

Abstract
Toll-like receptors (TLR) are pattern recognition receptors for highly conserved microbial molecular patterns. Activation of TLR is a pivotal step in the initiation of innate, inflammatory, and immune defense mechanisms. Recent findings indicate that G protein-coupled receptors (GPCR) may modulate TLR signaling, but it is unclear which GPCR are involved in this process. One such cooperation between GPCR and TLR can be attributed to the sphingosine 1-phosphate (S1P) receptor family. The S1P receptors (S1P1–5) are a family of GPCR with a high affinity for S1P, a serum-borne bioactive lipid associated with diverse biological activities such as inflammation and healing. In this study, we show that pro-inflammatory cytokine production, including IL-6 and IL-8, was increased with LPS and concomitant S1P stimulation. Furthermore, elevated cytokine production following LPS and S1P challenge in human gingival epithelial cells (HGEC) was significantly reduced when TLR4, S1P1 or S1P3 signaling was blocked. Our study also shows that S1P1 and S1P3 expression was induced by LPS in HGEC, and this elevated expression enhanced the influence of S1P in its cooperation with TLR4 to increase cytokine production. This cooperation between TLR4 and S1P1 or S1P3 demonstrates that TLR4 and GPCR can interact to enhance cytokine production in epithelial cells.
Keywords: Cytokines, Epithelial cells, S1P1, S1P3, TLR4
Introduction
Host defense against invading microbial pathogens is mediated by the host defense system comprising innate and adaptive immune responses [1]. Recognition of microorganisms is mediated by a set of germ-line-encoded receptors, referred to as pattern recognition receptors (PRR). Eleven members of the Toll-like receptor (TLR) family have been identified so far [2]. These receptors recognize highly conserved molecular patterns (microbial-associated molecular patterns) shared by a broad group of microorganisms [3] which contain lipopolysaccharide (LPS) and peptidoglycan. TLR4 was the first characterized TLR in humans [4] and recognizes bacterial LPS, an outer membrane component of gram-negative bacteria [5]. TLR4 is expressed by a variety of cell types, including leucocytes, fibroblasts, endothelial and epithelial cells [6, 7]. Receptor-ligand interactions are critical events in triggering the inflammatory and immune responses to eliminate pathogens through release of a wide range of cytokines, antimicrobial peptides and chemokines [8]. Pro-inflammatory cytokines induce inflammation, which together with released chemokines attract professional phagocytes to the site of microbial perturbation or invasion and thus aid in the maintenance of the epithelial barrier function [9]. This barrier is crucial in preventing invasion and dissemination of both pathogenic and commensal microorganisms, particularly in oral and gastrointestinal mucosa which are permanently exposed to high bacterial loads.
Inflammatory cytokines are now appreciated as having a role in the wound healing process [10] as well as for their inflammatory and immune regulatory function in cells [11]. It was shown that the inflammatory cytokines, granulocyte/macrophage colony-stimulating factor (GM-CSF) [12] and tumor necrosis factor (TNF)-α, can modulate collagen synthesis in cultured fibroblasts [13]. More interestingly, it has been suggested that IL-6 is a cytoprotective molecule [14]. On the other hand, the chemokine IL-8 is expressed by gingival epithelial cells [15] and is responsible for recruitment of bacteria-killing phagocytes such as neutrophils to the periodontal tissues [16]. Thus, inflammation and tissue healing are intertwined in that induction of pro-inflammatory cytokines is crucial for host defense mechanisms and plays a role in tissue homeostasis.
Considerable attention has been devoted to the diverse biological effects of sphingosine 1-phosphate (S1P), a bioactive sphingolipid derived from sphingomyelin released by activated platelets and other cells in response to a wide array of stimuli [17]. S1P regulates broad biological functions such as proliferation, migration, differentiation, cell-cell interaction, and apoptosis [18–20]. S1P receptors (S1P1–5) are part of the G protein-coupled receptor (GPCR) superfamily and are widely expressed in most tissues at different levels [21], suggesting that this receptor influences many cellular processes.
In this study, we demonstrate a novel cooperative mechanism between TLR4 and S1P1 or S1P3 signaling. S1P1 and S1P3 expression was significantly increased in primary human gingival epithelial cells (HGEC) following LPS-mediated TLR4 activation. Crucial biological responses, namely release of pro-inflammatory cytokines (IL-6 and IL-8), were enhanced in epithelial cells challenged with a combination of LPS and S1P. We also demonstrate that S1P1 or S1P3 signaling cooperates with TLR4 signaling. This enhanced innate immune response is significantly reduced in TLR4-diminished epithelial cells or when S1P1 or S1P3 signaling is blocked. This is the first demonstration of the cooperation between TLR4 and the S1P family of GPCR, whose purpose may be to enhance the epithelial cell barrier integrity while simultaneously triggering the inflammatory defensive responses following tissue damage by microbial insult.
Results
Primary human gingival epithelial cells
TLR4 deficiencies may detrimentally reduce the innate immune response to pathogenic bacteria [6], and mice lacking TLR signaling have increased morbidity [14]. We have isolated a bank of more than 40 primary HGEC cultures and have determined their levels of TLR4 mRNA (by real-time PCR) and protein (by fluorescence-activated cell sorting; FACS). We have detected multiple HGEC that show TLR4 expression that is significantly increased or does not increase beyond the basal level at both the gene and protein levels after LPS treatment; we define these as TLR4-normal or –diminished HGEC, respectively. These TLR4-diminished cells are typically but not exclusively related to carriage of the Asp299Gly polymorphism in the TLR4 gene [6]. In the present study, we utilized HGEC with either TLR4-normal (HGEC-15, -16 and -17) or -diminished (HGEC-9, -11 and -12) cellular TLR4 expression derived from healthy gingival tissues isolated from six subjects free of obvious oral and systemic diseases.
