

Abstract
Doxorubicin (DOX) is used for treating various cancers. Its clinical use is, however, limited by its dose-limiting cardiomyopathy. The exact mechanism of DOX-induced cardiomyopathy still remains unknown. The goals were to investigate the molecular mechanism of DOX-induced cardiomyopathy and cardioprotection by mitoquinone (Mito-Q), a triphenylphosphonium-conjugated analog of coenzyme Q, using a rat model. Rats were treated with DOX, Mito-Q, and DOX plus Mito-Q for 12 weeks. The left ventricular function as measured by two-dimensional echocardiography decreased in DOX-treated rats but was preserved during Mito-Q plus DOX treatment. Using low-temperature ex vivo electron paramagnetic resonance (EPR), a time-dependent decrease in heme signal was detected in heart tissues isolated from rats administered with a cumulative dose of DOX. DOX attenuated the EPR signals characteristic of the exchange interaction between cytochrome c oxidase (CcO)-Fe(III) heme a3 and CuB. DOX and Mito-Q together restored these EPR signals and the CcO activity in heart tissues. DOX strongly downregulated the stable expression of the CcO subunits II and Va and had a slight inhibitory effect on CcO subunit I gene expression. Mito-Q restored CcO subunit II and Va expressions in DOX-treated rats. These results suggest a novel cardioprotection mechanism by Mito-Q during DOX-induced cardiomyopathy involving CcO.
Introduction
Doxorubicin (DOX), an anthracycline quinone antibiotic, is widely used in the treatment of several cancers, including breast cancer, Hodgkin's disease, and leukemia (1,2). However, its clinical use is limited by a dose-dependent, acute and chronic cardiotoxicity that is cumulative and irreversible (3–5). The cardiac dysfunctions, i.e., irreversible congestive heart failure, arrhythmias, and cardiomyopathy, are the major cause of mortality in cancer patients treated with DOX (6,7). Heart problems in DOX-treated cancer patients do not manifest until several years after cessation of chemotherapy (8,9). Children treated for leukemia with DOX develop heart conditions (e.g., impaired left ventricular contractility and late congestive heart failure) many years after DOX chemotherapy (10,11). Estimates indicate that children who undergo DOX chemotherapy are nearly eight times more susceptible to develop heart problems later in life (11). The exact mechanism of DOX-induced belated cardiotoxicity still remains unknown, although multiple mechanisms have been proposed, including oxidative stress, mitochondrial DNA damage, intracellular calcium overload, cytokine release, and disturbance of myocardial adrenergic function (12,13).
Recent research shows that DOX treatment induces apoptosis in myocytes and endothelial cells (14–16). Apoptosis in myocardium eventually leads to cardiomyopathy due to a systematic reduction in the number of cardiomyocytes (17). DOX also causes acute toxicity, including endothelial dysfunction (18). The target organ of DOX toxicity is the myocardium enriched with mitochondria. Prior reports suggest that one of the reasons why heart muscle is more susceptible to oxidant-induced damage is due to the presence of low levels of antioxidant enzymes, such as catalase, in myocytes (19,20). DOX accumulates over time into mitochondria. The intramitochondrial concentration of DOX was reported to be nearly two orders of magnitude higher than its extracellular concentration in the culture media (21,22). Thus, at clinically relevant plasma concentrations (0.5-1 μM), the intramitochondrial concentration of DOX should be ∼50-100 μM. The selective toxicity of DOX to the heart was attributed to the selective damage to cardiac mitochondria (20).
Previously it has been shown that coenzyme-Q (Co-Q) administration prevents the onset and progression of DOX-induced cardiomyopathy (23). Co-Q administration resulted in improved EKG changes and survival rates (23). It was proposed that the lipophilic aglycone metabolite of DOX presumably penetrates the inner membrane, displacing Co-Q, which leads to the acute rise in the plasma Co-Q levels in patients during DOX chemotherapy. Although numerous antioxidant therapies have previously been developed to combat DOX toxicity (24), most of these agents were not particularly effective. In addition, these antioxidants were not specifically targeted to mitochondria of cells. Recent developments in organic syntheses have made it possible to specifically target mitochondria (25–27). Antioxidants such as vitamin-E and coenzyme-Q were coupled to the triphenylphosphonium cation, and the resulting compounds (Mito-E and Mito-Q) were preferentially taken up by the mitochondria (26,27). The driving force responsible for mitochondrial accumulation of lipophilic triphenylphosphonium cations is the large membrane potential of 150-180 mV (negative inside) across the mitochondrial inner membrane (26,27). Mito-Q was shown to accumulate 50- to 100-fold within mitochondria (27).
To more closely mimic DOX-induced cardiomyopathy in the clinic, lower doses of DOX were chronically administered to Sprague-Dawley rats once a week (2.5 mg/kg), and 2D echocardiography was performed to assess the morphologic and functional changes in the left ventricle. This model is consistent with the previously established model of chronic DOX-induced cardiomyopathy (28). The cardiac tissues are abundant in electron paramagnetic resonance (EPR) active binuclear and mononuclear Cu, Fe centers and various bi-, tri- and tetranuclear Fe-S clusters (29) (Fig. 1). In this study, we have examined levels of mitochondrial cytochrome c oxidase subunits and electron transfer iron sulfur clusters using an established rat model that mimics the lower doses of DOX used to study DOX-induced cardiomyopathy in the clinic. Treatments with DOX or DOX plus Mito-Q, a coenzyme-Q analog, were compared to assess the cardioprotective effects of Mito-Q. The electronic states of cytochrome c oxidase components were monitored by ex vivo low-temperature (liquid helium) X-band EPR.
Figure 1.
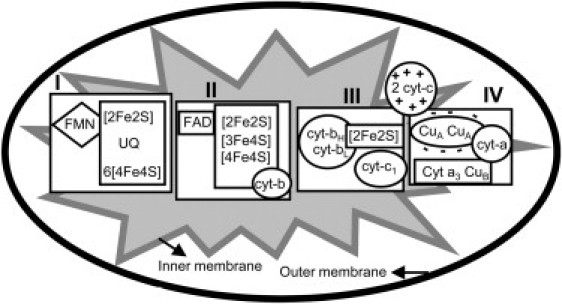
Mitochondrial respiratory chain iron-sulfur centers, cytochromes, and active metal centers involved in the electron transport process. (I) NADH dehydrogenase, (II) Succinate dehydrogenase, (III) Cytochrome c reductase, and (IV) Cytochrome c oxidase.
Our data revealed that DOX treatment alters exchange coupling between high spin heme a3 and CuB in complex IV, but leaves iron sulfur clusters unaffected. After prolonged treatment with DOX, levels of subunits II and Va were decreased relative to other cytochrome c oxidase (CcO) subunits. Mito-Q attenuated the effects of DOX treatment.
Materials and Methods
Materials
DOX hydrochloride, rotenone, diethylenetriaminepentaacetic acid, cytochrome c, antimycin, EDTA, tris, ubiquinone-1, potassium cyanide, and NADH were purchased from Sigma Chemical (St. Louis, MO). Mitoquinone (Mito-Q10) was synthesized according to the published method with some modifications (30). A major difference between our procedure and the published procedure (30) is the synthesis of the intermediate bromoidebenone from idebenone (Fig. S1 in the Supporting Material). The published route for bromoidebenone involved the use of carbon tetrabromide and triphenyl phosphine. Bovine CcO was obtained as a gift from Dr. Helmut Beinert (Department of Biochemistry, University of Wisconsin, Madison).
