

Abstract
Background and purpose:
In endothelial dysfunction, signalling by nitric oxide (NO) is impaired because of the oxidation and subsequent loss of the soluble guanylyl cyclase (sGC) haem. The sGC activator 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid (BAY 58-2667) is a haem-mimetic able to bind with high affinity to sGC when the native haem (the NO binding site) is removed and it also protects sGC from ubiquitin-triggered degradation. Here we investigate whether this protection is a unique feature of BAY 58-2667 or a general characteristic of haem-site ligands such as the haem-independent sGC activator 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt (HMR 1766), the haem-mimetic Zn-protoporphyrin IX (Zn-PPIX) or the haem-dependent sGC stimulator 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine (BAY 41-2272).
Experimental approach:
The sGC inhibitor 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) was used to induce oxidation-induced degradation of sGC. Activity and protein levels of sGC were measured in a Chinese hamster ovary cell line as well as in primary porcine endothelial cells. Cells expressing mutant sGC were used to elucidate the molecular mechanism underlying the effects observed.
Key results:
Oxidation-induced sGC degradation was prevented by BAY 58-2667 and Zn-PPIX in both cell types. In contrast, the structurally unrelated sGC activator, HMR 1766, and the sGC stimulator, BAY 41-2272, did not protect. Similarly, the constitutively haem-free sGC mutant β1H105F was stabilized by BAY 58-2667 and Zn-PPIX.
Conclusions:
The ability of BAY 58-2667 not only to activate but also to stabilize oxidized/haem-free sGC represents a unique example of bimodal target interaction and distinguishes this structural class from non-stabilizing sGC activators and sGC stimulators such as HMR 1766 and BAY 41-2272, respectively.
Keywords: soluble guanylyl cyclase, BAY 58-2667, HMR 1766, BAY 41-2272, oxidative stress, nitric oxide, cGMP, haem
Introduction
The second messenger cyclic guanosine monophosphate (cGMP), synthesized from guanosine triphosphate (GTP) by soluble guanylyl cyclase (sGC), is a key regulator of vascular smooth-muscle cell relaxation and inhibition of platelet aggregation (Lucas et al., 2000). sGC is a heterodimeric enzyme consisting of an α and a haem-containing β subunit, which represents the intracellular receptor for the gaseous messenger, nitric oxide (NO). However, other mechanisms of sGC regulation have been described such as membrane association or binding to the chaperone heat shock protein 90 (HSP90; Zabel et al., 2002; Agullóet al., 2005; Nedvetsky et al., 2008). The prosthetic haem group is non-covalently bound to the β1 subunit via the proximal haem ligand H105 (Wedel et al., 1994; Zhao et al., 1998) and the haem-binding motif Y-x-S-x-R (Pellicena et al., 2004; Schmidt et al., 2004; 2005; Ma et al., 2007). Binding of NO to the haem group activates sGC and results in a considerable increase (up to 200-fold) in the formation of cGMP (Ignarro et al., 1982). In turn, cGMP affects various downstream targets such as protein kinases, cyclic nucleotide-gated channels or phosphodiesterases (Lucas et al., 2000; Feil et al., 2003), making the NO-sGC-cGMP signalling one of the most important vasoprotective signalling pathways.
One of the crucial prerequisites of the NO-mediated sGC activation is the presence of the reduced haem moiety. Its oxidation or loss renders the enzyme insensitive to NO. Oxidative stress, a hallmark of many cardiovascular diseases, impairs the NO-cGMP signalling (Melichar et al., 2004). Proposed mechanisms include direct chemical scavenging of NO by reactive oxygen species (ROS) such as O2−, resulting in a reduced bioavailability of NO and, in parallel, the formation of the strong oxidant peroxynitrite (ONOO−). In turn, this reactive intermediate is able to further inhibit NO signalling by oxidizing the sGC prosthetic haem group to its NO-insensitive Fe3+ state (Gladwin, 2006; Stasch et al., 2006; Chirkov and Horowitz, 2007). In addition to this acute inactivation of sGC, oxidation of the haem group facilitates the degradation of the enzyme (Stasch et al., 2006; Meurer et al., 2007). Oxidation-induced impairment of protective NO-cGMP signalling is likely to contribute to endothelial dysfunction in different vascular diseases such as arterial hypertension, atherosclerosis, heart failure and erectile dysfunction (Evgenov et al., 2006; Kemp-Harper and Feil, 2008).
For more than a century, provision of NO, as via NO-releasing organic nitrates, has been a major therapeutical approach for the treatment of cardiovascular diseases. However, this class of drugs suffers from several drawbacks including the development of tolerance and, unlike endogenous NO, the lack of any antithrombotic effect on platelets. Moreover, blood vessels suffering from oxidative stress conditions become increasingly unresponsive to NO, a situation that is further aggravated by the fact that organic nitrates increase oxidative stress and have been shown to directly oxidize sGC (Artz et al., 2002; Munzel et al., 2005; 2007).
As an alternative therapeutic approach, two structurally distinct classes of NO-independent, sGC-activating compounds have been discovered, with the potential to overcome some if not all of the above-mentioned shortcomings (Evgenov et al., 2006). Haem-dependent sGC stimulators, including 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine (BAY 41-2272), show a strong synergism with NO but lose their ability to stimulate sGC once the prosthetic haem group is oxidized or lost (Stasch et al., 2001). In contrast, haem-independent sGC activators (e.g. BAY 58-2667) activate the NO-insensitive oxidized/haem-free form of the enzyme (Stasch et al., 2002a) via binding at the enzyme's haem pocket (Schmidt et al., 2004; Roy et al., 2008). Although this mechanism of binding is generally accepted it is still unclear whether BAY 58-2667 is able to actively compete with the weakly bound haem moiety or if the compound binds solely to the haem-free sGC after the enzyme has lost its oxidized prosthetic group as suggested very recently by Roy et al.
By activating sGC and thereby increasing the amount of released cGMP, BAY 58-2667 has different vascular effects. BAY 58-2667 lowers systemic blood pressure, has beneficial effects in hypertension-induced cardiac hypertrophy and inhibits platelet aggregation. In experimental pulmonary hypertension, treatment with BAY 58-2667 leads to a reduction of ventricular systolic pressure and selective pulmonary vasodilatation. Furthermore, BAY 58-2667 decreases the load on the heart and increases cardiac output as well as renal blood flow in experimental congestive heart failure. Preload- and afterload-reducing effects of BAY 58-2667 have been observed in a phase I clinical study and in a phase II clinical trial with patients suffering from acute decompensated heart failure (see Evgenov et al., 2006; Schmidt et al., 2009).
In addition to its activating effect, BAY 58-2667 is able to rescue the oxidation-impaired sGC from enhanced ubiquitin-mediated degradation, thus accumulating its receptor in a positive feedback loop (Stasch et al., 2006; Meurer et al., 2007). Compounds mimicking the porphyrinic structure of haem, for example zinc-protoporphyrin IX (Zn-PPIX) and BAY 58-2667, protect sGC from oxidation-induced degradation (Stasch et al., 2006). With respect to the structurally unrelated sGC activator, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt (HMR 1766), competition with different porphyrins suggests an interaction with the enzyme's haem pocket, as shown for BAY 58-2667 (Schindler et al., 2006; Stasch et al., 2006). This raises the possibility that protection against oxidation-induced degradation is a general feature of haem-independent sGC activators. To test this hypothesis and to further substantiate the mechanisms leading to the observed stabilization of sGC protein levels, we investigated the effects of both compounds and the high affinity metallo-porphyrin, Zn-PPIX, under normal and haem-oxidizing conditions. As the sGC stimulator BAY 41-2272 does not bind to the haem pocket, a stabilizing effect on sGC protein levels was not anticipated, making this compound suitable as negative control.
Experiments were conducted by using two cell models, primary porcine endothelial cells (ECs) and Chinese hamster ovary (CHO) cells expressing wild-type (WT) sGC or the constitutive haem-free sGC mutants β1H105F and β1Y135A/R139A. Our findings suggest that BAY 58-2667-like compounds have a unique structural ability to reassemble the spatial structure of the haem moiety within sGC and that this feature allows to prevent sGC degradation in a hitherto not reported drug-induced positive feedback loop on the expression level of its therapeutic target protein.