S1P and LPS cooperatively enhance inflammatory cytokine production
To examine the role of the TLR4 receptor in the cooperation with S1P receptors, we used TLR4-normal and -diminished primary gingival epithelial cells challenged with LPS in the presence or absence of S1P for 24 h. The secretion of the cytokines including IL-6 (Fig. 1A) and IL-8 (Fig. 1B) by TLR4-normal HGEC (gray bar), was increased in the presence of both LPS and S1P. However, neither LPS nor S1P alone could markedly induce these cytokines: there was only a minor increase in IL-8 and IL-6 production after individual LPS or S1P treatments, but these increases were not statistically significant. In addition, LPS and S1P costimulation was unable to increase the cytokine production in TLR4-diminished HGEC (Fig. 1A, B, white bar), indicating that TLR4, LPS and S1P are needed to up-regulate cytokine production in these cells. Moreover, we found that the other TLR4-normal but not -diminished cells resulted in increased production of cytokines after LPS + S1P stimuli (data not shown). It is well known that exogenous S1P can simultaneously increase its own synthesis. It has been shown that the intracellular S1P synthesis is regulated by sphingosine kinase (SK) [22]. We therefore inhibited intracellular synthesis of S1P by a pharmacological inhibitor against SK. Initially, we titrated the inhibitor concentration and found that 1 μM of SK inhibitor appeared to be the optimum dose (Fig. 1C). We next challenged the cells with LPS + S1P in the presence or absence of the SK inhibitor. We found that the SK inhibitor did not reduce the production of IL-6 as compared to LPS + S1P-challenged cells (Fig. 1D). In addition to IL-6, we also found that the elevated IL-8 production was not ablated in the cells challenged with LPS + S1P in the presence of SK inhibitor (data not shown).
Figure 1.

Cytokine production following S1P/LPS stimuli in epithelial cells. Primary HGEC were challenged with LPS (1 μg/mL), S1P (100 nM), or a combination of LPS + S1P for 24 h at 37°C. Induction of IL-6 (A) and IL-8 (B) was determined in TLR4-normal and TLR4-diminished epithelial cell culture supernatants by Luminex following S1P, LPS or LPS + S1P challenge. TLR4-normal cells were challenged with 1, 2, or 4 μM SK inhibitor (inh) or medium only for 24 h. Then IL-6 production was determined in the challenged groups (C). The cells were challenged with LPS + S1P in the presence or absence of SK (inh) for 24 h. Subsequently, IL-6 production was measured by ELISA (D). Data are presented as the mean ± standard deviation (SD) of triplicate determinations, from one of three independent sets of cell cultures that yielded similar findings. Statistically significant (p <0.05) induction of cytokine release is indicated by an asterisk in TLR4-normal cells (NS; not statistically significant from basal level, LPS or S1P challenge).
Next, we examined whether S1P-related enhancement of TLR4 signaling also occurs with other TLR. The TLR4-normal epithelial cells were challenged with FSL-1 (a specific agonist for TLR2), S1P, or a combination of S1P and FSL-1 for 24 h. The cells challenged with FSL-1 but not S1P alone were able to induce IL-6 (Fig. 2A) and IL-8 (Fig. 2B), whereas adding exogenous S1P to the cells did not further increase the cytokine production. We also found that S1P did not cooperate with different concentrations of FSL-1, such as 0.1, 0.25, or 0.5 μg/mL (data not shown). Together, these data indicate that LPS but not FSL-1 cooperates with exogenous S1P to up-regulate inflammatory cytokine production in primary HGEC.
Figure 2.
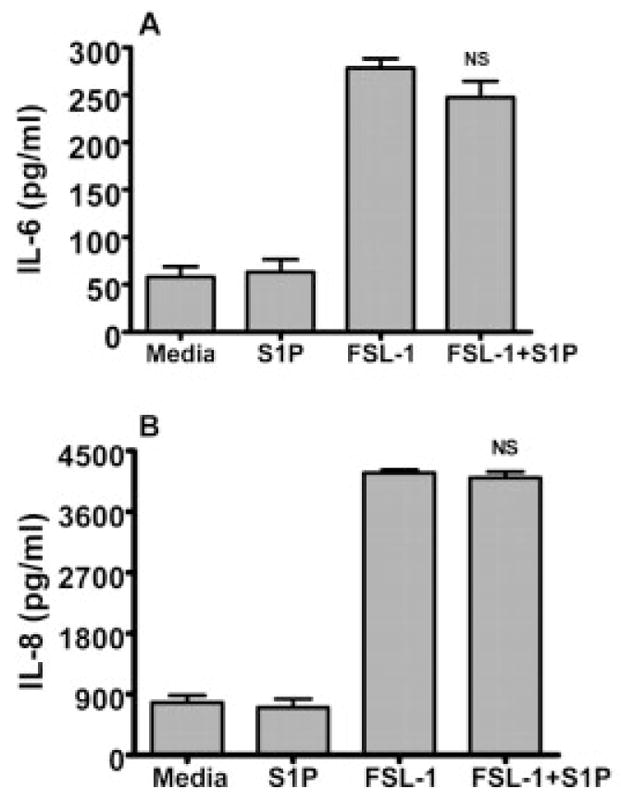
S1P does not cooperate with TLR2 for the production of cytokines. HGEC were challenged with S1P (100 nM), FSL-1 (1 μg/mL) or a combination of FSL-1 + S1P for 24 h at 37°C. The production of IL-6 (A) and IL-8 (B) was assayed by Luminex following FSL-1, S1P or FSL-1 + S1P challenge for 24 h. Data are presented as the mean ± SD of triplicate determinations from one of three independent sets of cell cultures that yielded similar findings (NS; not statistically significant from FSL-1 challenge).
