained PMAT activity toward cationic substrates (Fig. 6), showed no activity toward uridine (Fig. 7a). In addition, other plasma membrane-expressed PMAT mutants (e.g. D91A and T220A), which carry the equivalent ENT1 residues, showed no uptake activity toward uridine (data not shown). These data provided further evidence that Glu206 plays an essential role for the cation selectivity of PMAT, and this negatively charged residue may serve as a filter to exclude uncharged molecules such as the nucleosides. However, when its equivalent residue in ENT1, Gln180, was replaced with Glu or Asp, the resulting mutants behaved like WT ENT1, transporting uridine (Fig. 7b) but not MPP+ (data not shown). These data suggest that whereas Glu206 is essential for the cation selectivity in PMAT, changing the equivalent residue in ENT1, Gln180, to a negatively charged residue is not sufficient to make ENT1 accommodate an organic cation as a substrate.
FIGURE 7. Uridine uptake by wild type and mutant transporters of PMAT (a) and hENT1 (b).

The uptake of [3H]uridine (5 μM) was performed at 37 °C for 1 min. Each value represents the mean ± S.D. (n = 3). *, p < 0.05 significantly different from uptake in cells expressing the wild type transporter.
Effect of Mutations at Thr220
The above study identified that Glu206 in TM5 is essential for the cation selectivity of PMAT. Another residue, Thr220, whose alanine mutant showed reduced transport activity toward organic cations, is also located in TM5. Compared with WT PMAT, the plasma membrane expression level of T220A did not decrease as suggested by both confocal microscopy imaging and cell surface biotinylation and Western analysis (Figs. 4 and 5). Therefore, the reduced activity of this mutant indicates that Thr220 may also participate in interactions with substrates. To further elucidate the functional role of Thr220, we generated two additional mutants, T220S and T220I. The serine (Ser) mutation conserved the hydroxyl group of threonine (Thr) but shortened the side chain by one carbon length. The isoleucine (Ile) mutation eliminated the hydroxyl group and also increased the side chain volume. When stably expressed in MDCK cells, both mutants exhibited plasma membrane expression (Fig. 5a) with cell surface expression levels comparable with that of WT PMAT (Fig. 5b). Like T220A, T220S exhibited a significantly decreased activity (Fig. 6). Kinetic analysis of T220A and T220S mutants showed that the reduced activity was mainly due to a decrease in Vmax (Table 2), suggesting that substitution of Thr220 may have affected the turnover rates of the transporter. Mutant T220I exhibited normal plasma membrane localization but had no uptake activity toward all three tested substrates, suggesting that substitution of Thr220 by a bulky side chain abolished transporter activity (Fig. 6).
Helical Wheel Analysis
To further elucidate the role of TM5, in which Glu206 and Thr220 are contained, helical wheel analysis was performed using a fixed angle of 100°. A distinct amphipathic pattern was observed, in which residues Glu206 and Thr220 are clustered closely in the center of the hydrophilic face (Fig. 8). Such a distinct amphipathic pattern was not observed for other TMs. These data indicate that TM5 may participate in the formation of the substrate permeation pathway in PMAT.
FIGURE 8. Helical wheel analysis of TM5 in PMAT.

The transmembrane domain is assumed to be a standard α-helix and each residue is plotted every 100° around the center of a circle. The figure shows the projection of the positions of the residues on a plane perpendicular to the helical axis. Hydrophobic residues are shown in white, whereas hydrophilic residues are shown in blue. The hydrophobicity and hydrophilicity are assigned according to the consensus scale of Eisenberg et al. (20).
DISCUSSION
Several major findings are presented in this study. First, our PMAT and hENT1 chimera studies clearly suggest that the critical functional regions in PMAT are contained in TM1–6. Second, a negatively charged residue located in TM5, Glu206, has been identified as a key molecular determinant for the organic cation selectivity of PMAT. Finally, our data suggest that TM5 is a vital component of the substrate permeation pathway in PMAT.
Our observation that chimera T1–6 maintained the substrate specificity of PMAT (Fig. 1) provides strong evidence that TM1–6 contains the key domains for substrate recognition/translocation. Whereas it is possible that some TMs in the C-terminal half of PMAT may also participate in the formation of the substrate permeation pathway, the functional property of chimera T1–6 suggested that the major site(s) determining the organic cation selectivity is located within TM1–6. The importance of this region is further supported by our mutagenesis data, which identified that key residues for substrate recognition are located in TM5. Our data are consistent with results from chimeric studies of the ENTs, which also showed that the N-terminal half (TM1–6) of ENT1 and -2 contains critical domains for substrate/inhibitor recognition (21, 22). Together, these results indicate that despite the distinct substrate selectivity and low sequence homology, PMATs and the ENTs share a similar protein modular organization. In contrast, a series of studies on the OCTs suggested that the C-terminal half of the OCTs play a more important role in substrate selectivity (24–26). Thus, despite the large similarity in substrate selectivity shared by PMAT and OCTs, the structural mechanisms governing their organic cation transport functions may be fundamentally different.
