

Abstract
The mouse model of liver ischemia and reperfusion injury has proven to be valuable for our understanding of the role that reactive oxygen and nitrogen metabolites play in postischemic tissue injury. This methods paper provides a detailed protocol for inducing partial liver ischemia followed by reperfusion. Liver ischemia is induced in anesthetized mice by cross-clamping the hepatic artery and portal vein for varying lengths of time resulting in deprivation of blood flow to approximately of 70% of the liver. Restoration of blood flow to the ischemic lobes enhances superoxide production concomitant with a rapid and marked decrease in the bioavailability of nitric oxide resulting in alterations in the redox state of the liver in favor of a more oxidative environment. This hepatocellular oxidative stress induces the activation of oxidant-sensitive transcription factors followed by the upregulation of pro-inflammatory cytokines and mediators that ultimately lead to liver injury. This model can be induced in any strain or sex of mouse and requires 1-2 months of practice to become proficient in the surgery and animal manipulation. The role of different reactive metabolites of oxygen and nitrogen may be evaluated using genetically-engineered mice as well as selective molecular, cellular and/or pharmacological agents.
Keywords: free radicals, NFkB, neutrophils, cytokines
Introduction
Few aspects of vascular pathobiology have garnered more attention over the past 3 decades than has ischemia and reperfusion injury. Indeed, it has been almost 30 years since Granger, Rutili and McCord first reported that much of the post-ischemic tissue damage observed in a feline model of intestinal ischemia and reperfusion (I/R) was mediated directly or indirectly by the superoxide anion radical (1). Since that publication, a voluminous literature has accumulated implicating both reactive oxygen and nitrogen species as important mediators and modulators of post-ischemic tissue injury in a number of different organ systems including the gut, liver, heart, kidney, brain and lung to mention just a few (2) One organ system that has produced some of the most detailed information regarding the mechanisms by which reactive metabolites of oxygen and nitrogen mediate or modulate tissue damage following I/R is the liver. It is well appreciated by the clinical community that liver I/R injury is a major complication associated with liver transplantation and resectional surgery as well as hemorrhagic or endotoxin shock and thermal injury. A large body of experimental data using rat or mouse models of I/R suggests that post-ischemic tissue injury is initiated by rapid alterations in the generation and/or steady-state levels of reactive oxygen and nitrogen species (3-5). One of the earliest events associated with reperfusion of ischemic tissue is a remarkable dysfunction of sinusoidal endothelial cells (SECs) characterized by profound decreases in the steady state levels of endothelial cell nitric oxide synthase (eNOS)-derived nitric oxide (NO) (6). This decline in NO bioavailability occurs within the first few minutes of reperfusion and appears to be due to decreased synthesis of NO, enhanced inactivation of NO by the overproduction of certain reactive oxygen species (ROS) or both. Co-incident with the fall in NO production is an equally rapid increase in the production of ROS such as superoxide (O2-) and hydrogen peroxide (H2O2) (5;7-11). Hepatocyte-associated xanthine oxidase, mitochondria and Kupffer cell- and/or sinusoidal endothelial cell-associated NADPH oxidase have all been implicated as important generators of ROS in the acute or early phase of I/R-induced ischemic liver (5;7-12). Because NO is well-known to interact with and decompose certain ROS such as O2-, hydroxyl radical (OH·), and ferryl hemoproteins, I/R-induced over production of ROS coupled to decreased steady state levels of NO results in a rapid alteration in the redox state of the different cells within the liver in favor of a more oxidative environment (13-15).
Early studies suggested that post-ischemic liver injury was mediated directly by the overproduction of injurious oxidizing agents derived from oxygen ie ROS. However, more recent data suggest that oxidative stress may injure the tissue indirectly by activating redox-sensitive transcription factors such as NF-kB and activating protein-1 (AP-1) thereby up-regulating a variety of potentially injurious cytokines, chemokines and pro-inflammatory mediators (5;10;16)(17). Inherent in this hypothesis is the prediction that exogenous antioxidants or NO may attenuate post-ischemic tissue injury via their ability to prevent or attenuate transcription factor activation and the consequent expression of the different injurious mediators. Indeed, a number of different studies, using pharmacologic interventions or genetic approaches have demonstrated protective effects with administration of certain nonenzymatic or enzymatic antioxidants prior to the induction of liver ischemia (9;10;18-20). Furthermore, the mouse model of liver I/R has been especially informative in demonstrating that endogenous or exogenous NO protects the liver from post-ischemic injury by normalizing the redox potential of the liver, attenuating the transcriptional activation of a number of injurious and pro-inflammatory genes, limiting hypoxic insult and/or by interfering with platelet- and leukocyte-endothelial cell interactions (18;21-24)
Because one cannot completely recapitulate the complexity of the physiological setting in vitro, it is becoming increasingly important for investigators who wish to study the normo- and/or patho-biology of reactive oxygen and nitrogen metabolism to work in a physiologically-relevant environment. Thus, the objective of this methods paper is to provide a detailed protocol describing the mouse model of I/R. It is our hope that this model will be useful for investigators interested in exploring the many different molecular, cellular and physiological mechanisms associated with vascular pathobiology, free radical metabolism and inflammation in vivo.