TLR4 signaling up-regulates S1P receptors
To understand the mechanism underlying S1P and LPS cooperation, we tested whether S1P receptor gene and protein expression are induced by TLR4 stimulation in HGEC. TLR4-normal cells were challenged with 100, 200, 500, 1000 and 2000 ng/mL LPS. Our findings showed that S1P1 was markedly induced by 500 ng/mL LPS and maximized at 1000 ng/mL LPS (Fig. 3A). We next challenged the cells with 1000 ng/mL LPS for 0.5, 1, 2, 4, 6, 8 and 24 h. S1P1 gene expression was significantly detected 4 h after LPS stimulation and the expression of S1P1 was up-regulated in a time-dependent manner (Fig. 3B). However, TLR4 expression and that of other TLR including TLR1, 2 and 6 could not be induced in the cells following S1P challenge for 24 h (data not shown).
Figure 3.

S1P1 is induced by LPS in a time- and dose-dependent fashion in epithelial cells. TLR4-normal cells were challenged with 100, 200, 500, 1000 and 2000 ng/mL of protein-free E. coli LPS or medium only for 24 h at 37°C (A). TLR4-normal cells were challenged with 1 μg/mL of protein-free E. coli LPS or medium only for 0.5, 1, 2, 4, 6, 8 or 24 h at 37°C (B). Following the challenge assay, relative expression of S1P1 was determined by real-time PCR. The ratio of S1P1 was normalized to GAPDH mRNA in cells following LPS challenge. Statistically significant (p <0.05) expression of S1P1 is indicated by an asterisk.
We next examined the expression of other S1P receptors, namely S1P2, S1P3, S1P4 and S1P5, in TLR4-normal cells. We found that the epithelial cells expressed all receptors except S1P4 and that S1P2 and S1P3 expression was slightly increased in the cells challenged with LPS for 24 h (Fig. 4A). However, the expression of S1P1 was greater than that of either S1P2 or S1P3. More interestingly, the S1P3 expression, similar to S1P1, was reduced in TLR4-diminished cells challenged with LPS (data not shown). We then determined if the increase in S1P1 is only specific for LPS signaling. We therefore challenged the epithelial cells (TLR4-normal) for 24 h with TLR4 (highly purified E. coli LPS), TLR2 (FSL-1) agonists or human recombinant IL-1β for IL-1R, which shares structural and signaling pathway similarities with TLR [23]. In TLR4-normal cells, S1P1 mRNA expression was significantly increased by LPS but not by IL-1β or FSL-1 stimulation (Fig. 4B). Subsequently, we determined S1P1 expression in both cell types following LPS challenge. We found that the TLR4 and S1P1 expression increased in TLR4-normal epithelial cells but not in TLR4-diminished cells after LPS stimuli (Fig. 4C). To confirm our results at the protein level, the S1P1 protein was determined by immunoblotting in cells challenged with LPS for 24 h. The induction of S1P1 was increased by more than fourfold (Fig. 4D, E). We also confirmed TLR4 expression at the protein level by FACS in both cells types. The basal expression of TLR4 in TLR4-normal cells was slightly higher than that of TLR4-diminished cells. However, TLR4 induction was dramatically increased in TLR4-normal cells but not in TLR4-diminished cells after LPS challenge for 24 h (Fig. 4F). We measured the expression of TLR4, S1P1 and S1P3 expression in other TLR4-normal and diminished cells and found similar results to those shown above. TLR4 and S1P1 or S1P3 expression was induced in other TLR4-normal but not in TLR4-diminished cells (data not shown).
Figure 4.

S1P1 is induced by TLR4 ligation in human epithelial cells. TLR4-normal HGEC were challenged with LPS (1 μg/mL) for 24 h. Relative expression of S1P1–5 was determined by real-time PCR after normalizing their expression to GAPDH (A). TLR4-normal HGEC were challenged with protein-free E. coli LPS (1 μg/mL), TLR2 agonist (FSL-1, 1 μg/mL) or IL-1β (5 ng/mL) for 24 h at 37°C. Real-time PCR was performed and the ratio of S1P1 was normalized to GAPDH mRNA in TLR4-normal gingival epithelial cells following LPS, FSL-1 or IL-1β challenge (B). The ratios of TLR4 and S1P1 were normalized to GAPDH in TLR4-normal and TLR4-diminished gingival epithelial cells after LPS challenge (C). Protein expression of S1P1 in TLR4-normal epithelial cells following LPS challenge was detected by Western blotting after S1P1 immunoprecipitation (IgH and IgL were included for specificity, the control β-actin is not shown) (D) and band intensity was determined using the ratio of the increase of S1P1 over endogenous control (β-Actin) (E). Both cells types (TLR-4-normal and TLR-4-diminished) were stained with PE-conjugated monoclonal anti-human TLR4 antibody or its isotype control (IgG2a, data not shown) for 20 min at 4°C. The stained cells were analyzed by flow cytometry using a BD FACSCalibur and CellQuest software (F). Data presented are the means ± SD of triplicate determinations from one of three independent sets of cell cultures that yielded similar findings. Statistically significant (p <0.05) expression of TLR4 or S1P1 is indicated by an asterisk.
TLR4 is required for pro-inflammatory cytokine production
To examine the precise role of TLR4 in LPS + S1P-induced cytokine production, expression of TLR4 was reduced using the siRNA technique. Initially, we determined the TLR4 expression in cells challenged with LPS in the presence of S1P. TLR4 expression was up-regulated in cells challenged with LPS + S1P after irrelevant gene silencing (siLaminin); however, its expression was significantly reduced in cells with siRNA knocked down for TLR4, challenged with LPS + S1P (Fig. 5A). Next, we determined cytokine induction in the challenged cells after TLR4 gene knockdown. The production of IL-8 (Fig. 5B) and IL-6 (Fig. 5C) was elevated in sham-silenced cells (siLaminin) following LPS + S1P stimuli; however, the same stimuli had no effect on the TLR4 knockdown cells. These data clearly reveal that TLR4 ligation is indispensable for the enhanced pro-inflammatory cytokine production following LPS and S1P challenge.