Animal care and treatment protocols
All animal procedures were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee, and the investigation conforms to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Male Sprague-Dawley rats weighing 250-260 g were used because females may possess cardiac protective effects because of estrogen, along with increased levels of telomerase activity, which could increase the tissue regeneration capacity. Animals were housed in the Biocontainment Suite and had free access to a diet consisting of normal rat chow (Harlan Teklad, Madison, WI) and water ad libitum.
Animals were randomly assigned to four different treatment groups: control (n = 16), DOX (n = 11), DOX plus Mito-Q (n = 12), and Mito-Q (n = 19) (Fig. S2). In experiment I, rats were injected intravenously 2.5 mg/kg per week with doxorubicin hydrochloride dissolved in saline, and control groups received the same volume of saline (0.9% sodium chloride solution). In experiment II, DOX (2.5 mg/kg per week) and Mito-Q (5.0 mg/kg by intraperitoneal injection, twice a week) were given. In experiment III, Mito-Q alone (5.0 mg/kg) was administered under similar conditions. During this period, rat body weight and echocardiograms were obtained at baseline, 4, 8, 10 and 12 weeks after initiation of treatment. After 4, 8, 10 and 12 weeks of treatment, animals were euthanized with an intraperitoneal injection of sodium pentobarbital (90 mg/100 g body weight). The heart was excised immediately and stored in liquid nitrogen for histology and EPR measurements. Tissues for histology were fixed in zinc-formaldehyde solution (Fisher Scientific, Pittsburgh, PA).
EPR measurements
The X-band EPR of cardiac tissues were recorded at liquid helium temperatures on a Bruker E500 ELEXYS spectrometer with 100 kHz field modulation, equipped with an Oxford Instrument ESR-9 helium flow cryostat and a DM-0101 cavity. For EPR measurements, heart tissues were excised and immersed in liquid nitrogen, minced to appropriate sizes under liquid nitrogen, and then transferred to a four mm quartz EPR tube (Wilmad-Lab Glass, Buena, NJ). The heart tissue weight in each EPR experiment was ∼0.3 g. The tissue packing factor was not perceived to be a major problem, as reloading of tissues in the same EPR tube (with a different packing) resulted in EPR spectra with a 5% variation in signal intensity. Bovine heart CcO (30 μM) present in 10% dodecyl maltoside containing 100 mM potassium/HEPES, pH 7.0, was used as a standard. Spectrometer conditions were as follows: microwave frequency, 9.635 GHz; modulation frequency, 100 kHz; modulation amplitude, 10 G; receiver gain, 85 dB; time constant, 0.01 s; conversion time, 0.08 s; sweep time, 83.9 s. EPR spectra were obtained over the temperature range 4-50 K using an incident microwave power of 5 mW and a modulation amplitude of 10 G. The spectrometer was calibrated with the radical 1,1-diphenyl-2-picrylhydrazyl, exhibiting an EPR signal centered at g = 2.0036.
EPR simulations
EPR spectra for an exchange coupled two-spin system were successfully fitted using the spin Hamiltonian shown in Eq.1 in the “Results” section. All computer simulations of the EPR spectra were calculated from diagonalization of this equation with XSophe software. A matrix diagonalization method was used to obtain the eigenvalues and eigenvectors. For a dinuclear Fe(III) (heme a3) CuB(II) center that is antiferromagnetically coupled, where S1 = 5/2 and S2 = 1/2, a 12 × 12 energy matrix was obtained when all twelve spin transitions were included (i.e., (2S1 + 1) × (2S2 + 1) = 12). Microwave power saturation and variable temperature EPR spectra are shown (Figs. S4 and S5).
Activities of NADH-quinone reductase (complex I), CcO and aconitase
The activities of complex I were assayed as reported at 30°C as a decrease of absorption at 340 nm with 200 μM NADH and 100 μM ubiquinone-1 in the presence of 1.5 mM KCN. The activity of CcO was measured spectrophotometrically by following the oxidation of ferrocytochrome c at 550 nm (31). The standard assay mixture contained the following: 0.25 M sucrose, 50 mM Tris/Cl -(pH 8.0), 0.2 mM potassium EDTA, and the enzyme preparation (mitochondria ∼10 μg protein/ml). Aconitase activity was determined spectrophotometrically by monitoring the disappearance of cis-aconitate as reported (32).
Caspase activity measurements
The excised heart was washed in ice-cold Krebs-Henseleit buffer solution containing NaCl 118 mM, KCl 4.75 mM, KH2PO4 1.19 mM, MgSO4 1.19 mM, CaCl2 2.54 mM, NaHCO3 25 mM, EDTA 0.5 mM, and glucose 11 mM and immediately frozen in liquid nitrogen. Frozen tissue was subsequently ground into a powder, using a mortar and pestle, and resuspended with ice-cold lysis buffer (50 mM HEPES, pH 7.4, 0.1% CHAPS, 5 mM DTT, 2 mM EDTA, 2 mM EGTA, Triton X-100, 0.1%) containing 1 mM phenylmethylsulfonyl fluoride and 10 μg/ml aprotinin and leupeptin. Homogenates were centrifuged at 14,000 × g for 10 min and the supernatants were used. Proteins (100 μg) were diluted with the assay buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10 mM DTT, 2 mM EDTA, 2 mM EGTA, Triton X-100, 0.1%, and incubated at 25°C with DEVD-pNA (acetyl Asp-Glu-Val-Asp paranitroanilide), ac-LEHD pNA (acetyl Leu-Glu-His-Asp pNA), and ac-IETD pNA (acetyl-Ile-Glu-Thr-Asp-pNA) as substrates (each 200 μM final concentration). The absorbance at 405 nm of the released pNA was monitored in a spectrophotometer and quantitated using a suitable standard (33).
Immunoblot analysis for cytochrome c oxidase subunit content
Proteins were subjected to electrophoresis on 15% polyacrylamide gels as described by Kotamraju (33). The conditions for Western blot analysis and immunodetection of proteins were the same as described previously (34). Specific antibody interactions were tested by probing with monoclonal antibodies to CcO subunits I, II, IV and Vb obtained from Mitosciences (Eugene, OR). Secondary antibody was detected and quantitated by incubating the membrane with West Femto Super Signal (Pierce, IL). Blots were quantified with an Image Scanner STORM (Molecular Dynamics, Sunnyvale, CA).
Echocardiography
Transthoracic two-dimensional echocardiography (Vivid 7 Echocardiograph, General Electric, Waukesha, WI) was performed with the use of a linear array 11 MHz transducer at baseline, 4, 8, 10, and 12 weeks on all animals anesthetized by using 0.25 mg/kg medtomidine and 75 mg/kg ketamine. The chest of the animal was shaved for the procedure. The parasternal short axis view at the mid left ventricular level was used, verified by the presence of prominent papillary muscles. B-mode 2-dimensional echocardiography was performed. Offline analyses were performed using Echopac workstation (General Electric, Waukesha, WI). Myocardial radial strain, an intrinsic measure of myocardial mechanics, was obtained using Q analysis software (General Electric, Waukesha, WI) as reported previously (31). Briefly, the endocardial border was traced and the epicardial border was delineated manually. The software automatically computes the radial strain (percent change of myocardial segment length toward or away from the center of the heart in short axis view) from six equidistant segments of the left ventricle. The global radial strain was the average of the six segmental radial strains The data from rats that survived >8 weeks were used for analysis.