To further identify key residues in PMAT, we employed a site-directed mutagenesis approach based on multiple sequence alignment. We mainly focused on negatively charged residues (Glu or Asp) because we have previously demonstrated that carrying a positive charge is essential for a compound to be recognized by PMAT (8). Of the 33 negatively charged Glu or Asp in PMAT, 10 residues were selected for mutational analysis based on multiple sequence alignment with the ENTs. Of these 10 candidate residues, only Glu206 was identified to be essential for the organic cation transport activity of PMAT. Other residues were either not absolutely required for the transport activity (Asp91, Glu128, Glu227, Glu242, and Glu375) or appeared to have affected membrane trafficking (Asp107 and Asp163) or protein stability (Asp154 and Asp336). In contrast, changing Glu206 to Gln, Ala, or Arg led to a complete loss of organic cation transport activity without affecting plasma membrane expression (Figs. 4 – 6). Furthermore, changing Glu206 to a charge conserved residue (Asp) restored organic cation transport activity (Fig. 6). More interestingly, whereas mutant E206Q, which possesses the equivalent residue in ENT1, completely lost transport activity toward MPP+, it gained the ability to transport the ENT substrate, uridine (Fig. 7a). These results strongly suggest that Glu206 is essential for the cation selectivity of PMAT and may function as the charge sensor for cationic substrates. Recently, Barnes et al. (17) reported that PMAT is able to transport adenosine, a nucleoside, at acidic pH. Because adenosine can be protonated at the nitrogen atom in the imidazole ring at low pH, this may “camouflage” adenosine to be cationic and recognized by residue Glu206.
It is possible that Glu206 may not directly interact with the substrate but is more involved in a generalized function such as protein folding. Studies of the human organic anion transporter 1 (hOAT1) indicated that a negatively charged glutamate residue (Glu506) in hOAT1 is critical for its transport activity possibly by maintaining the correct tertiary structure of the transporter through salt-bridge formation (27). However, the observation that mutants E206Q and E206A gained the ability to transport uridine suggests that Glu206 is more involved in substrate selectivity than protein folding. If mutations at Glu206 disrupted the tertiary structure of PMAT, one would expect that mutants E206Q and E206A should have lost transport activity toward all substrates. Additionally, these mutants showed normal plasma membrane expression, indicating that they were able to exit the endoplasmic reticulum, which often retains and degrades proteins that are not folded properly (28). Thus, our data suggested a specific role for Glu206 in determining the cation selectivity of PMAT.
In addition to Glu206, Thr220 in TM5 also plays an important role in transporter function. Mutation of Thr220 to smaller residues (Ala or Ser) greatly reduced PMAT activity, and replacement with a bulky residue (Ile) abolished transport activity without affecting cell surface expression. Whereas our data clearly demonstrated that the size of the side chain at position 220 is critical for PMAT function, the specific role of Thr220 is less clear. Thr220 may directly interact with substrate through hydrogen bonding and/or hydrophobic interactions, or it may simply contribute to the control of an appropriate volume of the binding pocket.
Both Glu206 and Thr220 are located on TM5, suggesting that this TM is a key component of the substrate permeation pathway. The importance of TM5 is further supported by helical wheel analysis, which revealed a distinct amphipathic pattern and suggested that one side of TM5 may face an aqueous pore for substrate recognition and translocation. Residues Glu206 and Thr220 are located in the center of the hydrophilic face, and their side chains may protrude into the pore and interact with the substrates. However, we have no experimental evidence regarding how the TM helices are packed to form the substrate permeation pathway. Therefore, the exact role of TM5 in substrate recognition and translocation requires further investigation.
In summary, our studies revealed the key structural and molecular elements that underlie the transport function of PMAT. Furthermore, the integrated information gained from our study provides unique insights into the structure and function relationships of transporters in the SLC29 family.
Footnotes
This work was supported by National Institutes of Health Grant GM66233.
The abbreviations used are: SLC, solute carrier; PMAT, plasma membrane monoamine transporter; ENT, equilibrative nucleoside transporter; OCT, organic cation transporter; YFP, yellow fluorescent protein; MPP+, 1-methyl-4-phenylpyridinium; MDCK, Madin-Darby canine kidney; WT, wild type.