Figure 5.

TLR4 cooperation is required for cytokine production in HGEC. The TLR4-normal epithelial cells were transfected with 100 pmol of anti-sense RNA to TLR4 or laminin, an irrelevant gene silencer; 3 μL of the transfection reagent FuGene 6 was diluted using 95 μL of serum-free media, and 100 pmol of siTLR4 or siLaminin was added and incubated at room temperature for 15 min. The reaction was carried out overnight and the medium was replaced with fresh medium. After 48 h of silencing, the cells were challenged with LPS (1 μg/mL) in the presence of S1P (100 nM) for 24 h. The expression of TLR4, and S1P1 following the silencing, was determined by real-time PCR (A). The protein induction of IL-8 (B) and IL-6 (C) was determined in culture supernatants by Luminex. Data are presented as the mean ± SD of triplicate determinations. Statistically significantly (p <0.05) induced or reduced cytokine production is indicated by two asterisks or an asterisk, respectively.
S1P1 or S1P3 is required for cytokine induction
Since S1P is a ligand for all of the S1P family receptors and we found that S1P1,2,3,5 are expressed by HGEC as shown Fig. 1A, we examined which of the S1P receptors cooperated with TLR4 in human epithelial cells. Initially, the cells were challenged with 100, 200 or 400 nM S1P to determine the optimum dose for the assay. We found that 100 nM S1P was the optimum dose to increase the production of cytokines in the presence of LPS in epithelial cells (data not shown). We knocked down S1P1 receptor expression to examine the specific role of S1P1 in S1P/LPS-induced inflammatory cytokine production. The S1P1 mRNA expression was abrogated in epithelial cells challenged with LPS + S1P for 24 h after knocking down S1P1 by the siRNA (Fig. 6A). The cytokines including IL-8 (Fig. 6B) and IL-6 (Fig. 6C) were significantly reduced at the protein level in the cells challenged with S1P and LPS as compared to scrambled siRNA. Alternatively, we confirmed the specificity between S1P1 and LPS; the cells were challenged with an S1P1-specific agonist, SEW, in the presence or absence of LPS for 24 h. IL-6 was assayed as a representative pro-inflammatory cytokine as shown above. IL-6 production was significantly increased by LPS and SEW but neither SEW nor LPS increased the cytokine induction (Fig. 6D). Furthermore, we chemically blocked S1P1 signaling with pertussis toxin to test its effect with LPS signaling (since it has been shown that S1P1 couples exclusively to Gi/o proteins [24]). S1P1 induces signaling through Gi/o, which is pertussis toxin sensitive [25]. Elevated IL-8 (Fig. 6E) and IL-6 (Fig. 6F) levels were ablated in the cells challenged with LPS and S1P after blocking Gi/o with pertussis toxin. However, our results showed that the cytokine production was not completely abrogated in the cells challenged with LPS + S1P after reducing S1P1 expression, suggesting that there are other S1P receptors involved in the cytokine production. It has already been established that S1P1 and S1P3 have similar positive effects on the cells [26]. We thus blocked S1P3 signaling with the pharmacological inhibitor suramin [27]. The optimal dose for suramin was determined prior to the challenge. We observed that a dose of 0.4 mg/mL of suramin neither increased nor decreased the IL-6 production as compared to basal induction (Fig. 7A). Most interestingly, we found that elevated IL-6 (Fig. 7B) and IL-8 (Fig. 7C) induction was significantly reduced in the cells treated with LPS + S1P in the presence of suramin. Taken together, this data demonstrates that S1P1 and/or S1P3 signaling cooperates with TLR4 signaling to increase pro-inflammatory cytokine production in HGEC.
Figure 6.

S1P1 is specifically involved in the production of cytokines in gingival epithelial cells. TLR4-normal epithelial cells were silenced for S1P1 or a scramble (scr) gene. The reaction was carried out as described in the Materials and methods section. After 48 h of silencing, the cells were challenged with LPS (1 μg/mL) in the presence of S1P (100 nM) for 24 h. The expression of S1P1 was determined by real-time PCR performed with an ABI 7500 system following the silencing (A). The cytokines IL-8 (B) and IL-6 (C) were determined in culture supernatants by Luminex technology. TLR4-normal epithelial cells were challenged with LPS (1 μg/mL), SEW (3 μM), or a combination of LPS + SEW for 24 h at 37°C. Production of IL-6 was determined in the cell culture supernatants by ELISA following SEW, LPS or LPS + SEW stimuli (D). The cells were incubated with pertussis toxin (PTx) prior to LPS and S1P challenge. The induction of IL-6 (E) and IL-8 (F) was determined in culture supernatants by Luminex. Data are shown as means ± SD of triplicate determinations. Statistical significance, declared at p <0.05 for molecule induction or reduction, is shown by asterisks or an asterisk, respectively.
Figure 7.

S1P3 is also involved in the cytokine production. TLR4-normal cells were treated with 0.2, 0.4, 0.5, 1, or 2 mg/mL suramin or medium only for 8 h. Subsequently, the production of IL-6 was determined by ELISA (A). The cells were challenged with LPS + S1P for 8 h after 1 h of suramin (0.4 mg/mL) treatment. The IL-6 (B) and IL-8 (C) induction was measured by ELISA. Statistical significance was declared at p <0.05, and cytokine induction or reduction is shown by an asterisk or asterisks, respectively.