Methods
Cell culture
Primary ECs were obtained from fresh porcine aortae by collagenase detachment as previously described (Stasch et al., 2002b). Briefly, aortae were freed from surrounding tissue, cut open and mounted on a framework with the intima facing upwards. An amount of 20 mL sterile 0.14% collagenase solution (Biochrom AG, Berlin, Germany) was poured onto the aorta's luminal surface, and the aorta was incubated for 15 min. ECs were scraped from the tissue and cultured until confluent.
cGMP reporter cells were generated and cultured as previously described (Schmidt et al., 2004; Wunder et al., 2005). Briefly, the cGMP reporter cells consist of CHO cells stably transfected with the cGMP-gated Ca2+-channel CNG2 and aequorin, which translates increasing levels of intracellular Ca2+ into bioluminescence. In addition, these cells have been stably transfected with WT sGC (α1 and β1 subunits of the rat lung enzyme), α1/β1H105F sGC or α1/β1Y135A/R139A sGC (Becker et al., 1999; Schmidt et al., 2004).
Western blotting
Cells were seeded in six-well plates, grown until confluent and subsequently incubated with 10 µmol·L−1 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) alone or combined with 0.01–10 µmol·L−1 BAY 58-2667, 10 µmol·L−1 BAY 41-2272, 5 µmol·L−1 Zn-PPIX or 10 µmol·L−1 HMR 1766 respectively. After 24 h, cells were harvested and lysed, and protein was extracted as described earlier (Rothkegel et al., 2007). An amount of 15–30 µg of total protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. The individual sGC subunits were detected by using polyclonal antibodies directed against specific epitopes of the α1 subunit (Sigma, Steinheim, Germany) and the β1 subunit (Cayman Chemical Company, Ann Arbor, MI, USA). Actin was used as loading control by using commercially available antibodies (Sigma). Detection was performed by the ECL method (Amersham/GE Healthcare, Buckinghamshire, UK). Protein levels were determined by densitometric analysis of the specific protein bands (GS-800 Calibrated Densitometer, Quantity One Analysis Software, BioRad, Munich, Germany). Values were normalized to the respective control of sGC, which was set to 100% as well as to the respective actin ratio. Data shown in Figures 3 and 8 were obtained in independent sets of experiments using different batches of cells.
Figure 3.

Effects of 10 µmol·L−1 BAY 58-2667, 10 µmol·L−1 BAY 41-2272, 10 µmol·L−1 HMR 1766 and 5 µmol·L−1 Zn-PPIX on sGC protein levels under normal and haem-oxidizing conditions in cGMP reporter cells. Haem oxidation was achieved by pre-incubating cells with 10 µmol·L−1 ODQ for 24 h. (A) Representative Western blots of α1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. α1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. (B) Representative Western blots of β1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. β1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. sGC protein levels are expressed as percentage of the respective control which was set as 100% (means ± SEM of 5–32 independent experiments). *P < 0.05, **P < 0.01, ***P < 0.005: Student's t-test. BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667,4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl [benzoic] acid; HMR, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
Figure 8.

Effects of BAY 58-2667 on protein levels under normal and oxidative conditions of WT, α1/β1H105F sGC and α1/β1Y135A/R139A sGC. cGMP reporter cells were incubated with 10 µmol·L−1 ODQ or 10 µmol·L−1 BAY 58-2667 alone or combined for 24 h as indicated. (A) Representative Western blots of α1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. α1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. (B) Representative Western blots of β1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. β1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. sGC protein levels are normalized to the respective control, which was set as 100% (means ± SEM of three to eight independent experiments). *P < 0.05, **P < 0.01, ***P < 0.005: Student's t-test. BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; SEM, standard error or the mean; sGC, soluble guanylyl cyclase; WT, wildtype.
sGC activity assays
cGMP concentrations of ECs were determined by a commercially available radio-immuno assay kit (IBL, Hamburg, Germany; Stasch et al., 2002a; Schmidt, 2009). In the cGMP reporter cell line, sGC activity was determined 48 h after seeding (Schmidt et al., 2004; Wunder et al., 2005). Briefly, cells were incubated with increasing concentrations of the respective test substances for 10 min. Subsequently, 10 mmol·L−1 CaCl2 was added, and the resulting bioluminescence directly correlated with intracellular cGMP concentrations (Wunder et al., 2005). Values were expressed as relative light units (RLUs).
The activity of haem-free recombinant rat sGC was assayed via the formation of [32P]-cGMP from [α-32P]-GTP in the presence of Mg2+(Hoenicka et al., 1999; Schmidt, 2009). Removal of the haem group was achieved by adding 2% Tween-20 to the reaction buffer, as previously described (Foerster et al., 1996; Schmidt et al., 2003). sGC was incubated with 100 nmol·L−1 BAY 58-2667 or 100 µmol·L−1 HMR 1766, which resulted in similar fold stimulation. These fixed concentrations of sGC activators were combined with increasing concentrations of Zn-PPIX.
Receptor binding assay
Homologous and heterologous competition binding studies were performed by using a receptor binding assay, as described previously (Schmidt et al., 2003). An amount of 1 µg sGC was incubated with 100 nmol·L−13H-BAY 58-2667 and increasing concentrations of unlabelled BAY 58-2667, HMR 1766 or Zn-PPIX respectively. Free and bound radioligands were separated via 96-well filter plates coated with polyvinylpyrrolidone. Bound radioactivity was determined by scintillation counting. Non-specific binding was measured by the addition of a 1000-fold excess of unlabelled BAY 58-2667 and subtracted from total binding in every individual assay.
Statistics
Data are presented as means ± standard error of the mean (SEM). GraphPad Prism software version 4.02 (GraphPad Software Inc., San Diego, CA, USA) was used for curve fitting and calculation of EC50 or IC50 values. Ninety-five per cent confidence intervals of EC50 and half maximal inhibitory concentration (IC50) values are given in parentheses. Statistical comparisons were performed by using the paired Student's t-test.
Materials
BAY 58-2667, BAY 41-2272 and HMR 1766 were synthesized as described (Figure 1; Straub et al., 2001; Stasch et al., 2002b; Schindler et al., 2006). Tritium labelling of BAY 58-2667 was performed as described (Shu and Heys, 2000). ODQ was purchased from Tocris Bioscience (Avonmouth, UK); Zn-PPIX (zinc-3,18-divinyl-2,7,13,17-tetramethylporphine-8,12-dipropionic acid), from Sigma. All other chemicals were of analytical grade and obtained from Sigma.
Figure 1.
Chemical structures of the sGC activators BAY 58-2667 and HMR 1766, the sGC stimulator BAY 41-2272 and the haem pocket antagonist zinc protoporphyrin IX (Zn-PPIX). BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
Results
Inhibition of sGC activity by Zn-PPIX
To validate the hypothesis that HMR 1766 interacts with the sGC haem pocket as shown for BAY 58-2667, purified recombinant haem-free sGC was incubated with concentrations of BAY 58-2667 or HMR 1766 that activated the enzyme to a similar extent (Figure 2A). BAY 58-2667 (100 nmol·L−1) activated the enzyme 69.5-fold (reflecting a specific activity of 10.2 µmol cGMP·mg−1·min−1). At a concentration of 100 µM, HMR 1766 induced a comparable 72.9-fold activation (to 13.6 µmol cGMP·mg−1·min−1). The addition of increasing concentrations of Zn-PPIX resulted in an inhibition of activated sGC with IC50 values of either 4.8 (2.2–10.2) nmol·L−1 (for BAY 58-2667-activated sGC) or 2.2 (0.9–5.3) nmol·L−1 (for HMR 1766-activated sGC), indicating that both compounds interacted with the sGC haem pocket.
Figure 2.