Statistical analysis
For radial strain, repeated analyses of variance with treatment and time as covariants was used (Sigmastat 3.5, Systat Software Richmond, VA). For pairwise analysis, Holm-Sidak method was used. Significant p-value was set at p = 0.05.
Results
Mito-Q administration prevents DOX-induced cardiac dysfunction
Initially, the cardiac function after treatment with DOX, with and without Mito-Q coadministration, was monitored by physiological parameters and by 2D echocardiography. As shown in Fig. S2, animals were divided into four groups. After 10 weeks of DOX treatment, animals became weak and lethargic and the skin color became pale as compared to control rats. The most prominent feature in DOX-treated animals was the development of an enlarged abdomen and ascites after eight weeks of DOX treatment; postsacrifice, a significant amount of peritoneal fluid was noted. In addition, the body weight of the DOX-treated rats was significantly lower than that of the control (Fig. 2). After 10 weeks of treatment, the heart weight of DOX-treated rats was considerably less than that of the control. In contrast, the body weight and the heart weight of DOX plus Mito-Q treated rats were similar to those of control and Mito-Q-treated rats. Prolonged treatment with Mito-Q partially reversed the adverse side effects caused by DOX administration (Fig. 2).
Figure 2.

(A) Changes in the heart weights of rats after 12 weeks of treatment with DOX, Mito-Q, and DOX plus Mito-Q. (B) Changes in the body weights of rats undergoing DOX treatment as shown in Fig. S2.
DOX treatment diminished myocardial function. Myocardial strain is a measure of intrinsic myocardial mechanics (31). Two-dimensional strain echocardiography serves as a sensitive measure of myocardial dysfunction in both human and rodent models and is superior to other conventional measures of ventricular function. The administration of DOX caused a reduction in cardiac function assessed by echocardiography (31). There was a progressive reduction in global radial strain starting at eight weeks after DOX treatment (Fig. 3). Coadministration of Mito-Q prevented the progressive decline in global radial strain seen with DOX treatment alone.
Figure 3.

(A) Endsystolic 2D B-mode images at mid-ventricular level: (1) Control rat, (2) DOX treatment for 12 weeks, (3) DOX plus Mito-Q treatment for 12 weeks, and (4) Mito-Q treatment for 12 weeks. (B) Anatomical M-mode through the anterior and interior walls. (C) Graph showing the radial strain in one cardiac cycle of 6 equidistant regions of the myocardium in the short axis view. Global radial strain was computed as the average of all six segments.
Ex vivo low temperature EPR spectra of control heart tissues: Effects of DOX and DOX plus Mito-Q
To evaluate the status of redox-active components of heart tissue, rats were sacrificed after defined treatment protocols. As shown in Fig. S2, rats were sacrificed after 4, 8, 10, and 12 weeks of treatment protocols, and hearts were removed and frozen for EPR analysis. The EPR spectra of control rat heart tissues were recorded at 15 K (Fig. 4 A). The most clearly discernible signals are due to the high spin heme at g = 6.0, assigned to heme a3 (Fea33+CuB1+ state of CcO), high spin cytochrome c, and a trinuclear 3Fe-4S cluster signal at g = 2.015 (35). At a higher gain, other EPR absorptions were noticeable (Fig. 4 A, dashed line). Based on spectral simulations, the composite EPR spectrum was deconvoluted into several different species (Fig. S3). As cardiac tissues are enriched in mitochondria, the signals arising from the binuclear [2Fe-2S] cluster (N1), complex II or III, and three tetranuclear [4Fe-4S] clusters (N2, N3 and N4) were also detected. The EPR parameters obtained from computer simulations are in agreement with those previously reported for complex I, II and III iron-sulfur clusters (35). The spectrum (Fig. 4 A) also consists of a low spin heme signal at gz = 3.0 [heme a, cytochrome c] and at gz = 3.9-3.2 (cyt-bH and cyt-bL), respectively (36,37). The simulations of these signals were achieved by using a large line width parameter, suggesting that these absorptions are either a magnetically coupled system or that there is strain broadening. The resonance at gz = 2.16 was assigned to a dinuclear CuACuA center of complex IV (38). The CuACuA signal at g = 2.02 was obscured by the strong 3Fe-4S signal. In addition, an EPR signal detected at g = 4.3 was tentatively attributed to a rhombic iron (III). The experimental and simulated EPR spectra of authentic CcO enzyme are shown in Fig. S6.
Figure 4.
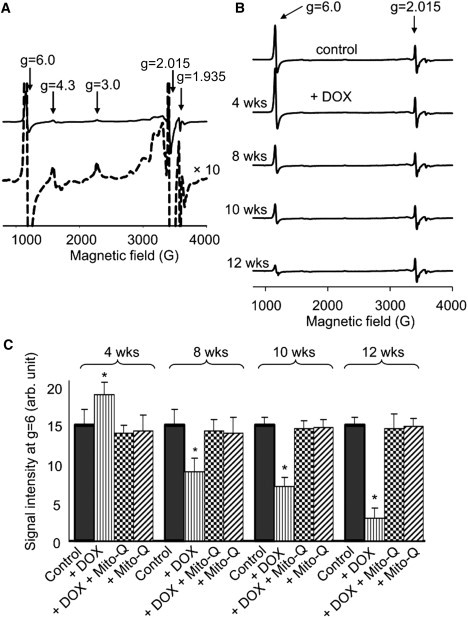
(A) (Top) X-band EPR spectra of control heart tissues from rats. EPR were performed at 15 K using a 5 mW microwave power. (Bottom) Same as above but at a higher gain. (B) Time course EPR spectra obtained from cardiac tissues isolated from rats treated with DOX at 0, 4, 8, 10 and 12 weeks. EPR spectra were recorded at 10 K using a 5 mW microwave power. (C) EPR signal intensities of g = 6.0 signal. Values are given as mean ± SE. ∗p<0.05 versus controls. Statistical analysis was performed by using Student's unpaired t-test.
Fig. 4 B shows the effect of DOX on the EPR spectra of heart tissues with time. Heart tissues were isolated from rats after 4, 8, 10, and 12 weeks of DOX treatment. Initially, the EPR intensity of the g = 6 signal (assigned to heme a3 of CcO) after four weeks of DOX treatment increased. Thereafter, the intensity of this signal (g = 6) decreased with increasing duration of DOX treatment. However, the EPR signals (g = 2.013) corresponding to the Fe-S clusters of complex I, II and III were not significantly affected by DOX treatment under the same conditions. In the presence of DOX and Mito-Q, the EPR signal intensities at g = 6 obtained from heart tissues after 4, 8, 10, and 12 weeks of DOX treatment were similar to those obtained from control tissues. These results suggest that a prolonged treatment with DOX disrupts CcO in myocardial tissues and that the protective effect of Mito-Q in DOX-treated rats could be related to restoration of the signal intensity of heme a3 of CcO in heart tissues (Fig. 4 C).