References
- 1.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. Pflugers Arch. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 2.Saier MH., Jr Microbiol Mol Biol Rev. 2000;64:354–411. doi: 10.1128/mmbr.64.2.354-411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright EM, Loo DD, Hirayama BA, Turk E. Physiology (Bethesda) 2004;19:370–376. doi: 10.1152/physiol.00026.2004. [DOI] [PubMed] [Google Scholar]
- 4.Kong W, Engel K, Wang J. Curr Drug Metab. 2004;5:63–84. doi: 10.2174/1389200043489162. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. Pflugers Arch. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin SA, Yao SY, Hyde RJ, Ng AM, Foppolo S, Barnes K, Ritzel MW, Cass CE, Young JD. J Biol Chem. 2005;280:15880–15887. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- 7.Acimovic Y, Coe IR. Mol Biol Evol. 2002;19:2199–2210. doi: 10.1093/oxfordjournals.molbev.a004044. [DOI] [PubMed] [Google Scholar]
- 8.Engel K, Wang J. Mol Pharmacol. 2005;68:1397–1407. doi: 10.1124/mol.105.016832. [DOI] [PubMed] [Google Scholar]
- 9.Engel K, Zhou M, Wang J. J Biol Chem. 2004;279:50042–50049. doi: 10.1074/jbc.M407913200. [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Urban TJ, Leabman MK, Fujita K, Giacomini KM. J Pharm Sci. 2006;95:25–36. doi: 10.1002/jps.20536. [DOI] [PubMed] [Google Scholar]
- 11.Koepsell H, Endou H. Pflugers Arch. 2004;447:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- 12.Wright SH, Dantzler WH. Physiol Rev. 2004;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Brett CM, Giacomini KM. Annu Rev Pharmacol Toxicol. 1998;38:431–460. doi: 10.1146/annurev.pharmtox.38.1.431. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M, Engel K, Wang J. Biochem Pharmacol. 2007;73:147–154. doi: 10.1016/j.bcp.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia L, Engel K, Zhou M, Wang J. Am J Physiol. 2006 in press. [Google Scholar]
- 16.Sundaram M, Yao SY, Ingram JC, Berry ZA, Abidi F, Cass CE, Baldwin SA, Young JD. J Biol Chem. 2001;276:45270 – 45275. doi: 10.1074/jbc.M107169200. [DOI] [PubMed] [Google Scholar]
- 17.Barnes K, Dobrzynski H, Foppolo S, Beal PR, Ismat F, Scullion ER, Sun L, Tellez J, Ritzel MW, Claycomb WC, Cass CE, Young JD, Billeter-Clark R, Boyett MR, Baldwin SA. Circ Res. 2006;99:510–519. doi: 10.1161/01.RES.0000238359.18495.42. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Giacomini KM. J Biol Chem. 1997;272:28845–28848. doi: 10.1074/jbc.272.46.28845. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Giacomini KM. J Biol Chem. 1999;274:2298–2302. doi: 10.1074/jbc.274.4.2298. [DOI] [PubMed] [Google Scholar]
- 20.Eisenberg D, Wilcox W, McLachlan AD. J Cell Biochem. 1986;31:11–17. doi: 10.1002/jcb.240310103. [DOI] [PubMed] [Google Scholar]
- 21.Sundaram M, Yao SY, Ng AM, Griffiths M, Cass CE, Baldwin SA, Young JD. J Biol Chem. 1998;273:21519–21525. doi: 10.1074/jbc.273.34.21519. [DOI] [PubMed] [Google Scholar]
- 22.Yao SY, Ng AM, Vickers MF, Sundaram M, Cass CE, Baldwin SA, Young JD. J Biol Chem. 2002;277:24938–24948. doi: 10.1074/jbc.M200966200. [DOI] [PubMed] [Google Scholar]
- 23.Lai Y, Bakken AH, Unadkat JD. J Biol Chem. 2002;277:37711–37717. doi: 10.1074/jbc.M204986200. [DOI] [PubMed] [Google Scholar]
- 24.Popp C, Gorboulev V, Muller TD, Gorbunov D, Shatskaya N, Koepsell H. Mol Pharmacol. 2005;67:1600–1611. doi: 10.1124/mol.104.008839. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Shirahatti NV, Mahadevan D, Wright SH. J Biol Chem. 2005;280:34813–34822. doi: 10.1074/jbc.M506342200. [DOI] [PubMed] [Google Scholar]
- 26.Gorboulev V, Shatskaya N, Volk C, Koepsell H. Mol Pharmacol. 2005;67:1612–1619. doi: 10.1124/mol.104.008821. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Tanaka K, Sun AQ, You G. J Biol Chem. 2006;281:31178–31183. doi: 10.1074/jbc.M605664200. [DOI] [PubMed] [Google Scholar]
- 28.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 3. Garland Publishing, Inc.; New York: 2002. [Google Scholar]