ERK1/2 activation was increased by LPS and S1P
It has also been reported that the extracellular signal-regulated kinase 1/2 (ERK1/2) involves both TLR4 [28] and S1P1 signaling in the mammalian cell signaling pathways [29]. Thus, we determined whether the elevated ERK1/2 activation by LPS is increased with S1P in the cells. Initially, we determined cytokine production in the cells challenged with LPS and S1P after blocking activation of ERK1/2 with U0126. The increased IL-6 (Fig. 8A) and IL-8 (Fig. 8B) production was abrogated with the pharmacological inhibitor U0126. We next examined the activation level of ERK1/2 after LPS or LPS + S1P challenge. The cells were challenged with LPS in the presence or absence of S1P for 30 min and, alternatively, the cells were challenged with LPS + S1P for 30 min after blocking ERK1/2 phosphorylation with U0126 for 2 h. LPS-challenged cells were slightly increased in ERK1/2 phosphorylation (pERK1/2), but LPS + S1P-challenged cells markedly increased the level of pERK1/2. Moreover, U0126 dramatically abrogated pERK1/2 levels in the cells following LPS + S1P treatment (Fig. 8C). In addition, the pERK1/2 level was not significantly increased in cells challenged with S1P as compared to media only (data not shown). This data demonstrates that S1P enhances the TLR4 signaling pathway by increasing the level of ERK1/2 phosphorylation and that activation of the S1P1 and S1P3 signaling pathways is required for this cooperative enhancement of the pro-inflammatory cytokine production in epithelial cells.
Figure 8.
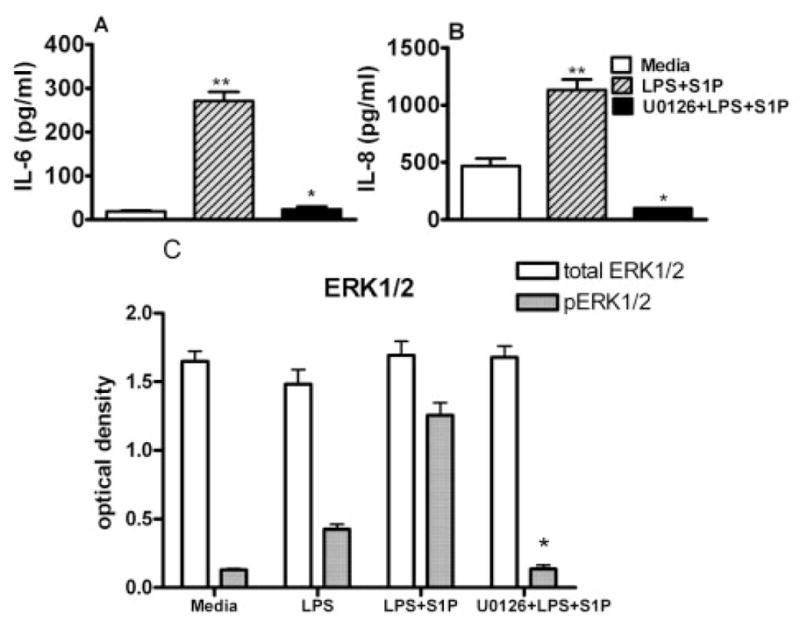
ERK1/2 activation was enhanced by S1P in LPS-challenged cells. The cells were incubated with inhibitory reagent against ERK1/2 (U0126) prior to LPS and S1P challenge. The induction of IL-6 (A) and IL-8 (B) was determined in culture supernatants by Luminex technology. The level of phosphorylated ERK1/2 was determined by FACE (Active Motif), as described in the Material and methods section, in cells following LPS or S1P/LPS challenge (C). Data are presented as means ± SD of triplicate determinations. Statistically significant (p <0.05) cytokine induction is indicated by two asterisks, and significantly reduced phosho-ERK/12 or induction of cytokines is indicated by an asterisk.
Discussion
Oral mucosal epithelial cells are considered part of the innate immune system since they can induce inflammatory cytokines and express all TLR except TLR8 [6]. TLR play a critical role in the induction of innate, inflammatory and immune responses [11]. In this study, we have shown that the production of inflammatory cytokines including IL-6 and IL-8 was increased by LPS in the presence of S1P. Furthermore, these increases occurred in TLR4-normal epithelial cells but not in TLR4-diminished cells. We also showed that expression of S1P1 and S1P3 was reduced in TLR4-diminished cells compared to TLR4-normal cells, and suggest that LPS induction of S1P1 and/or S1P3 influences the production of inflammatory cytokines in epithelial cells. Accumulating evidence suggests that alternative receptors including GPCR [30] are involved inTLR4 signaling. Medzhitov and coworkers suggest that GPCR play a critical role in the initiation of TLR signaling [30]; however, it is unclear which GPCR are involved. Our data show that the inflammatory cytokine production was only moderately increased by LPS-stimulated TLR4 activation, but significantly induced by the combined LPS and S1P challenge of epithelial cells (Fig. 1).