(A) Inhibition of BAY 58-2667 or HMR 1766-induced sGC activation by Zn-PPIX. Activity was measured by formation of [32P]-cGMP from [α-32P]-GTP. Isolated sGC was incubated with 100 nmol·L−1 BAY 58-2667 or 100 µmol·L−1 HMR 1766 and increasing concentrations of Zn-PPIX. Data are shown as means ± SEM from five independent experiments performed in duplicate. (B) Comparison of the competition binding of BAY 58-2667, HMR 1766 and Zn-PPIX. Displacement of 100 nmol·L−13H-BAY 58-2667 was studied in a receptor binding assay. BAY 58-2667 showed an average non-specific binding of 717 dpm and maximal values of 4338 dpm. HMR 1766 had an average non-specific binding of 616 dpm and maximal values of 4149 dpm. Data are means ± SEM from three to five independent experiments performed in duplicate. BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; SEM, standard error of the mean; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
Competition binding of BAY 58-2667 and HMR 1766
A receptor binding assay using 3H-labelled BAY 58-2667 was used to further investigate whether BAY 58-2667 and HMR 1766 directly compete for the same binding site (Figure 2B). Radioactively labelled BAY 58-2667 was incubated with increasing concentration of unlabelled BAY 58-2667, HMR 1766 or Zn-PPIX. Unlabelled BAY 58-2667 displayed an IC50 of 200 (140.1–281.4) nmol·L−1. Based on a binding constant (KD) of 13.4 nmol·L−1 (Schmidt et al., 2003), a Ki of 23 nmol·L−1 was estimated. Zn-PPIX displaced 3H-BAY 58-2667 with an IC50 of 2.9 (1.3–6.3) nmol·L−1. HMR 1766 competed with 3H-BAY 58-2667 only at very high concentrations of ≥10 µmol·L−1.
Levels of sGC protein in cGMP reporter cells under normal and haem-oxidizing conditions
sGC degradation was induced by incubating cells for 24 h with the sGC inhibitor ODQ (Garthwaite et al., 1995; Olesen et al., 1998; Zhao et al., 2000). Under these conditions, sGC protein levels decreased by 59.7 and 35.6% for the α1 and β1 sGC subunit respectively (Figure 3). Control experiments in which cells were incubated with ODQ for only 30 min did not induce any significant changes in sGC protein levels (data not shown). Co-incubation with the sGC activator, BAY 58-2667, prevented the ODQ-induced decrease in sGC protein levels for both subunits, concentration dependently with a minimal effective concentration of 10 nmol·L−1 (Figure 3, Table 1). sGC protein levels of cells treated with BAY 58-2667 alone (i.e. without haem oxidation by ODQ) remained constant (Figure 3, Table 1). In contrast, the structurally unrelated sGC activator HMR 1766 showed no alteration in sGC protein levels, neither in control nor ODQ-treated reporter cells (Figure 3). Conversely, exposure of cGMP reporter cells to the competitive haem pocket antagonist Zn-PPIX led to a slight reduction of sGC protein levels. However, upon haem oxidation, sGC protein levels in Zn-PPIX-treated cells were higher than in cells exposed to ODQ alone. As expected, the sGC stimulator BAY 41-2272 had no effect on sGC protein levels, neither under control nor under haem-oxidizing conditions.
Table 1.
Effects of increasing concentrations of BAY 58-2667 on sGC protein levels under normal and haem-oxidizing conditions in cGMP reporter cells stably transfected with WT sGC
| cGMP reporter cells |
α1 sGC |
β1 sGC |
||
|---|---|---|---|---|
| BAY 58-2667 |
ODQ (10 µmol·L−1) |
ODQ (10 µmol·L−1) |
||
| [µmol·L−1] | − | + | − | + |
| – | 100 | 60 ± 4*** | 100 | 36 ± 3*** |
| 0.01 | 88 ± 7***†† | 65 ± 5***# | 79 ± 5***††† | 38 ± 10***†† |
| 0.1 | 93 ± 15††† | 76 ± 8*** | 73 ± 10***††† | 63 ± 13***††† |
| 1 | 94 ± 8***††† | 94 ± 6***††† | 92 ± 25††† | 88 ± 33††† |
| 10 | 100 ± 9††† | 95 ± 11††† | 116 ± 10††† | 97 ± 9††† |
Values are expressed as % control (no ODQ or BAY 58-2667) and are means ± SEM of 3–32 independent experiments.
P < 0.005: Student's t-test (indicated sample vs. control);
P < 0.01,
P < 0.005: Student's t-test (indicated sample vs. ODQ treated control);
P < 0.05: Student's t-test (substance treatment vs. substance plus ODQ treatment).
BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; cGMP, cyclic guanosine monophosphate; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; SEM, standard error of the mean; sGC, soluble guanylyl cyclase; WT, wild type.
Effects of haem oxidation on sGC protein levels in ECs
Long-term (24 h, Figure 4) but not short-term (30 min, data not shown) incubation with ODQ decreased sGC α1 and β1 levels by 57.8 and 46.0% respectively. Unlike the results from the cGMP reporter cell line, BAY 58-2667 induced a concentration-dependent increase of the β1 subunit beyond control even in the absence of ODQ, whereas α1 protein levels remained unchanged (Figure 4, Table 2). Similar results were obtained in ODQ-treated ECs, where BAY 58-2667 increased β1 protein levels beyond the amounts observed in untreated controls, whereas α1 levels were stabilized at the level of untreated controls (Table 2, Figure 4). Similar to BAY 58-2667, exposure of ECs to Zn-PPIX resulted in increased sGC β1 protein levels both under control conditions and upon haem oxidation beyond the levels of the respective controls. And even upon haem oxidation, α1 protein levels of Zn-PPIX-treated cells were higher than in cells treated with only ODQ. Conversely, neither the NO-independent sGC agonists BAY 41-2272 nor HMR 1766 relevantly increased the levels of either sGC subunit in ECs under any of the tested conditions.
Figure 4.
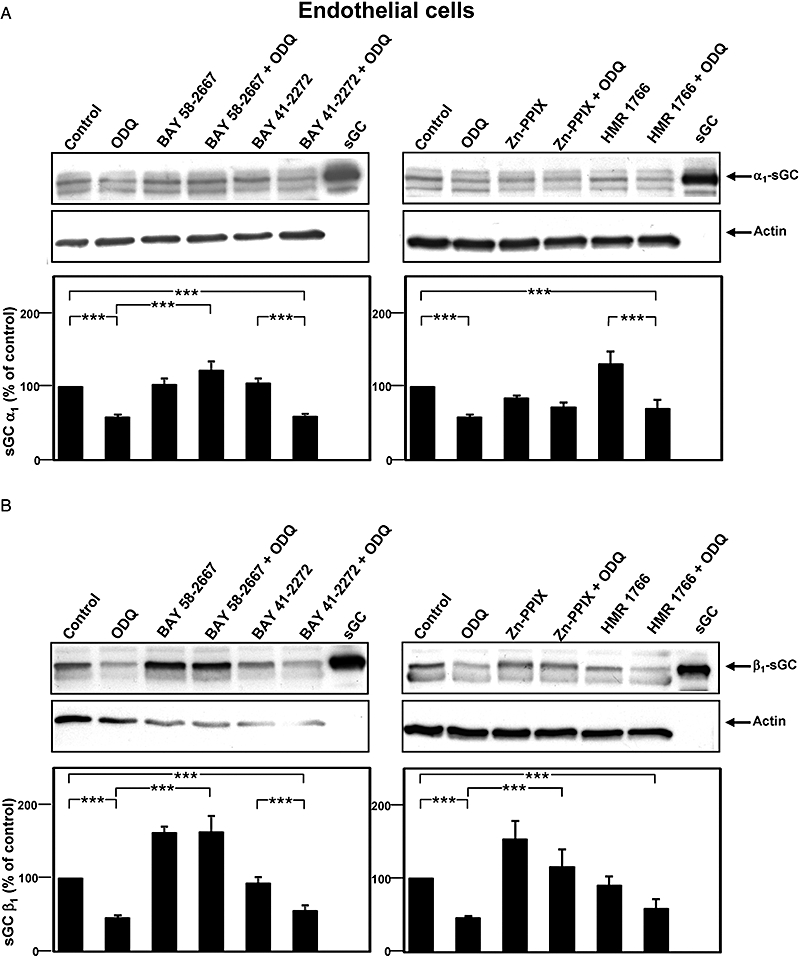
Effects of 10 µmol·L−1 BAY 58-2667, 10 µmol·L−1 BAY 41-2272, 10 µmol·L−1 HMR 1766 and 5 µmol·L−1 Zn-PPIX on sGC protein levels under normal and haem-oxidizing conditions in endothelial cells. Haem oxidation was achieved by incubation of the cells with 10 µmol·L−1 ODQ for 24 h. (A) Representative Western blots of α1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. α1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. (B) Representative Western blots of β1 sGC and actin as a loading control (upper panel). Purified rat sGC was used as control. β1 sGC protein levels as determined by densitometric measurement are shown in the lower panel. sGC protein levels are expressed as percentage of the respective control which was set as 100% (means ± SEM of 5–29 independent experiments). *P < 0.05, **P < 0.01, ***P < 0.005: Student's t-test. BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
Table 2.