Effect of DOX and Mito-Q on exchange-coupled resonances
Heme a3 and CuB sites are in close proximity, and thus exhibit exchange coupling. Fig. 5 shows the EPR spectra of control heart tissues obtained at a higher microwave power. At low temperatures (= 10 K) and at a higher microwave power (120 mW), the EPR signals at gz = 3.0 arising from the low spin Fe(III) of heme a and cytochrome c were partially saturated for analysis of the exchange coupled resonances. The exchanged coupled spectra were best simulated (Fig. 5 B) based on the weak magnetic exchange interaction between high spin Fe(III) heme and Cu(II) of CcO, described by an effective spin-Hamiltonian involving an exchange coupling tensor J (Eq. 1) (37):
| (1) |
where the first term represents the local spin-Hamiltonian for the S = 5/2 heme Fe(III) ion and contains Zeeman, axial (D), and rhombic (E) zero-field splitting terms, respectively. The second term is the Zeeman spin-Hamiltonian for S = 1/2 CuB(II). When the exchange interaction is weak, | J | « | D |, the axial zero-field splitting at the heme splits the Fe(III) levels into three Kramers doublets, MsFe = ±5/2, ±3/2 and ±1/2, separated in energy by 2D and 4D, respectively. The lowest doublet, MsFe = ±1/2, has typical D values of 5-7 cm−1. Exchange coupling between each of these three Kramers pairs and the single Kramers pair of CuB(II) S = 1/2 gives rise to three sets of states, each consisting of four levels with the components of the lowest set characterized by MS values of 0, 0, +1, and −1. An excellent simulation based on the weak exchange coupled model as shown in Fig. 5 (inset) was obtained using reported zero-field splitting values, D = 5 cm−1 and E = 0.03 cm−1, | J | is ∼1 cm−1 and d = 5.0 Å (the distance between Fea3 and CuB) (39).
Figure 5.
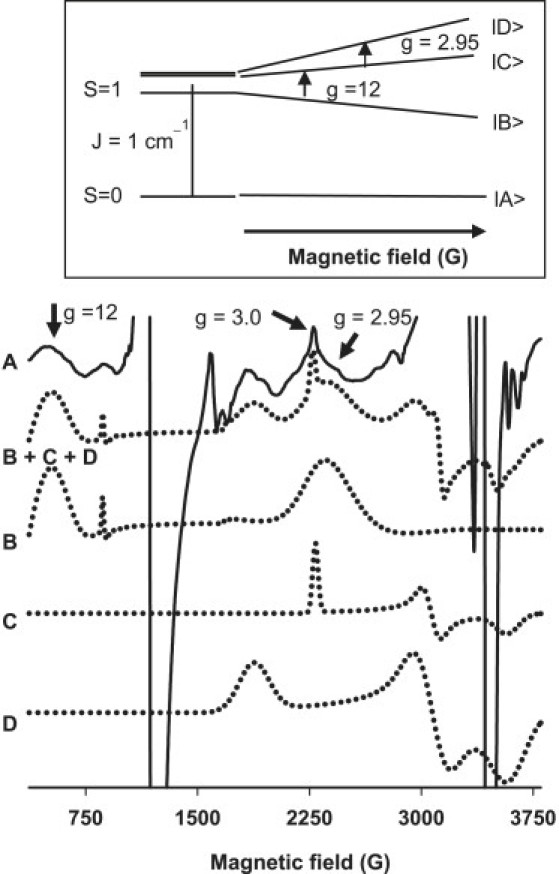
Fig. 6 A (signal marked ↓) shows the inhibitory effects of DOX treatment for 12 weeks on the exchange coupled transitions observed at higher powers in heart tissues. Upon treatment with Mito-Q for 12 weeks, the broad signals due to heme a3/CuB of CcO were partially restored in heart tissues. However, DOX and DOX plus Mito-Q had no effect on the EPR signals observed at g = 2.015 that are characteristic of oxidatively-inactivated iron sulfur centers (Fig. 6 B).
Figure 6.
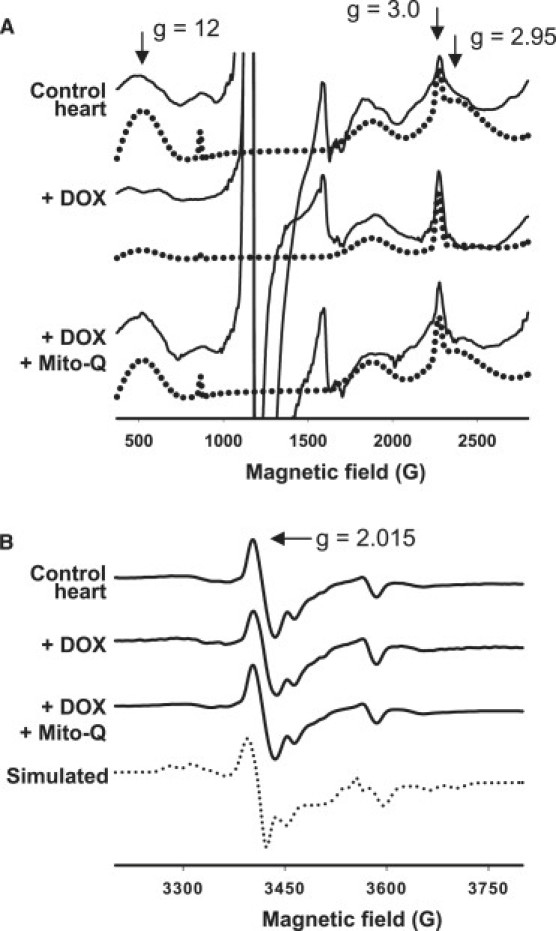
(A) The exchange coupled EPR spectra of heart tissues obtained from control rats, DOX-treated rats for 12 weeks, and DOX plus Mito-Q-treated rats for 12 weeks. Spectra were obtained at 10K using a microwave power of 100 mW, (solid line) experimental; (dotted line) simulated. (B) Effect of DOX / Mito-Q on iron-sulfur cluster EPR spectra recorded at 10 K using a 5 mW microwave power.
Effects of DOX and Mito-Q on CcO subunit expressions
Immunoblot analysis of four subunits of CcO was performed in heart tissues obtained from control, DOX-treated, Mito-Q-treated, and DOX plus Mito-Q-treated rats. Fig. 7 shows the levels of subunits I, II, IV and Va detected in total heart homogenates. Note that there is almost constant expression of CcO IV with time. However, the expressions of subunits II and Va were more severely affected with nearly a 65-70% decrease in their levels after DOX treatment for 12 weeks as compared to controls. Administering Mito-Q to DOX-treated rats significantly improved the expression of the CcO subunits. However, DOX treatment for four or eight weeks did not exhibit any significant changes in CcO on subunit levels in heart tissues (Fig. 7).
Figure 7.