This biological effect observed for LPS and S1P challenge did not occur following the combination of the specific TLR2 agonist (FSL-1) with S1P challenge (Fig. 2). We also report that stimulating either the IL-R1, which shares signaling and structural similarities with TLR4 [23], or TLR2 did not result in increased S1P1 expression (Fig. 4A). In contrast, the S1P1 expression increased following LPS or S1P stimulation for 24 h (Fig. 4D). Moreover, we found that other S1P family receptors including S1P2, S1P3 and S1P5 were expressed by human epithelial cells (Fig. 4A). Although S1P1 and S1P2 have opposing effects [31], they are both triggered by S1P, and the resulting cellular response depends on the relative concentrations of these receptors. Costimulation with LPS alters the ratio of S1P1 to S1P2 in favor of S1P1 and explains why LPS and S1P together increase cytokine production. Our study demonstrated that cytokine levels were reduced but not abrogated in the cells challenged with LPS + S1P after silencing TLR4 (Fig. 5), which suggests that LPS could be recognized by other receptors in addition to TLR4 [32], such as RP105 [32, 33]. Despite finding that our primary gingival epithelial cells also express RP105 (data not shown), the present study shows that TLR4 is an important receptor cooperating with S1P receptors to enhance the production of IL-6 or IL-8, as shown in Fig. 5. We found that the elevated cytokine induction was not completely abrogated in the cells challenged with LPS + S1P after reducing S1P1 expression, suggesting that other S1P family receptors are involved in their induction. We therefore blocked S1P3 signaling, as S1P1 and S1P3 are reported to have similar functions [26], and found that S1P3 signaling also plays a role in cytokine production following challenge with LPS + S1P (Fig. 7). Together, the data demonstrates that S1P1 and S1P3 cooperate with LPS signaling to enhance the production of pro-inflammatory cytokines in human epithelial cells (Fig. 6, 7).
Our current and previous results consistently reveal that IL-8 induction was greater than that of IL-6 in epithelial cells [6, 15]. It is well known that IL-8 is a critical chemokine for neutrophil recruitment, and epithelial cells need professional phagocyte assistance to combat bacterial challenge, which may explain the greater IL-8 production compared to IL-6. Our results showed that MAPK activity such as ERK1/2 was minimally increased in the cells challenged with LPS; however, its activation was significantly increased with LPS in the presence of S1P, suggesting that TLR4 signaling up-regulates the function of S1P receptors, including S1P1 and S1P3 expression, to enhance cytokine increase. We confirmed that ERK1/2 plays an important role in the regulation of cytokines in LPS + S1P-challenged cells, since blocking ERK1/2 activity reduced the cytokine levels (Fig. 8).
It is our observation that primary HGEC express more TLR2 than TLR4 [6]; thus, the TLR4 receptor may require other coreceptors and adaptor molecules to induce significant amounts of pro-inflammatory cytokines. Another reason could be the lack of outer-membrane CD14 in human epithelial cells since we have previously shown that membrane-bound CD14 is not expressed by these cells [15] and CD14 has been shown to be required for LPS recognition [34]. So, TLR4 signaling requires other molecules including S1P1 and/or S1P3 to increase the cytokine production.
In conclusion, remarkable progress has been made in identifying TLR4 ligands and how the receptors activate intracellular pathways by recruiting multiple adaptor molecules. Here, we report that TLR4 signaling requires additional mediators such as the S1P1 and/or S1P3 GPCR receptors to increase cytokine production. Overall, our results were consistent with previous work showing that GPCR have a role in LPS signaling [30]. Analysis of how TLR4 cooperates with S1P1 and S1P3 is still in progress. This is the first report showing that LPS-induced TLR4 signaling cooperates with the S1P1 and S1P3 network to induce pro-inflammatory cytokines in HGEC.
Materials and methods
Cell isolation and culture
HGEC, with University of Louisville ethical board approval, were obtained from healthy patients after third molar extraction. The gingiva was treated with 0.025% trypsin and 0.01% EDTA overnight at 4°C and HGEC were isolated as previously described [35]. Briefly, the cell suspension was centrifuged at 120 × g for 5 min, and the pellet was suspended in K-SFM medium (Invitrogen) containing 10 μg/mL insulin, 5 μg/mL transferrin, 10 μM 2-mercaptoethanol, 10 μM 2-aminoethanol, 10 mM sodium selenite, 50 μg/mL bovine pituitary extract, 100 U/mL penicillin/streptomycin and 50 ng/mL fungizone. The cells were seeded in 60-mm plastic tissue culture plates coated with type-I collagen, and incubated with 5% CO2 and 95% air at 37°C. When the cells reached subconfluence, they were harvested and subcultured as described [15].
Challenge assays
Primary epithelial cultures were harvested at the fourth passage, seeded at a density of 0.5 × 105 cells/6-well culture plate coated with type-I collagen, and maintained in 2 mL medium. When they reached 100% confluence, the cells were washed twice with fresh medium and 2 mL medium was added. The cells were challenged with E. coli LPS (1 μg/mL; Invivogen), S1P (100 nM; Biomol), SEW (3 μM; BioMol) or FSL-1 (1 μg/mL; InvivoGen). To examine the effect of ERK1/2, cells were pretreated with the ERK1/2 inhibitor, U0126 (25 μM; Cell Signal), for 2 h prior to LPS and S1P stimulation. Additionally, the cells were pretreated with pertussis toxin (100 ng/mL; Calbiochem), an inhibitor of Gi/o heterotrimeric G protein, for 2 h. Subsequently, the medium was replaced with fresh medium containing 10 ng/mL pertussis toxin and stimulated with LPS in the presence of S1P. To exclude intracellular S1P synthesized by S1P, the TLR4-normal cells were treated with an SK inhibitor (1 μM; Calbiochem) 1 h prior to the LPS + S1P challenge. In order to examine the function of S1P3 signaling in the cytokine induction, TLR4-normal cells were treated with suramin sodium (0.4 mg/mL; Biomol) 1 h prior to the assay; then the cells were challenged with LPS + S1P for 8 h. The culture supernatant was then collected. The secreted cytokines were quantified by Luminex 100 technology using a multiplex of cytokines including IL-6 and IL-8 (Upstate).
Real-time PCR
Total RNA was extracted from cultured cells by using TRIzol® reagent (Invitrogen). Of total RNA samples, 10 μg was used to perform first-strand cDNA synthesis using the High-Capacity cDNA Archive kit (Applied Biosystems). Real-time PCR was performed by using 100 ng cDNA with an ABI 7500 system (Applied Biosystems) in the presence of TaqMan DNA polymerase. The sense and anti-sense primers used to detect the gene expression of human S1P1–5, TLR4 and GAPDH were purchased from Applied Biosystems. The qPCR reaction was performed by using a universal PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions.