Effects of increasing concentrations of BAY 58-2667 on sGC protein levels under normal and haem-oxidizing conditions in endothelial cells
| ECs |
α1 sGC |
β1 sGC |
||
|---|---|---|---|---|
| BAY 58-2667 |
ODQ (10 µmol·L−1) |
ODQ (10 µmol·L−1) |
||
| [µmol·L−1] | − | + | − | + |
| – | 100 | 58 ± 4*** | 100 | 46 ± 3*** |
| 0.01 | 108 ± 3***††† | 60 ± 8***### | 106 ± 5***††† | 53 ± 5***### |
| 0.1 | 99 ± 11†† | 64 ± 15*** | 111 ± 11**††† | 74 ± 9***†††# |
| 1 | 100 ± 9††† | 92 ± 9**†† | 138 ± 13***††† | 129 ± 17***††† |
| 10 | 103 ± 9††† | 122 ± 12***††† | 162 ± 24***††† | 163 ± 22***††† |
Values are expressed as % control (no ODQ or BAY 58-2667) and are means ± SEM of 4–29 independent experiments.
P < 0.01,
P < 0.005: Student's t-test (indicated sample vs. control);
P < 0.01,
P < 0.005: Student's t-test (indicated sample vs. ODQ treated control);
P < 0.05,
P < 0.005: Student's t-test (substance treatment vs. substance plus ODQ treatment).
BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; cGMP, cyclic guanosine monophosphate; EC, endothelial cell; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; SEM, standard error of the mean; sGC, soluble guanylyl cyclase.
sGC activity in cGMP reporter cells
For the effects of the haem oxidant ODQ in cGMP reporter cells to be examined, two approaches were tested, namely a long-term incubation (24 h) to reduce sGC protein levels, as well as a short-term incubation (10 min) with ODQ to oxidize the sGC haem group and inhibit activity without affecting the enzyme's protein level. Moreover, long-term treated cells were additionally short-term treated to test whether the BAY 58-2667-induced activity of residual sGC can be further enhanced by acute oxidation. Figure 5 summarizes the results of these protocols on the subsequent sGC activation by BAY 58-2667, BAY 41-2272, Zn-PPIX or HMR 1766 respectively.
Figure 5.

Concentration response curves of wildtype sGC in cGMP reporter cells incubated with increasing concentrations of BAY 58-2667 (A), BAY 41-2272 (B), HMR 1766 (C) or Zn-PPIX (D) alone, in combination with 10 µmol·L−1 ODQ for 10 min, after 24 h pretreatment with 10 µmol·L−1 ODQ and pretreatment with additional treatment with 10 µmol·L−1 ODQ for 10 min. Data are means ± SEM from 7–19 independent experiments performed in quadruplicate. sGC activation is represented as x-fold stimulation compared with non-stimulated control. Following basal activities were measured: (A) 10 min BAY 58-2667 1091 RLUs; 10 min BAY 58-2667 + ODQ 867 RLUs; 24 h ODQ/10 min BAY 58-2667 1441 RLUs; 24 h ODQ/10 min BAY 58-2667 + ODQ 866 RLUs. (B) 10 min BAY 41-2272 980 RLUs; 10 min BAY 41-2272 + ODQ 1800 RLUs; 24 h ODQ/10 min BAY 41-2272 1069 RLUs; 24 h ODQ/10 min BAY 41-2272 + ODQ 584 RLUs. (C) 10 min HMR 1766 1528 RLUs; 10 min HMR 1766 + ODQ 1380 RLUs; 24 h ODQ/10 min HMR 1766 1411 RLUs; 24 h ODQ/10 min HMR 1766 + ODQ 856 RLUs. (D) 10 min Zn-PPIX 1130 RLUs; 10 min Zn-PPIX + ODQ 1131 RLUs; 24 h ODQ/10 min Zn-PPIX 1750 RLUs. BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; cGMP, cyclic guanosine monophosphate; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; RLU, relative light unit; SEM, standard error of the mean; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
BAY 58-2667 (30 µmol·L−1) activated sGC up to 73-fold with an EC50 value of 23.3 (13.1–41.3) nmol·L−1. Short-term exposure to ODQ enhanced sGC activation to 129-fold with a decrease in the EC50 to 9.1 (4.6–17.9) nmol·L−1 (Figure 5A). Long-term incubation with ODQ resulted in only 78-fold sGC activation by BAY 58-2667 with a slightly higher EC50 of 15.1 (9.7–23.6) nmol·L−1. When long-term incubated cells were additionally exposed to a second short-term application of ODQ, sensitivity to BAY 58-2667 was similar [EC50 9.9 (4.0–24.5) nmol·L−1, 82-fold activation].
BAY 41-2272 (30 µmol·L−1) stimulated sGC up to 115-fold with an EC50 of 596 (364.7–973.5) nmol·L−1. Short-term treatment with ODQ (10 min) reduced sGC stimulation to 23-fold with a corresponding increase in the EC50 to 831 (414.8–1665) nmol·L−1 (Figure 5B). Long-term haem oxidation resulted in a further decrease in sGC activity [maximal stimulation 17-fold, EC50 value of 580 (352.4–953.7) nmol·L−1]. Additional acute oxidation of long-term treated cells led to a similar EC50 value of 607 (197.1–1868) nmol·L−1 for BAY 41-2272-induced sGC activity as in cells that has only long-term treatment. As expected for a full antagonist, Zn-PPIX had no effect on sGC activity, neither under haem-oxidizing nor under control conditions (Figure 5D).
Treatment with increasing concentrations of HMR 1766 resulted in an up to 557-fold activation with an EC50 value of 8.8 (6.6–11.8) µmol·L−1 (Figure 5C). Surprisingly, acute ODQ did not increase HMR 1766-induced activation, and long-term ODQ even slightly diminished HMR 1766-induced sGC activation (Figure 5D), which was also not changed by additional acute ODQ exposure [EC50 13.5 (6.5–27.9) µmol·L−1].
sGC activity in ECs
In ECs, sGC activity was determined by measuring cGMP accumulation via radio-immuno assay upon incubating cells with increasing concentrations of BAY 58-2667, Zn-PPIX, HMR 1766 or BAY 41-2272 under normal or haem-oxidizing conditions (Figure 6) respectively. BAY 58-2667 showed a flat concentration response curve, and the maximal activation was only eightfold with an EC50 value of 0.3 (0.03–2.3) µmol·L−1. Haem oxidation potentiated BAY 58-2667-induced sGC activation up to 134-fold at the highest applied concentration of 10 µmol·L−1, with an EC50 value of 0.2 (0.05–0.7) µmol·L−1 (Figure 6A).
Figure 6.

Effects of BAY 58-2667, BAY 41-2272, HMR 1766 and Zn-PPIX on sGC activity under normal and haem-oxidizing conditions. Endothelial cells were treated with different concentrations of BAY 58-2667 (A), HMR 1766 (C), Zn-PPIX (D) for 30 min or BAY 41-2272 (B) for 15 min with or without 24 h ODQ (10 µmol·L−1) pretreatment. sGC activity was determined by measurement of cGMP accumulation via radio-immunoassay. Data are expressed as x-fold stimulation of control (means ± SEM of 8–14 independent experiments). The basal cGMP contents was as follows: (in fmol cGMP per well): (A) BAY 58-2667 409; BAY 58-2667 + ODQ 24 h 19; (B) BAY 41-2272 484; BAY 41-2272 + ODQ 24 h 26; (C) HMR 1766 241; HMR 1766 + ODQ 24 h 16; (D) Zn-PPIX 408; Zn-PPIX + ODQ 24 h 19. BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; cGMP, cyclic guanosine monophosphate; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; sGC, soluble guanylyl cyclase; Zn-PPIX, zinc-protoporphyrin IX.