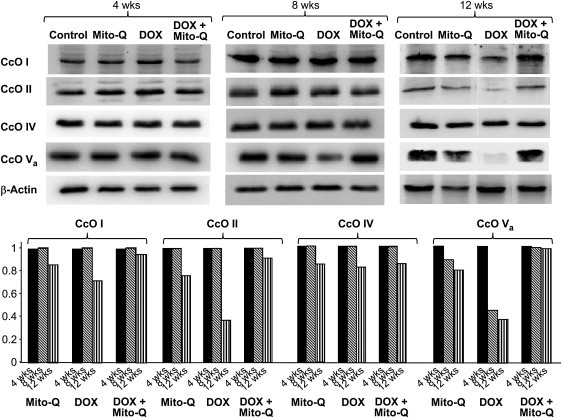
Immunoblot analyses of CcO subunits (I-Va) isolated from rat hearts after 4, 8, and 12 weeks of DOX and DOX plus Mito-Q. Experimental protocols are shown in Fig. S2.
Mito-Q inhibits DOX-mediated fibrosis and apoptosis
The cardiac tissues stained with trichrome blue showed no apparent histological differences between control and Mito-Q alone treated animals (Fig. S8). However, in the DOX-treated animals, substantial fibrosis was observed. The cardiac sections from animals treated with Mito-Q plus DOX showed significantly less fibrosis compared to the DOX group (Fig. S8). The caspase-3 activity was significantly higher in the DOX group compared to controls after treatment with DOX for eight weeks (Fig. S9). The caspase-3 activity was diminished in the DOX plus Mito-Q group compared to the DOX group. The complex IV activity was determined in heart mitochondria isolated from control, DOX-treated, and DOX plus Mito-Q-treated rats. DOX treatment caused a decrease in complex IV activity by ∼60% (Fig. S9). There was no significant decrease in the complex IV activity in rats fed with Mito-Q alone and Mito-Q significantly restored the complex IV activity in rats treated with DOX (p < 0.05). From these results, we can conclude that Mito-Q specifically affects the pathway by which DOX causes a decrease in complex IV activity.
Discussion
Mitochondrial dysfunction was implicated in DOX cardiotoxicity. The exact mechanisms of DOX inhibition of mitochondrial electron transport and oxidative phosphorylation are not clearly understood. Mitochondria-rich cardiac tissue consists of many complexes, I-IV, with several metal center proteins viz., Fe-S clusters, heme and binuclear Cu(II) and Fe(III)-Cu(II) in their active sites. Here, we have used the low temperature ex vivo X-band EPR to monitor the changes in mitochondrial proteins induced by DOX during cardiomyopathy and cardioprotection by Mito-Q.
The EPR spectra of heart tissues obtained at liquid helium temperatures recapitulate the evidence for exchange coupling previously reported in isolated CcO preparations. The EPR results from this study show that chronic treatment with DOX decreased the high spin heme a3 and uncoupled the exchange-coupled heme a3 to CuB in CcO enzyme in rat hearts. Concomitantly, the CcO activity and the catalytic subunit expressions (CcO II and CcO Va) were decreased. Mito-Q restored the signal intensity of heme a3 and the signal for coupled heme a3 and CuB of CcO in heart tissues, as well as the heart function and CcO activity in DOX-treated rats. These results suggest a novel mechanism involving CcO inhibition and cardiac dysfunction during DOX treatment.
Effect of DOX and Mito-Q on EPR spectra of cardiac tissue
EPR spectral simulations of cardiac tissue from rats revealed the presence of 4Fe-4S, 3Fe-4S, heme (high and low spin), dinuclear Cu(II) and Fe(III)Cu(II) centers in the mitochondrial electron transport chain. A comparison of EPR spectra of CcO from heart tissue with purified CcO (Fig. S6) reveals that the g = 6, 3, and 2.16 signals are assigned to heme a3 (high spin), heme a (low spin) and dinuclear CuA of CcO. Upon DOX treatment, the g = 6 signal initially increases due to partial reduction of the binuclear site, Fe(III)Cu(II) of CcO to Fe(III)Cu(I) plus the oxidized high spin signal from cytochrome c (40). After four weeks, the g = 6 signal progressively decreased. However, the characteristic signals due to Fe-S clusters viz., 3Fe-4S, 4Fe-4S, 2Fe-2S, heme of complexes II and III and the nonspecific iron signal at g = 4.3 of the DOX-treated group were found to be similar to control cardiac tissues. Thus, the active metal centers of complexes I–III as detected by EPR were not affected by DOX treatment in this chronic model. Mito-Q treatment restored the g = 6.0 signal intensity. The signal assigned to cytochrome c1 (gz = 3.9), cytochrome bH (gz = 3.4) and cytochrome bL (gz = 3.75) of complex-III were not affected by DOX or by DOX plus Mito-Q treatment. These results reveal that complex III hemes and iron-sulfur clusters detected by EPR were unaffected by DOX treatment in this chronic rat model of cardiomyopathy.
Effect of DOX and Mito-Q on exchange-coupled resonances between Fea3CuB
Previous EPR results indicate that CcO with the binuclear active center, Fea3CuB, show exchange-coupled resonances at g = 2.95 and 12 at higher microwave power (40). The microwave power dependence on EPR signals from control, DOX- and Mito-Q-treated cardiac tissues was similar. With increasing microwave power, the peak intensity at g = 2.95 and 12 became more pronounced. The broad resonances observed from cardiac tissues in Fig. 5 were similar to previous reports wherein CcO was reduced by sodium dithionite for 2 s (40,41). Computer simulations of exchange-coupled EPR spectra using the reported parameters of CcO based on the weak exchange coupling model (41,42) gave a reasonably good fit. Upon DOX treatment, the intensities of signals at g = 2.95 and 12 were diminished, similar to that of the g = 6 signal. At 10-12 weeks after DOX treatment, these signals were significantly diminished. However, in the presence of Mito-Q, these signals were partially restored. The EPR results are in agreement with the observed decrease in the enzyme activity of CcO after a prolonged treatment with DOX and the subsequent restoration of the CcO activity during Mito-Q treatment.
DOX-induced suppression of CcO subunit expressions
The complex IV of the mitochondrial respiratory chain consists of 13 polypeptide chains (43). The three larger, catalytic subunits (CcO I, CcO II, and CcO III) are encoded by the mitochondrial DNA, and the remaining smaller subunits (IV-VIII) are encoded by nuclear DNA (43) (Fig S10). Mutations in the mitochondrial and nuclear genes encoding polypeptides of the respiratory chain often lead to cardiomyopathy (44). DOX treatment was reported to inhibit the CcO activity in cardiac tissues and leukemia cells (45). DOX decreased the expression of mitochondrially-encoded CcO subunits, CcO I and CcO II (46–48). In another report, overexpression of metallothionein protein was shown to mitigate DOX-induced cardiotoxicity (49). Cardioprotection was accompanied by upregulation of CcO Va subunit (49). The in vivo data show that CcO II and CcO Va subunits were greatly diminished in heart tissues after 12 weeks of DOX treatment (Fig. 7). There was a 25% reduction in CcO I expression. Mito-Q administration totally restored the expressions of CcO I, CcO II, and CcO Va. Previously, dexrazoxane administration was reported to restore mitochondrial DNA-encoded CcO subunit II in DOX-treated rats (50). As previously mentioned, both CcO subunits I and II are mitochondrially-encoded, whereas the CcO Va is encoded by the nuclear DNA. This suggests that DOX-induced cardiotoxicity may involve alterations in both mitochondrial and nuclear genomes and that Mito-Q inhibits the oxidative mitochondrial and nuclear DNA damage.