Silencing TLR4 and S1P1
Primary epithelial cultures at the fourth passage were harvested, seeded at a density of 0.5 × 105 cells/6-well culture plate coated with type-I collagen, and maintained in 2 mL medium until they reached 50–70% confluence. Subsequently, the epithelial cells were transfected with 100 pmol of siRNA to TLR4 (Dharmacon), siRNA to laminin (Dharmacon) or 1 μg of lentiviral vector carrying siRNA to S1P1 [36] with FuGene 6 reagent (Roche). Briefly, 3 μL FuGene 6 was added to 95 μL of serum-free medium, followed by the addition of 100 pmol anti-sense RNA. The transfection mixture was incubated at room temperature for 15 min and then added to the cell suspension for transfection overnight. After 48 h, the cells were challenged with LPS + S1P for 24 h following the transfection.
Western blot analysis
HGEC (8 × 106 cells) in 75-T flasks were pretreated with LPS (1 μg/mL) or medium only for 4 h. At the indicated time points, cells were washed with cold PBS and then lysed on ice for 30 min in 100 μL RIPA lysis buffer (Sigma-Aldrich) containing protease (Roche) and phosphatase inhibitors (Sigma-Aldrich). The whole-cell lysate was passed three times through a 30-gauge needle and then incubated on ice for an additional 30 min. Cell debris was pelleted by centrifugation, and the supernatants were collected and stored at −80°C until assayed. Of total cellular protein, 25 μg was suspended in lithium dodecyl sulfate (LDS) buffer, heated for 10 min at 70°C, resolved by LDS-PAGE and then transferred to polyvinylidene difluoride membranes using the NuPAGE system (Invitrogen). All Western blotting reagents were supplied by Invitrogen except for the Chemiluminescence kit (Pierce). Primary antibodies (anti-phosphorylated and anti-total ERK1/2) and secondary antibody (anti-rabbit IgG; Cell Signaling) were used at 1: 1000 and 1: 2000, respectively, Probing and visualization of immunoreactive bands were performed by following the manufacturer’s protocol. Densitometer scans of the blots were performed using the Kodak Image 4000M.
LPS-induced S1P1 expression
Proteins extracted from cells treated with or without LPS for 24 h were immunoprecipitated with monoclonal S1P1 antibody (a gift from C.-Y. Lin, Georgetown University). The immunocomplexes were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. After blocking with 5% nonfat milk, the membranes were blotted with anti-human S1P1 antibody. Subsequently, the nitrocellulose membranes were incubated with HRP-conjugated secondary antibody (Pierce) and visualized using the ECL method (Amersham Biosciences).
Phosphorylated ERK1/2 levels
The cells were seeded into collagen-coated 96-well plates and challenged with LPS + S1P in the presence or absence of 25 μM U0126, which was incubated for 2 h prior to the challenge assay, or with LPS alone for 30 min. Next, the challenged cells were rapidly fixed with 100 μL 8% formaldehyde to preserve activation-specific protein modifications. The phospho-ERK1/2 level was determined with Fast Activated Cell-based ELISA (FACE; Active Motif) by following the company’s protocol.
Flow cytometry
TLR4-normal and -diminished gingival epithelial cells were washed three times with PBS and 1 × 106 cells were stained with 0.5 μg phycoerythrin (PE) conjugated to anti-human TLR4 antibody or to isotype control, mouse IgG1 (eBioscience, San Diego, CA), in 100 μL total staining buffer. The cells were analyzed by flow cytometry using a BD FACSCalibur and the CellQuest software.
Acknowledgments
This work was supported by United States Public Health Service, National Institutes of Health, NIDCR grant DE017384 to D.F.K.
Abbreviations
- GPCR
G protein-coupled receptors
- HGEC
human gingival epithelial cell
- pERK1/2
ERK1/2 phosphorylation
- S1P
sphingosine 1-phosphate
- S1P1–5
S1P receptors 1–5
- SK
sphingosine kinase
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Rimoldi M, Chieppa M, Salucci V, Avogadri F, Sonzogni A, Sampietro GM, Nespoli A, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol. 2005;6:507–514. doi: 10.1038/ni1192. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Kaisho T, Akira S. Critical roles of Toll-like receptors in host defense. Crit Rev Immunol. 2000;20:393–405. [PubMed] [Google Scholar]
- 4.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the DrosophilaToll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 6.Kinane DF, Shiba H, Stathopoulou PG, Zhao H, Lappin DF, Singh A, Eskan MA, et al. Gingival epithelial cells heterozygous for Toll-like receptor 4 polymorphisms Asp299Gly and Thr399Ile are hypo-responsive to Porphyromonas gingivalis. Genes Immun. 2006;7:190–200. doi: 10.1038/sj.gene.6364282. [DOI] [PubMed] [Google Scholar]
- 7.Kaisho T, Akira S. Regulation of dendritic cell function through Toll-like receptors. Curr Mol Med. 2003;3:373–385. doi: 10.2174/1566524033479726. [DOI] [PubMed] [Google Scholar]
- 8.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 9.Nagler-Anderson C. Man the barrier! Strategic defences in the intestinal mucosa. Nat Rev Immunol. 2001;1:59–67. doi: 10.1038/35095573. [DOI] [PubMed] [Google Scholar]