BAY 41-2272 increased cGMP levels up to 123-fold at the highest tested concentration of 10 µmol·L−1, which was reduced to 86-fold after long-term ODQ treatment. The corresponding EC50 value was shifted from 737 (326.1–1667) to 946 (411.8–2175) nmol·L−1 respectively (Figure 6B). Incubating ECs with HMR 1766 lead to a maximal activation of 20-fold, and this was increased to 749-fold upon haem oxidation. EC50 values were 1261 (0.0–28545) and 810 (0.0–419) µmol·L−1 respectively (Figure 6C).
Activities of mutant β1H105F and β1Y135A/R139A sGCs
The activity of sGC containing either β1H105F or β1Y135A/R139A was determined in cGMP reporter cells stably transfected with expression vectors encoding for the respective enzyme mutants. WT, β1H105F and β1Y135A/R139A sGCs were incubated with increasing concentrations of BAY 58-2667 or HMR 1766 alone or combination with short-term ODQ treatment (Figure 7). WT sGC was activated up to 17-fold by 30 µmol·L−1 BAY 58-2667, with an EC50 of 0.2 (0.02–1.1) µmol·L−1, and this activation was increased to 64-fold with an EC50 of 2.0 (0.4–9.3) µmol·L−1 by addition of 10 µmol·L−1 ODQ (Figure 7A). Incubation of these WT sGC expressing cells with HMR 1766 resulted in a maximal activation of 230-fold by 30 µmol·L−1 HMR 1766, with an EC50 of 4.2 (1.6–10.8) µmol·L−1. Co-incubation with ODQ increased the activation to 328-fold with an EC50 of 1.5 (0.7–3.0) µmol·L−1 (Figure 7B).
Figure 7.

Concentration response curves of cGMP reporter cells stably transfected with WT (A, B) α1/β1H105F sGC (C, D) or α1/β1Y135A/R139A sGC (E, F) incubated with increasing concentrations of BAY 58-2667 (A, C, E) or HMR 1766 (B, D, F) alone or in combination with short-term ODQ (10 µmol·L−1) treatment. Data are means ± SEM from four to eight independent experiments performed in duplicate. sGC activation is represented as x-fold stimulation compared with non-stimulated control. Following basal activities from which x-fold stimulation was calculated were measured: (A) BAY 58-2667 2986 RLUs; BAY 58-2667 + ODQ 10 min 3078 RLUs; (B) HMR 1766 2957 RLUs; HMR 1766 + ODQ 10 min 2654 RLUs; (C) BAY 58-2667 16796 RLUs; BAY 58-2667 + ODQ 10 min 18266 RLUs; (D) HMR 1766 16624 RLUs; HMR 1766 + ODQ 10 min 19589 RLUs; (E) BAY 58-2667 3626 RLUs, BAY 58-2667 + ODQ 10 min 3822 RLUs; (F) HMR 1766 3494 RLUs; HMR 1766 + ODQ 10 min 3498 RLUs. BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; cGMP, cyclic guanosine monophosphate; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt; ODQ, 1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one; RLU, relative light unit; sGC, soluble guanylyl cyclase; WT, wildtype.
Incubation of cGMP reporter cells expressing the haem-free sGC mutant β1H105F with BAY 58-2667 resulted in a maximal activation of 45-fold, which was only slightly increased to 55-fold by addition of ODQ [determined EC50 were 11.6 (3.2–41.8) nmol·L−1 and 7.6 (1.8–31.2) nmol·L−1 respectively (Figure 7C)]. The highest concentration of HMR 1766 (30 µmol·L−1) activated β1H105F cells up to 95-fold [EC50 0.8 (0.5–1.1) µmol·L−1] and 100-fold [EC50 0.8 (0.4–1.8) µmol·L−1] by the addition of ODQ (Figure 7D). The double mutant β1Y135A/R139A was activated neither by BAY 58-2667 nor by BAY 58-2667 in combination with ODQ (Figure 7E) and only slightly activated by HMR 1766 (5-fold) or HMR 1766 and ODQ (6-fold; Figure 7F).
sGC protein levels of β1H105F sGC and β1Y135A/R139A sGC
Incubation of the WT sGC expressing cell line with ODQ for 24 h decreased sGC protein levels by 49% (α1) and 61% (β1) (Figure 8). BAY 58-2667 was able to rescue sGC α1 protein to control levels, whereas sGC β1 was increased beyond control. This observation was not affected by additional ODQ. In the cell line expressing the haem-free mutant β1H105F, oxidation did not result in reduced protein levels of both subunits. The addition of BAY 58-2667 both under control and haem-oxidizing conditions strongly increased β1H105F protein levels with weaker effects on α1 sGC. In cells expressing haem-free β1Y135A/R139A, sGC protein levels remained unchanged both under control and haem-oxidizing conditions, and BAY 58-2667 had no effect on the protein levels of either subunit (Figure 8).
Discussion
The NO-cGMP pathway plays a key role in the cardiovascular system, and its impairment is associated with different cardiovascular diseases. Recent findings suggest that increased levels of oxidative stress as observed under pathophysiological conditions can lead to oxidation or even loss of the sGC haem group, rendering the enzyme insensitive to NO and prone to ubiquitin-mediated degradation (Stasch et al., 2006; Meurer et al., 2007; Xia et al., 2007). These results might explain at least partially the observed reduction of sGC protein levels in different animal models of cardiovascular diseases (Ruetten et al., 1999; Kagota et al., 2001; Melichar et al., 2004).
Occupation of the sGC haem pocket with high affinity metallo-porphyrins or compounds resembling the spatial structure and charge distribution of haem such as BAY 58-2667 is able to prevent the oxidation-induced degradation of sGC (Stasch et al., 2006; Meurer et al., 2007). In parallel, Schindler et al., (2006) identified another haem-independent sGC activator that, although structurally unrelated, shares some characteristics with BAY 58-2667 such as the activation of haem-free sGC. The first evidence suggested an interaction of HMR 1766 with the haem pocket, as described for metallo-porphyrins and BAY 58-2667. However, whether this compound binds to the sGC haem site and if it is able to prevent the oxidation-induced sGC degradation as shown for Zn-PPIX and BAY 58-2667 have not been investigated yet. To clarify this open question and to shed light on the general mechanism of sGC stabilization, the present study investigated the molecular requirements of sGC activation and stabilization using the sGC activators, BAY 58-2667 and HMR 1766, and the haem pocket antagonist, Zn-PPIX, on sGC activity and protein levels in two cell systems expressing WT and mutant sGC.
To validate a putative interaction of HMR 1766 with the sGC haem pocket, sGC activity and competition binding assays were performed with BAY 58-2667, HMR 1766 and Zn-PPIX. Activity assays with purified sGC showed unequivocally that the high-affinity metallo-porphyrin is able to inhibit BAY 58-2667 and HMR 1766-induced sGC activation, suggesting an interaction of HMR 1766 with the haem-binding site as shown for BAY 58-2667 (Schmidt et al., 2004; Schindler et al., 2006; Stasch et al., 2006). The competition binding assays performed here support this view, as unlabelled BAY 58-2667 and Zn-PPIX displaced 3H-BAY 58-2667 from the enzyme. The results obtained with HMR 1766 were less clear; the relative low affinity of this compound prevented full displacement of radioactively labelled BAY 58-2667 from the enzyme. Nevertheless, at high micromolar concentrations, HMR 1766 reduced 3H-BAY 58-2667 binding to 67%, suggesting that HMR 1766, BAY 58-2667 and Zn-PPIX bind to the same or at least partially overlapping binding sites. The concentration of HMR 1766 used for this study is higher than the concentration used by Schindler et al. but was chosen to achieve similar sGC activation by HMR 1766 and BAY 58-2667. This difference in sGC sensitivity might be due to the facts that we used recombinant rat sGC expressed in and purified from a baculovirus/Sf9 insect cell system, instead of native bovine sGC. Heterologous expression in insect cells might result in an sGC that lacks certain post-translational modifications such as phosphorylation, which might affect sGC activity (Meurer et al., 2005).