Alternatively, the binding of anthracyclines (daunomycin and doxorubicin) to CcO has been reported in cancer cells (51). In that study, the investigators suggest that CcO is a potential target site for anthracyclines and that this interaction with CcO is a plausible mechanism of DOX toxicity (52). CcO is also the target site of the antitumor retinoid drug, N-(4-hydroxyphenyl) retinamide (53). This chemotherapeutic drug was shown to decrease the activity of CcO by downregulating the CcO subunit III (53). The data presented here (Fig. S4) show that the power saturation of low spin heme a is affected by the addition of DOX, suggesting that the environment surrounding heme a was altered. Our EPR results also show that the heme a3 coupled to CuB signal intensity in heart tissues was altered during DOX treatment and restored by Mito-Q (Figs. 4 and 6).
CcO catalyzes the electron transfer from cytochrome c to oxygen, forming water and creating the necessary electrochemical potential difference for ATP synthesis via proton pumping. Subunit 1 of CcO contains heme a, heme a3, and CuB. Heme a3 and CuB form a binuclear center, the site where molecular oxygen is reduced to water. The CuA site in CcO subunit II contains two copper atoms. Cytochrome c binds to the negatively-charged region in subunit II and transfers the electrons to CuA located in subunit II (54). Electrons from CuA are subsequently transferred to heme a and ultimately to the binuclear center (Fig. S10). Whether DOX binds to the negatively-charged region of CcO subunit II (the binding site of lysine-rich, positively-charged domain of cytochrome c) and alters the electron distribution from cytochrome c in CcO subunit II remains unknown. Mito-Q could compete for the same binding site, thereby inhibiting CcO/DOX interaction and sustaining the electron-transport in CcO. This hypothetical scenario is analogous to bacterial quinol oxidases where ubiquinone/ubiquinol mediates electron-transfer from the quinol oxidation site to the low-spin heme B (55). The optical and EPR data obtained in a model system consisting of DOX, hemin, and Mito-Q/hemin (Fig. S7) suggest the interaction of DOX and Mito-Q with the heme (Fig. S7). At present, we do not fully understand the mechanism of suppression of CcO subunit expression. Previously, CcO subunit Va was shown to play an important role in the mitochondrial electron transfer process (49). Ongoing research indicates that Mito-Q enhances the antitumor activity of DOX in breast cancer cells (MCF-7 and MD-MAB-231) but protects against DOX toxicity in rat cardiomyocytes (H9c2). These results strongly suggest that Mito-Q is a favorable drug candidate for combination therapy with DOX in breast cancer treatment (56).
In summary, we have shown here that the ex vivo low-temperature EPR technique could be used as an effective analytical tool to investigate the changes in the mitochondrial heme proteins and heme centers in myocardial tissues during DOX-induced cardiomyopathy. The EPR data indicate that a prolonged treatment with DOX uncouples the heme a3/CuB dinuclear active center of subunit I of CcO that is involved in oxygen binding. In agreement with this, the CcO activity and the expressions of CcO subunits (CcO II and CcO Va) decreased during DOX treatment but were largely restored by Mito-Q. Thus, our data demonstrating selective changes to the CcO active center by DOX and restoration of CcO subunit expressions by Mito-Q may have mechanistic relevance in ameliorating DOX-induced cardiotoxicity.
Acknowledgments
This study is dedicated to the memory of Helmut Beinert, who pioneered EPR research of iron sulfur proteins and cytochrome c oxidase.
This work was supported by grants R01 CA077822 and R01 HL073056 from the National Institutes of Health, and a grant from the Cancer Center of the Medical College of Wisconsin. C.K. expresses thanks to the Managing Board of the Virudhunagar Hindu Nadars' Senthikumara Nadar College, Virudhunagar, India for granting him sabbatical leave.
Appendix A
Modified synthesis of Mito-Q
Mito-Q was originally synthesized by Murphy and co-workers (30). As this compound is not commercially available, we decided to synthesize it in-house. Although we obtained several milligrams of pure Mito-Q using the published procedures (30), the synthetic procedure was tedious and the reaction yields were considerably low. We have now modified the procedure, and with the new procedure developed in our laboratory, we can generate pure Mito-Q in gram quantities for in vivo experiments. Fig. S1 describes the modified procedure. The overall yield for the modified synthetic pathway is close to 80%. We made the following modifications to the published procedures: A major difference between our synthetic pathway of Mito-Q (Fig. S1) and the published synthetic route lies in the synthesis of the intermediate bromoidebenone from idebenone (steps 1 and 2). The published route for bromoidebenone involving carbon tetrabromide and triphenyl phosphine resulted in Mito-Q formation at a low yield that required extensive purification. The proposed method (steps 1 and 2 in Fig. S1) yielded a pure bromoidebenone derivative, and the yield is quantitative. Bromoidebenone was used in the subsequent steps without further purification. Because of these modifications, the overall yield of Mito-Q was 83%. High-performance liquid chromatography analysis of Mito-Q on a C-18 column using a mixture of water and acetonitrile (gradient flow of 60-80% at a flow-rate of 1 ml/min) showed a single peak at 14.5 min retention time.
Experimental protocol
DOX-induced cardiomyopathy in a rat model is shown in Fig. S2.
Microwave power and temperature-dependent EPR spectra
EPR spectra at different microwave powers ranging from 0.1 mW to 150 mW were investigated during DOX/Mito-Q-induced alterations in structural and magnetic interactions/surroundings around the metal spin center of interest. The EPR relaxation behavior of high and low spin heme and 3Fe-4S signals was analyzed (see Eq. S1) and the power saturation parameters listed in Table S2. Typical power saturation curves obtained for low spin heme for control heart tissue, DOX and Mito-Q treated samples are shown in Fig. S4. The P1/2 values of high spin heme and 3Fe4S were comparable in magnitude for native and DOX treated tissue samples, respectively. This indicates that their relaxation properties did not change significantly in the presence of DOX and DOX plus Mito-Q. However, the P1/2 values of low spin heme shows a threefold decrease as compared to the native state.
Fig. S5 shows the temperature-dependent EPR signals due to high spin heme (gz = 6.0), 3Fe-4S cluster (gz = 2.015), 4Fe-4S cluster (gz = 1.95) and low spin (gz = 3.0) for control heart tissue and the DOX-treated group, plotted as signal amplitude versus temperature. The relative peak heights plotted for the gz = 6.0 signals of control and the DOX-treated group versus 1/T give linear plots indicating that the signals obey the Curie law, (signal = a/T + b). The temperature dependence of signal at g = 2.015 is typical of the behavior of 3Fe4S clusters exhibiting high EPR signal intensity at 10 K, diminishing to base level at ∼35 K (57). Above 35 K, the residual signal remaining is only a small free radical at g = 2.003. Inset in Fig. S5 shows the Curie plot for control and DOX-treated samples for the signal at gz = 3.0, plotted as signal amplitude times temperature versus temperature.