- 10.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg. 2004;187:11S–16S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 11.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 12.Canturk NZ, Esen N, Vural B, Canturk Z, Kirkali G, Oktay G, Solakoglu S. The relationship between neutrophils and incisional wound healing. Skin Pharmacol Appl Skin Physiol. 2001;14:108–116. doi: 10.1159/000056340. [DOI] [PubMed] [Google Scholar]
- 13.Duncan MR, Berman B. Differential regulation of glycosaminoglycan, fibronectin, and collagenase production in cultured human dermal fibroblasts by interferon-alpha, -beta, and -gamma. Arch Dermatol Res. 1989;281:11–18. doi: 10.1007/BF00424266. [DOI] [PubMed] [Google Scholar]
- 14.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Eskan MA, Hajishengallis G, Kinane DF. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun. 2007;75:892–898. doi: 10.1128/IAI.01604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida Y, Shiba H, Komatsuzawa H, Hirono C, Ashikaga A, Fujita T, Kawaguchi H, et al. Irsogladine maleate influences the response of gap junctional intercellular communication and IL-8 of human gingival epithelial cells following periodontopathogenic bacterial challenge. Biochem Biophys Res Commun. 2005;333:502–507. doi: 10.1016/j.bbrc.2005.05.147. [DOI] [PubMed] [Google Scholar]
- 17.Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee H, Goetzl EJ, An S. Lysophosphatidic acid and sphingosine 1-phosphate stimulate endothelial cell wound healing. Am J Physiol Cell Physiol. 2000;278:C612–C618. doi: 10.1152/ajpcell.2000.278.3.C612. [DOI] [PubMed] [Google Scholar]
- 19.McGiffert C, Contos JJ, Friedman B, Chun J. Embryonic brain expression analysis of lysophospholipid receptor genes suggests roles for s1p(1) in neurogenesis and s1p(1–3) in angiogenesis. FEBS Lett. 2002;531:103–108. doi: 10.1016/s0014-5793(02)03404-x. [DOI] [PubMed] [Google Scholar]
- 20.Lee OH, Kim YM, Lee YM, Moon EJ, Lee DJ, Kim JH, Kim KW, Kwon YG. Sphingosine 1-phosphate induces angiogenesis: Its angiogenic action and signaling mechanism in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1999;264:743–750. doi: 10.1006/bbrc.1999.1586. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G, Contos JJ, Weiner JA, Fukushima N, Chun J. Comparative analysis of three murine G-protein coupled receptors activated by sphingosine-1-phosphate. Gene. 1999;227:89–99. doi: 10.1016/s0378-1119(98)00589-7. [DOI] [PubMed] [Google Scholar]
- 22.French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 24.Windh RT, Lee MJ, Hla T, An S, Barr AJ, Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J Biol Chem. 1999;274:27351–27358. doi: 10.1074/jbc.274.39.27351. [DOI] [PubMed] [Google Scholar]
- 25.Pyne S, Pyne NJ. The differential regulation of cyclic AMP by sphingomyelin-derived lipids and the modulation of sphingolipid-stimulated extracellular signal regulated kinase-2 in airway smooth muscle. Biochem J. 1996;315(Pt 3):917–923. doi: 10.1042/bj3150917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waeber C, Blondeau N, Salomone S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 2004;17:365–382. doi: 10.1358/dnp.2004.17.6.829028. [DOI] [PubMed] [Google Scholar]
- 27.Ancellin N, Hla T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J Biol Chem. 1999;274:18997–19002. doi: 10.1074/jbc.274.27.18997. [DOI] [PubMed] [Google Scholar]
- 28.Goral J, Kovacs EJ. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J Immunol. 2005;174:456–463. doi: 10.4049/jimmunol.174.1.456. [DOI] [PubMed] [Google Scholar]
- 29.Lee M-J, Evans M, Hla T. The inducible G protein-coupled receptor Edg-1 signals via the G(i)/mitogen-activated protein kinase pathway. J Biol Chem. 1996;271:11272–11279. doi: 10.1074/jbc.271.19.11272. [DOI] [PubMed] [Google Scholar]
- 30.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 31.Inoki I, Takuwa N, Sugimoto N, Yoshioka K, Takata S, Kaneko S, Takuwa Y. Negative regulation of endothelial morphogenesis and angiogenesis by S1P2 receptor. Biochem Biophys Res Commun. 2006;346:293–300. doi: 10.1016/j.bbrc.2006.05.119. [DOI] [PubMed] [Google Scholar]
- 32.Kimoto M, Nagasawa K, Miyake K. Role of TLR4/MD-2 and RP105/MD-1 in innate recognition of lipopolysaccharide. Scand J Infect Dis. 2003;35:568–572. doi: 10.1080/00365540310015700. [DOI] [PubMed] [Google Scholar]
- 33.Ogata H, Su I, Miyake K, Nagai Y, Akashi S, Mecklenbrauker I, Rajewsky K, et al. The Toll-like receptor protein RP105 regulates lipopolysaccharide signaling in B cells. J Exp Med. 2000;192:23–29. doi: 10.1084/jem.192.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akashi S, Ogata H, Kirikae F, Kirikae T, Kawasaki K, Nishijima M, Shimazu R, et al. Regulatory roles for CD14 and phosphatidylinositol in the signaling via toll-like receptor 4-MD-2. Biochem Biophys Res Commun. 2000;268:172–177. doi: 10.1006/bbrc.2000.2089. [DOI] [PubMed] [Google Scholar]
- 35.Shiba H, Venkatesh SG, Gorr SU, Barbieri G, Kurihara H, Kinane DF. Parotid secretory protein is expressed and inducible in human gingival keratinocytes. J Periodont Res. 2005;40:153–157. doi: 10.1111/j.1600-0765.2005.00781.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee JF, Zeng Q, Ozaki H, Wang L, Hand AR, Hla T, Wang E, Lee MJ. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J Biol Chem. 2006;281:29190–29200. doi: 10.1074/jbc.M604310200. [DOI] [PubMed] [Google Scholar]