Using Western blot analysis, the effect of these compounds on sGC protein levels upon haem oxidation by the sGC inhibitor, ODQ, was established. Long-term treatment of ECs and cGMP reporter cells with ODQ resulted in dramatically reduced protein levels of both sGC subunits, as observed for other primary cells (Stasch et al., 2006). This oxidation-induced degradation becomes prominent when the incubation time exceeds 2 h, whereas short-term incubations had no effect on sGC protein levels (Stasch et al., 2006). BAY 58-2667 and Zn-PPIX prevented this oxidation-induced decrease. In agreement with previous findings (Stasch et al., 2006), protein levels remained unchanged in cells treated with the sGC stimulator BAY 41-2272, suggesting that signalling events downstream of cGMP are not likely to be involved in sGC stabilization. Our results obtained with HMR 1766 were very surprising. Despite the fact that both BAY 58-2667 and HMR 1766 appear to interact with the sGC haem pocket, HMR 1766 did not show any protective effect on sGC protein levels.
The differences in protein levels under normal and haem-oxidizing conditions reflected the observed changes in sGC activity. Short-term incubation with 10 µmol·L−1 ODQ has been shown to potentiate BAY 58-2667-induced sGC activation at most, indicating that the majority of cellular sGC is converted into the oxidized/haem-free state, which can be activated by BAY 58-2667. As no impact on sGC protein levels has been reported for short-term ODQ incubations, the maximal BAY 58-2667-induced sGC activation upon short-term ODQ treatment compared with the combined long- and short-term ODQ incubation should reflect the decrease in sGC protein levels. Under this condition, a reduction of maximal sGC activation by BAY 58-2667 of 69% (129 to 82-fold activation) was observed, matching the observed reduction in sGC protein levels of 60% for α1 sGC and 36% of β1 sGC.
As shown in Figure 3, both BAY 41-2272 and HMR 1766 were unable to prevent oxidation-induced degradation of sGC. Therefore, we expected similar results for BAY 41-2272- or HMR 1766-induced sGC activity.
BAY 41-2272 activated sGC 115-fold under control conditions (Figure 5B). Combination of short- and long-term treatment reduced sGC activation to 7%. This is even lower than protein levels in ODQ and BAY 41-2272-treated cells, which were decreased by 57% (α1 sGC) and 43% (β1 sGC).
But, in contrast to BAY 41-2272, HMR 1766-induced sGC activity was unchanged or only slightly diminished compared with normal conditions. This discrepancy might be due to technical limitations, as the strong activation of sGC at high concentrations of HMR 1766 resulted in RLUs at the limit of detection. Although the addition of ODQ slightly increases the amount of RLUs, the expected HMR 1766 plus ODQ-induced maximum activation can presumably not be measured. Again, the highest concentration of HMR 1766 chosen for sGC activity measurements was higher than the concentrations used by Schindler et al. (2006). The differences in efficacy might be explained by the use of different cell lines or primary cells.
Comparison of Figures 5A and 6A shows that ECs were not stimulated to the same extend as cGMP reporter cells under control conditions. This might be due to the different cell types. In ECs, compared with cGMP reporter cells, the low BAY 58-2667-induced stimulation might reflect a small pool of naturally oxidized/haem-free sGC. On the other hand, this would argue for a bigger pool of haem-free/oxidized sGC in cGMP reporter cells compared with ECs. Mingone et al. (2006) showed that the levels of haem precursors (e.g. 5-amino-laevulinic acid) directly impact on haem synthesis and, as a result, in the relative amount of NO-sensitive, haem-containing sGC. As the cGMP reporter cells express sGC at very high levels, it might be possible that the native cellular haem synthesis is not able to match the needs of this artificially high expression, resulting in increased relative amounts of BAY 58-2667-sensitive, haem-free enzyme. Further studies applying haemin or 5-amino-laevulinic-acid might be able to shed light on this question. Wolin (2009) has shown that the haem precursor PPIX accumulates in vascular tissue incubated with 5-amino-laevulinic acid and thereby stimulates sGC and induces pulmonary artery relaxation.
In ECs, a dramatic increase in sGC activator-induced activation of sGC following long-term treatment with ODQ was observed. The haem-free state of sGC is preferentially targeted by sGC activators (Stasch et al., 2006; Roy et al., 2008). Moreover, haem loss in only a small proportion of sGC pool results in a dramatic increase of BAY 58-2667-induced sGC activity (Roy et al., 2008). In contrast, only a small decrease of BAY 41-2272-induced activation of ODQ pretreated sGC was observed, which might suggest a receptor reserve.
Protein levels of BAY 41-2272 and ODQ-treated ECs and cGMP reporter cells decreased by about 50% and reflected the measured sGC activity. In ECs, BAY 41-2272-induced activity was lowered by about 25%, and the corresponding EC50 was increased to about 25%. In cGMP reporter cells, sGC activity was diminished to an even greater extent.
In summary, BAY 58-2667 and Zn-PPIX but not BAY 41-2272 prevented sGC from oxidation-induced degradation. The measured sGC activity reflected the observed changes in protein levels. Importantly, the different sGC activators showed an unexpectedly different profile with respect to sGC protein and activity levels. Both haem-independent sGC activators, BAY 58-2667 and HMR 1766, induced sGC activation under normal and haem-oxidizing conditions, but only BAY 58-2667 was able to prevent oxidation-induced enzyme degradation. This molecular characteristic might be explained by different levels of haem mimicry and overlapping but not identical binding sites.
The sGC stabilizing ligands, BAY 58-2667 and Zn-PPIX, have the same chemical motif as haem and bind to the haem site with much higher affinity than the native prosthetic group. Furthermore, mutation analyses and structural models showed that both ligands interact with the haem-binding residues Y135 and R139 of the β1 subunit (Schmidt et al., 2004).
To investigate the interaction of the two haem-independent sGC activators, BAY 58-2667 and HMR 1766, the activation patterns of these compounds have been recorded with different sGC mutations that are known to effect haem-binding. Mutation of the axial haem ligand β1H105F has been shown to cause the expression of haem-free enzyme (Foerster et al., 1996), although this mutation did not preclude subsequent reconstitution of the enzyme with porphyrins (Schmidt et al., 2004). In contrast, the soluble mutant β1Y135A/R139A, which lacks the essential haem-binding residues, cannot be reconstituted with PPIX (Schmidt et al., 2004). Furthermore, Y135 and R139 were also identified as binding sites for BAY 58-2667 as the double mutant is no longer activated by BAY 58-2667 (Schmidt et al., 2004).
This double mutant α1/β1Y135A/R139A was used to determine whether both BAY 58-2667 and HMR 1766 activate sGC by binding to these residues within the haem binding motif (Schmidt et al., 2004).
When measuring sGC activity in cGMP reporter cells expressing these haem-free sGC variants, HMR 1766 and BAY 58-2667 apparently did not bind to the same residues. Although BAY 58-2667 is able to strongly activate the haem-free sGC mutant H105F, it was not able to induce any activation of the double mutant α1/β1Y135A/R139A, indicating that these residues are crucially important for BAY 58-2667 binding, as shown earlier (Schmidt et al., 2004). In contrast, HMR 1766 still activates the double mutant although its activation is diminished compared with WT sGC. These data clearly suggest that Y135 and R139 do not affect binding of HMR 1766 in the same way or extent as they affect binding of BAY 58-2667 or haem. Zhou et al. (2008) used docking simulations based on the sGC structure of Nostoc sp to identify putative regions through which HMR 1766 interacts with sGC. Contrary to our findings in living cells, they postulated Y135 and R139 as binding partners of HMR 1766, suggesting a BAY 58-2667-like binding mode.