Interaction between DOX or Mito-Q and hemin
Fig. S6 shows the experimental and simulated EPR spectra of CcO. Our assignment of the EPR spectra of myocardial tissues are supported by this data. Fig. S7, A and B support the proposal that both DOX and Mito-Q interact with the heme as seen by the EPR spectral (Fig. S7) and optical spectroscopic (Fig. S7, inset) changes.
DOX-mediated fibrosis and apoptosis: effect of Mito-Q
Figs. S8 and S9 show the effects of Mito-Q on DOX-induced fibrosis and apoptosis in cardiac tissues. As shown, Mito-Q inhibits DOX-induced fibrosis (Fig. S8) and apoptosis (Fig. S9 A) and restores the complex IV activity (Fig. S9 B) in heart tissues isolated from rats treated with DOX.
Supporting Material
References
- 1.Bristow M.R., Billingham M.E., Mason J.W., Daniels J.R. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat. Rep. 1978;62:873–879. [PubMed] [Google Scholar]
- 2.Minow R.A., Benjamin R.S., Gottlieb J.A. Adriamycin (NSC 123127) cardiomyopathy – an overview with determination of risk factors. Cancer Chemother. Rep. 1975;6:198–201. [Google Scholar]
- 3.Singal P.K., Iliskovic N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 4.Shan K., Lincoff A.M., Young J.B. Anthracycline-induced cardiotoxicity. Ann. Intern. Med. 1996;125:47–58. doi: 10.7326/0003-4819-125-1-199607010-00008. [DOI] [PubMed] [Google Scholar]
- 5.Jensen R.A. Doxorubicin cardiotoxicity: contractile changes after long-term treatment in the rat. J. Pharmacol. Exp. Ther. 1986;236:197–203. [PubMed] [Google Scholar]
- 6.Nousiainen T., Jantunen E., Vanninen E., Hartikainen J. Early decline in left ventricular ejection fraction predicts doxorubicin cardiotoxicity in lymphoma patients. Br. J. Cancer. 2002;86:1697–1700. doi: 10.1038/sj.bjc.6600346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gianni L., Salvatorelli E., Minotti G. Anthracycline cardiotoxicity in breast cancer patients: synergism with trastuzumab and taxanes. Cardiovasc. Toxicol. 2007;7:67–71. doi: 10.1007/s12012-007-0013-5. [DOI] [PubMed] [Google Scholar]
- 8.Sorensen K., Levitt G.A., Bull C., Dorup I., Sullivan I.D. Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer. 2003;97:1991–1998. doi: 10.1002/cncr.11274. [DOI] [PubMed] [Google Scholar]
- 9.Doyle J.J., Neugut A.I., Jacobson J.S., Grann V.R., Hershman D.L. Chemotherapy and cardiotoxicity in older breast cancer patients: a population-based study. J. Clin. Oncol. 2005;23:8597–8605. doi: 10.1200/JCO.2005.02.5841. [DOI] [PubMed] [Google Scholar]
- 10.Iarussi D., Indolfi P., Casale F., Martino V., DiTulliio M.T. Anthracycline-induced cardiotoxicity in children with cancer: strategies for prevention and management. Paediatr. Drugs. 2005;7:67–76. doi: 10.2165/00148581-200507020-00001. [DOI] [PubMed] [Google Scholar]
- 11.Lipshultz S.E., Lipsitz S.R., Sallen S.E., Dalton V.M., Mone S.M. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 12.Keizer H.G., Pinedo H.M., Schuurhuis G.J., Joenje H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 1990;47:219–231. doi: 10.1016/0163-7258(90)90088-j. [DOI] [PubMed] [Google Scholar]
- 13.Wallace K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003;93:105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 14.Sawyer D.B., Fuukazawa R., Arstall M.A., Kelly R.A. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ. Res. 1999;84:257–265. doi: 10.1161/01.res.84.3.257. [DOI] [PubMed] [Google Scholar]
- 15.Kotamraju S., Konorov E.A., Joseph J., Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 16.Kalyanaraman B., Joseph J., Kalivendi S., Wang S., Konorov E.A. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol. Cell. Biochem. 2002;234/235:119–124. [PubMed] [Google Scholar]
- 17.Haunstetter A., Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ. Res. 1998;82:1111–1129. doi: 10.1161/01.res.82.11.1111. [DOI] [PubMed] [Google Scholar]
- 18.Duquaine D., Hirsch G.A., Chakrabarti A., Han Z., Kehrer C. Rapid-onset endothelial dysfunction with adriamycin: evidence for a dysfunctional nitric oxide synthase. Vasc. Med. 2003;8:101–107. doi: 10.1191/1358863x03vm476oa. [DOI] [PubMed] [Google Scholar]
- 19.Simmons T.W., Jamall I.S. Relative importance of intracellular glutathione peroxidase and catalase in vivo for prevention of peroxidation to the heart. Cardiovasc. Res. 1989;23:774–779. doi: 10.1093/cvr/23.9.774. [DOI] [PubMed] [Google Scholar]
- 20.Davies K.J.A., Doroshow J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 21.Sarvazyan N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am. J. Physiol. 1996;271:H2079–H2085. doi: 10.1152/ajpheart.1996.271.5.H2079. (Heart Circ. Physiol. 40) [DOI] [PubMed] [Google Scholar]
- 22.Konorov E.A., Kennedy M.C., Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch. Biochem. Biophys. 1999;368:421–428. doi: 10.1006/abbi.1999.1337. [DOI] [PubMed] [Google Scholar]
- 23.Conklin K.A. Coenzyme q10 for prevention of anthracycline-induced cardiotoxicity. Integr. Cancer Ther. 2005;4:110–130. doi: 10.1177/1534735405276191. [DOI] [PubMed] [Google Scholar]
- 24.Dorr R.T. Cytoprotective agents for anthracyclines. Semin. Oncol. 1996;23:23–34. [PubMed] [Google Scholar]
- 25.Murphy M.P., Smith R.A.J. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 26.James M., Cocheme H.M., Smith R.A.J., Murphy M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 27.Dhanasekaran A., Kotamraju S., Karunakaran C., Kalivendi S.V., Thomas S. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic. Biol. Med. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Schwarz E.R., Pollick C., Dow J., Patterson M., Birnbaum Y. A small animal model of non-ischemic cardiomyopathy and its evaluation by transthoracic echocardiography. Cardiovasc. Res. 1998;39:216–223. doi: 10.1016/s0008-6363(98)00009-1. [DOI] [PubMed] [Google Scholar]
- 29.Orme-Johnson N.R., Hansen R.E., Beinert H. Electron paramagnetic resonance-detectable electron acceptors in beef heart mitochondria. Reduced diphosphopyridine nucleotide ubiquinone reductase segment of the electron transfer system. J. Biol. Chem. 1974;249:1922–1927. [PubMed] [Google Scholar]