Considering the results from the receptor binding assay, which showed that HMR 1766 competes with BAY 58-2667 only at high concentrations whereas it can be readily replaced by low amounts of Zn-PPIX, it becomes evident that BAY 58-2667 bears noticeably more resemblance to haem. In contrast, HMR 1766 seems to interact with different residues from those interacting with BAY 58-2667, although their binding sites might overlap at least partially.
Basing on these findings, we hypothesized that the different binding modes to the haem pocket might be responsible for the differences between HMR 1766 and BAY 58-2667 with respect to protection of sGC from oxidation-induced degradation. When using the same sGC mutants described above in Western blots, it became apparent that the protective function of BAY 58-2667 is also mediated by the haem-like occupation of the haem. This protection can only be observed for WT and β1H105F sGC, which are activated by BAY 58-2667, but not for β1Y135A/R139A sGC, which neither is activated by BAY 58-2667 nor can be reconstituted with PPIX (Schmidt et al., 2004). HMR 1766 was not used in this set of experiments because of its lack of protective or stabilizing properties, which was already demonstrated in WT-sGC-expressing cells, and HMR 1766-induced activity in Y135A/R139A sGC-expressing cells showed its different binding mode.
Another difference becomes obvious by comparing α1 and β1 sGC levels: Oxidation-induced degradation of the α1 subunit is not prevented by BAY 58-2667 and Zn-PPIX to the same extent as observed for the β1 subunit. Based on our data with β1H105F sGC, it is more likely that the β subunit is predominantly affected by ODQ. Incubation with ODQ does not lead to a significant decrease in sGC protein levels in cells expressing β1H105F, as observed in cells expressing WT sGC. In addition, the BAY 58-2667 activation profile of β1H105F expressing cells resembles the pattern of BAY 58-2667-induced sGC activity in ODQ treated WT cells. In agreement with Stasch et al. (2006), we assume that the physiological turnover of α1 sGC is not affected due to the lack of haem binding, and, thus, any changes that are mediated via the haem binding site do not apply to α1 sGC directly. The changes we observed for α1 sGC protein levels may rather be based on counter regulatory mechanisms as described for the α1 and α2 knockout mice (Mergia et al., 2006). Here, deletion of α subunits results in a concomitant decrease of β1 protein levels. Moreover, Friebe et al. (2007) demonstrated that β1 knockout mice lack the α1 subunit. We suppose that the observed changes underlie a similar mechanism of counter regulation, which results in decreased α1 protein levels when β1 subunits are depleted due to oxidation. However, instability of single α1 subunits cannot be an explanation for the decrease in α1 sGC protein levels, as homodimers can form and are stable (Zabel et al., 1999).
In summary, our results show that BAY 58-2667 and Zn-PPIX but neither BAY 41-2272 nor HMR 1766 prevent oxidation-induced degradation of sGC and decreased sGC activity. BAY 58-2667's protective effect depends on high-affinity binding to the haem-binding pocket in a manner that reassembles the native prosthetic group including the interaction with the haem binding motif Y-x-S-x-R. HMR 1766 lacks these properties, making it a distinct class of sGC activators. The results of this study are summarized in Figure 9. To our knowledge, BAY 58-2667 is thus the first pharmacological enzyme ligand, which, in addition to activating, also stabilizes its own target. As cardiovascular diseases are associated with increased levels of oxidative stress, it can be expected that the relative amount of haem-oxidized/haem-free sGC is increased under pathophysiological conditions. This view is in agreement to results obtained in a clinical trial with patients suffering from acute decompensated heart failure, which suggest the presence of a pool of haem-free/oxidized sGC in humans (Schmidt et al., 2009). This imbalance would be translated into decreased sGC protein levels due to accelerated degradation of the oxidation-impaired enzyme. The sGC stabilizing features of BAY 58-2667 described here might help to overcome this imbalance by preventing sGC from degradation and thus improving cardiovascular disease. Further pharmacological and clinical studies with sGC activators will provide more information on the in vivo efficacy and effects in the treatment of cardiovascular diseases.
Figure 9.

Model of the role of sGC's haem group and sGC targeting compounds in protection of sGC. Under haem-oxidizing conditions such as oxidative stress, binding of the sGC activator BAY 58-2667 stabilizes sGC and thereby protects sGC from degradation. In contrast, binding of the sGC activator HMR 1766 or the sGC stimulator BAY 41-2272 cannot stabilize sGC and therefore sGC like haem-free sGC is ubiquitinated and degraded. BAY 41-2272, 5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; BAY 58-2667, 4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid; sGC, soluble guanylyl cyclase; HMR 1766, 5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt.
Glossary
Abbreviations:
- BAY 41-2272
5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine
- BAY 58-2667
4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy]phenethyl}amino)methyl[benzoic]acid
- CHO
Chinese hamster ovary
- EC
endothelial cell
- HMR 1766
5-chloro-2-(5-chloro-thiophene-2-sulphonylamino-N-(4-(morpholine-4-sulphonyl)-phenyl)-benzamide sodium salt
- HSP90
heat shock protein 90
- ODQ
1H-(1,2,4)-oxadiazolo[4,3-a]quinoxalin-1-one
- ONOO−
peroxynitrite
- PPIX
protoporphyrin IX
- RLU
relative light unit
- ROS
reactive oxygen species
- sGC
soluble guanylyl cyclase
- WT
wildtype
- Zn-PPIX
zinc-protoporphyrin IX
Conflict of interest
LS Hoffmann, S Schaefer, Y Keim and JP Stasch are fulltime employees of Bayer HealthCare.
References
- Agulló L, Garcia-Dorado D, Escalona N, Ruiz-Meana M, Mirabet M, Inserte J, et al. Membrane association of nitric oxide-sensitive guanylyl cyclase in cardiomyocytes. Cardiovasc Res. 2005;68:65–74. doi: 10.1016/j.cardiores.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Artz JD, Schmidt B, McCracken JL, Marletta MA. Effects of nitroglycerin on soluble guanylyl cyclase: implications of nitrate tolerance. J Biol Chem. 2002;21:18253–18256. doi: 10.1074/jbc.C200170200. [DOI] [PubMed] [Google Scholar]
- Becker EM, Wunder F, Kast R, Robyr C, Hoenicka M, Gerzer R, et al. Generation and characterization of a stable soluble guanylate cyclase-overexpressing CHO cell line. Nitric Oxide. 1999;3:55–66. doi: 10.1006/niox.1999.0207. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Horowitz JD. Impaired tissue responsiveness to organic nitrates and nitric oxide: a new therapeutic frontier? Pharmacol Ther. 2007;116:287–305. doi: 10.1016/j.pharmthera.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HHHW, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907–916. doi: 10.1161/01.RES.0000100390.68771.CC. [DOI] [PubMed] [Google Scholar]
- Foerster J, Harteneck C, Malkewitz J, Schultz G, Koesling D. A functional heme-binding site of soluble guanylyl cyclase requires intact N-termini of α1 and β1 subunits. Eur J Biochem. 1996;240:380–386. doi: 10.1111/j.1432-1033.1996.0380h.x. [DOI] [PubMed] [Google Scholar]
- Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci USA. 2007;104:7699–7704. doi: 10.1073/pnas.0609778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- Gladwin MT. Deconstructing endothelial dysfunction: soluble guanylyl cyclase oxidation and the NO resistance syndrome. J Clin Invest. 2006;116:2330–2332. doi: 10.1172/JCI29807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenicka M, Becker EM, Apeler H, Sirichoke T, Schroder H, Gerzer R, et al. Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: stimulation by YC-1, nitric oxide, and carbon monoxide. J Mol Med. 1999;77:14–23. doi: 10.1007/s001090050292. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Wood KS, Wolin MS. Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc Natl Acad Sci USA. 1982;79:2870–2873. doi: 10.1073/pnas.79.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagota S, Tamashiro A, Yamaguchi Y, Sugiura R, Kuno T, Nakamura K, et al. Downregulation of vascular soluble guanylate cycalse induced by high salt intake in spontaneously hypertensive rats. Br J Pharmacol. 2001;134:737–744. doi: 10.1038/sj.bjp.0704300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp-Harper B, Feil R. Meeting report: cGMP matters. Sci Signal. 2008;1:12. doi: 10.1126/stke.19pe12. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melichar VO, Behr-Roussel D, Zabel U, Uttenthal LO, Rodrigo J, Rupin A, et al. Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late-stage atherosclerosis. Proc Natl Acad Sci USA. 2004;101:16671–16676. doi: 10.1073/pnas.0405509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergia E, Freibe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest. 2006;116:1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurer S, Pioch S, Gross S, Muller-Esterl W. Reactive oxygen species induce tyrosine phosphorylation of and Src kinase recruitment to NO-sensitive guanylyl cyclase. J Biol Chem. 2005;280:33149–33156. doi: 10.1074/jbc.M507565200. [DOI] [PubMed] [Google Scholar]
- Meurer S, Pabst T, Pioch S, Opitz N, Schmidt PM, Wagner K, et al. Oxidative stress induces CHIP-mediated ubiqitination and proteasomal degradation of soluble guanylyl cyclase. BMC Pharmacol. 2007;7(Suppl. 1):25. [Google Scholar]
- Mingone CJ, Gupte SA, Chow JL, Ahmad M, Abraham NG, Wolin MS. Protoporphyrin IX genenration from δ-aminolevulinic acid elicits pulmonary artery relaxation and soluble guanylate cyclase activation. Am J Physiol Lung Cell Mol Physiol. 2006;291:L337–L344. doi: 10.1152/ajplung.00482.2005. [DOI] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Munzel T, Genth-Zotz S, Hink U. Targeting heme-oxidized soluble guanylate cyclase: solution for all cardiorenal problems in heart failure? Hypertension. 2007;49:974–976. doi: 10.1161/HYPERTENSIONAHA.106.085456. [DOI] [PubMed] [Google Scholar]
- Nedvetsky PI, Meurer S, Opitz N, Nedvetskaya TY, Müller H, Schmidt HHHW. Heat shock protein 90 regulates stabilization rather than activation of soluble guanylate cyclase. FEBS Lett. 2008;582:327–331. doi: 10.1016/j.febslet.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Olesen SP, Drejer J, Axelsson O, Moldt P, Bang L, Nielsen-Kudsk JE, et al. Characterization of NS 2028 as a specific inhibitor of soluble guanylyl cyclase. Br J Pharmacol. 1998;123:299–309. doi: 10.1038/sj.bjp.0701603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena P, Karow DS, Boon E, Marletta M, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkegel C, Schmidt PM, Atkins DJ, Hoffmann LS, Schmidt HH, Schroder H, et al. Dimerization region of soluble guanylate cyclase characterized by bimolecular fluorescence complementation in vivo. Mol Pharmacol. 2007;72:1181–1190. doi: 10.1124/mol.107.036368. [DOI] [PubMed] [Google Scholar]
- Roy B, Mo E, Vernon J, Garthwaite J. Probing the presence of the ligand-binding haem in cellular nitric oxide receptors. Br J Pharmacol. 2008;153:1495–1504. doi: 10.1038/sj.bjp.0707687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetten H, Zabel U, Linz W, Schmidt HHHW. Downregulation of soluble guanylyl cylase in young and aging spontaneously hypertensive rats. Circ Res. 1999;85:534–541. doi: 10.1161/01.res.85.6.534. [DOI] [PubMed] [Google Scholar]
- Schindler U, Strobel H, Schonafinger K, Linz W, Lohn M, Martorana PA, et al. Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol. 2006;69:1260–1268. doi: 10.1124/mol.105.018747. [DOI] [PubMed] [Google Scholar]
- Schmidt HHHW, Schmidt PM, Stasch JP. NO- and haem-independent soluble guanylate cyclase activators. In: Schmidt HHHW, Hofmann F, Stasch JP, editors. cGMP: Generators, Effectors and Therapeutic Implications. Handbook of Experimental Pharmacology. Berlin: Springer; 2009. pp. 309–339. [DOI] [PubMed] [Google Scholar]
- Schmidt PM. Biochemical detection of cGMP from past to present: an overview. In: Schmidt HHHW, Hofmann F, Stasch JP, editors. cGMP: Generators, Effectors and Therapeutic Implications. Handbook of Experimental Pharmacology. Berlin: Springer; 2009. pp. 195–228. [Google Scholar]
- Schmidt PM, Schramm M, Schroder H, Stasch JP. Receptor binding assay for nitric oxide- and heme-independent activators of soluble guanylate cyclase. Anal Biochem. 2003;314:162–165. doi: 10.1016/s0003-2697(02)00660-7. [DOI] [PubMed] [Google Scholar]
- Schmidt PM, Schramm M, Schroder H, Wunder F, Stasch JP. Identification of residues crucially involved in the binding of the heme moiety of soluble guanylate cyclase. J Biol Chem. 2004;279:3025–3032. doi: 10.1074/jbc.M310141200. [DOI] [PubMed] [Google Scholar]
- Schmidt PM, Rothkegel C, Wunder F, Schroder H, Stasch JP. Residues stabilizing the heme moiety of the nitric oxide sensor soluble guanylate cyclase. Eur J Pharmacol. 2005;513:67–74. doi: 10.1016/j.ejphar.2005.02.046. [DOI] [PubMed] [Google Scholar]
- Shu AYL, Heys JR. Extension of organoiridium catalyzed hydrogen isotope exchange: photoaffinity labels and paclitaxel. In: Pleiss U, Voges R, editors. Proceedings of the 7th International Symposium 7. Dresden: John Wiley & Sons; 2000. pp. 68–70. [Google Scholar]
- Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Schmidt PM, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of of a new pharmacological principle. Br J Pharmacol. 2002a;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, Minuth T, et al. Pharmacological actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vitro studies. Br J Pharmacol. 2002b;135:333–343. doi: 10.1038/sj.bjp.0704484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, Arum Kumar HS, Meurer S, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub A, Stasch JP, Alonso-Alija C, Benet-Buchholz J, Ducke B, Feuerer A, et al. NO-independent stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett. 2001;11:781–784. doi: 10.1016/s0960-894x(01)00073-7. [DOI] [PubMed] [Google Scholar]
- Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, Böhme E, et al. Mutation of His-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1994;91:2592–2596. doi: 10.1073/pnas.91.7.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol. 2009;296:H539–H549. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunder F, Stasch JP, Hutter J, Alonso-Alija C, Huser J, Lohrmann E. A cell-based cGMP assay useful for ultra-high-throughput screening and identification of modulators of the nitric oxide/cGMP pathway. Anal Biochem. 2005;339:104–112. doi: 10.1016/j.ab.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Xia T, Dimitropoulou C, Zeng J, Antonova GN, Snead C, Venema RC, et al. The chaperone-dependent E3 ligase CHIP ubiquitinates and mediates proteasomal degradation of soluble guanylyl cyclase. Am J Physiol Heart Circ Physiol. 2007;293:H3080–H3087. doi: 10.1152/ajpheart.00579.2007. [DOI] [PubMed] [Google Scholar]
- Zabel U, Häusler C, Weeger M, Schmidt HHHW. Homodimerization of soluble guanylyl cyclase subuits. J Biol Chem. 1999;274:18149–18152. doi: 10.1074/jbc.274.26.18149. [DOI] [PubMed] [Google Scholar]
- Zabel U, Kleinschnitz C, Oh P, Smolenski A, Nedvetsky P, Kugler P, et al. Calcium-dependent membrane association sensitises soluble guanylyl cyclase to NO. Nature Cell Biol. 2002;4:307–311. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Schelvis JP, Babcock GT, Marletta MA. Identification of histidine 105 in the beta1 subunit of soluble guanylate cyclase as the heme proximal ligand. Biochemistry. 1998;37:4502–4509. doi: 10.1021/bi972686m. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Brandish PE, DiValentin M, Schelvis JP, Babcock GT, Marletta MA. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Pyriochou A, Kotanidou A, Dalkas G, van Eickels M, Spyroulias G, et al. Soluble guanylyl cyclase activation by HMR-1766 (ataciguat) in cells exposed to oxidative stress. Am J Physiol Heart Circ Physiol. 2008;295:H1763–H1771. doi: 10.1152/ajpheart.51.2008. [DOI] [PubMed] [Google Scholar]