- 30.Asin-Cayuela J., Manas A.R., James A.M., Smith R.A., Murphy M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 31.Migrino R.Q., Aggarwal D., Konorov E., Brahmbhatt T., Bright M. Early detection of doxorubicin cardiomyopathy using two-dimensional strain echocardiography. Ultrasound Med. Biol. 2007;34:208–214. doi: 10.1016/j.ultrasmedbio.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hausladen A., Fridovich I. Measuring nitric oxide and superoxide: rate constants for aconitase reactivity. Methods Enzymol. 1996;269:37–41. doi: 10.1016/s0076-6879(96)69007-7. [DOI] [PubMed] [Google Scholar]
- 33.Kotamraju S., Tampo Y., Keszler A., Chitambar C.R., Joseph J. Nitric oxide inhibits peroxide-induced transferrin receptor dependent iron uptake, oxidative damage and apoptosis in endothelial cells: role of ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 2003;100:10653–10658. doi: 10.1073/pnas.1933581100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vijayasarathy V., Damle S., Lenka N., Avadhani N.G. Tissue variant effects of heme inhibitors on the mouse cytochrome c oxidase gene expression and catalytic activity of the enzyme complex. Eur. J. Biochem. 1999;266:191–200. doi: 10.1046/j.1432-1327.1999.00843.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang D.C., Meinhardt S.W., Sackmann U., Weiss H., Ohnishi T. The iron-sulfur clusters in the two related forms of mitochondrial NADH: ubiquinone oxidoreductase made by Neurospora crassa. Eur. J. Biochem. 1991;197:257–264. doi: 10.1111/j.1432-1033.1991.tb15906.x. [DOI] [PubMed] [Google Scholar]
- 36.Ohnishi T., Schagger H., Meinhardt S.W., LoBrutto R., Link T.A. Spatial organization of the redox active centers in the bovine heart ubiquinol-cytochrome c oxidoreductase. J. Biol. Chem. 1989;264:735–744. [PubMed] [Google Scholar]
- 37.Gong X., Yu L., Xia D., Yu C.-A. Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex. J. Biol. Chem. 2005;280:9251–9257. doi: 10.1074/jbc.M409994200. [DOI] [PubMed] [Google Scholar]
- 38.Kroneck P.M., Antholine W.E., Kastrau D.H., Buse G., Steffens G.C. Multifrequency EPR evidence for a bimetallic center at the CuA site in cytochrome c oxidase. FEBS Lett. 1990;268:274–276. doi: 10.1016/0014-5793(90)81026-k. [DOI] [PubMed] [Google Scholar]
- 39.Hunter D.J.B., Oganesyan V.S., Salerno J.C., Butler C.S., Ingledew W.J. Angular dependences of perpendicular and parallel mode electron paramagnetic resonance of oxidized beef heart cytochrome c oxidase. Biophys. J. 2000;78:439–450. doi: 10.1016/S0006-3495(00)76606-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper C.E., Salerno J. Characterization of a novel g'=2.95 EPR signal from the binuclear center of mitochondrial cytochrome c oxidase. J. Biol. Chem. 1992;267:280–285. [PubMed] [Google Scholar]
- 41.Jancura D., Antalik M., Berka V., Palmer G., Fabian M. Filling the catalytic site of cytochrome c oxidase with electrons reduced CuB facilitates internal electron transfer to heme a3. J. Biol. Chem. 2006;281:20003–20010. doi: 10.1074/jbc.M602066200. [DOI] [PubMed] [Google Scholar]
- 42.Cheesman M.R., Oganesyan V.S., Watmough N.J., Butler C.S., Thomson A.J. The nature of the exchange coupling between high-spin Fe(III) heme o3 and CuB(II) in Escherichia coli quinol oxidase, cytochrome bo3: MCD and EPR studies. J. Am. Chem. Soc. 2004;126:4157–4166. doi: 10.1021/ja038858m. [DOI] [PubMed] [Google Scholar]
- 43.Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 44.Papadopoulou L.C., Sue C.M., Davidson M.M., Tanji K., Nishino I. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 45.Grandjean F., Bremaud L., Robert J., Ratinaud M.H. Alterations in the expression of cytochrome c oxidase subunits in doxorubicin-resistant leukemia K562 cells. Biochem. Pharmacol. 2002;63:823–831. doi: 10.1016/s0006-2952(01)00865-6. [DOI] [PubMed] [Google Scholar]
- 46.Lebrecht D., Setzer B., Ketelsen U.-P., Haberstroh J., Walker U.A. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- 47.Lebrecht D., Kokkori A., Ketelsen U.-P., Setzer B., Walker U.A. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J. Pathol. 2007;207:436–444. doi: 10.1002/path.1863. [DOI] [PubMed] [Google Scholar]
- 48.Papadopoulou L.C., Theophilidis G., Thomopoulos G.N., Tsiftsoglou A.S. Structural and functional impairment of mitochondria in adriamycin-induced cardiomyopathy in mice: suppression of cytochrome c oxidase II gene expression. Biochem. Pharmacol. 1999;57:481–489. doi: 10.1016/s0006-2952(98)00305-0. [DOI] [PubMed] [Google Scholar]
- 49.Merten K.E., Feng W., Zhang L., Pierce W., Cai J. Modulation of cytochrome c oxidase-va is possibly involved in metallothionein protection from doxorubicin cardiotoxicity. J. Pharmacol. Exp. Ther. 2005;315:1314–1319. doi: 10.1124/jpet.105.089763. [DOI] [PubMed] [Google Scholar]
- 50.Lebrecht D., Geist A., Ketelsen U.-P., Haberstroh J., Setzer B. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br. J. Pharmacol. 2007;151:771–778. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wheeler C., Robinson S.H., Tsiftsoglou A.S. Interactions of daunomycin (DN) with heme and hemoproteins: implications for the mode of action of anthracycline drugs. Blood. 1984;64(Suppl.):177a. [Google Scholar]
- 52.Papadopoulou L.C., Tsiftsoglou A.S. Mitochondrial cytochrome c oxidase as a target site for daunomycin in K-562 cells and heart tissue. Cancer Res. 1993;53:1072–1078. [PubMed] [Google Scholar]
- 53.You K.-R., Wen J., Lee S.-T., Kim D.-G. Cytochrome c oxidase subunit III: a molecular marker for N-(4-hydroxyphenyl)retinamise-induced oxidative stress in hepatoma cells. J. Biol. Chem. 2002;277:3870–3877. doi: 10.1074/jbc.M109284200. [DOI] [PubMed] [Google Scholar]
- 54.Iwata S., Ostermeier C., Ludwig B., Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 55.Sato-Watanabe M., Mogi T., Ogura T., Kitagawa T., Miyoshi H. Identification of a novel quinone-binding site in the cytochrome bo complex from Escherichia coli. J. Biol. Chem. 1994;269:28908–28912. [PubMed] [Google Scholar]
- 56.Rao V.A., Klein S., Zielonka J., Kalyanaraman B., Shacter E. The mitochondrially-targeted redox agent mitoquinone enhances doxorubicin-induced toxicity to breast cancer cells while protecting cardiac myocytes. Free Radic. Biol. Med. 2007;43(Suppl. 1):S59. [Google Scholar]
- 57.Agarwalla S., Stroud R.M., Gaffney B.J. Redox reactions of the iron-sulfur cluster in a ribosomal RNA methyltransferase, RumA: Optical and EPR studies. J. Biol. Chem. 2004;279:34123–34129. doi: 10.1074/jbc.M405702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.