Materials
Animals
8-12 week old wild type or genetically-engineered mice of the same sex. Important: Although the strain of the mouse is not critical for this model, gender differences may exist in response to liver ischemia and reperfusion injury. Therefore, it is recommended that at least 7 mice of the same sex and strain be used for each experimental group. All animals are maintained on a standard laboratory diet with free access to food and water until the time of the experiment. All experimental procedures must comply with the Guide for the Care and Use of Laboratory Animals (revised 1996), approved by the Council of The American Physiological Society, and with federal and state regulations.
Reagents
Ketamine (Ketaset III 100mg/mL; Fort Dodge Animal Health, Fort Dodge, IO cat. no. 44016)
Xylazine (Xylazine-20 Injection 20mg/mL; The Butler company, Columbus, OH. cat. no. WAB10945)
0.9% Sodium Chloride
Heparin Sodium (10,000 units/mL, Baxter, Deerfield, IL. cat. no. 462-274-01)
Sterile Alcohol Prep Pad (Fisher, Pittsburgh, PA. cat. no 06-669-62)
Povidone-Iodine Swabstick (Professional Disposables, Orangeburg, NY. cat. no. S41350)
10% phosphate-buffered Formalin
Hematoxylin
Eosin
Trizol (Invitrogen, Carlsbad, CA cat. no. 15596-018)
DNAse (Invitrogen, Carlsbad, CA)
MuLV reverse trascriptase (Applied Biosystems, Foster City, CA)
Mouse IL-1 alpha/IL-1F1 ELISA Kit (Quantikine, Minneapolis, MN, cat. no. MLA00)
Mouse TNF-alpha/TNFSF1A ELISA Kit (Quantikine, Minneapolis, MN, cat. no. MTA00).
Surgical Instruments
Tissue Scissors (blunt, 11.5cm; Fine Science Tools, Foster City, CA. cat. no. 14072-10)
Hemostats (Micro-Mosquito, Straight, 12cm; Fine Science Tools, Foster City, CA. cat. no. 13010-12)
Atraumatic clip (Micro Serrefine, Jaw Length 6mm, Jaw Width 1mm, Total Length 17mm, Jaw Pressure 110gr; Fine Science Tools, Foster City, CA. cat. no. 18055-05)
Micro clip applicator with lock (Total Length 15cm; Fine Science Tools, Foster City, CA. cat. no. 18056-14)
Equipment
25 gauge needle
1mL syringe
Gause Sponges (Fisher, Pittsburgh, PA. Cat. no. 22-362-178)
Cotton-Tipped Swabs (Fisher, Pittsburgh, PA. cat. no. 23-400-100)
Suture with needle (4-0 silk, C-3, 45cm, Ethicon, Somerville NJ. cat. no. 736G)
Serum Separator Tube (Becton Dickson, Franklin Lakes, NJ. cat. no. 365956)
JB4 plastic mounting media (Polysciences, Warrington, PA).
Heat lamp and Temperature controller
Electric razor
BioRad iCycler
Methods
Surgery
Fasted (16-18 hours) mice are anesthetized with ketamine (100mg/kg) and xylazine (10mg/kg) by intraperitoneal (ip) injection using a 25 gauge needle into the lower right quadrant of the mouse.
Once the animal is anesthetized, it is immobilize by taping the animal’s legs and arms to a clean flat surface (e.g. washable plastic) with the abdomen facing up. The abdomen is then shaved up to the xiphoid process and is cleaned by swabbing the skin with a 70% ethanol solution followed by a commercially-available betadine preparation to prevent the introduction of bacteria during the surgical procedure.
A small pair of scissors are then used to open the abdomen at the midline to expose the abdominal contents. This incision should be approximately 3 cm, beginning at the mid-adomen and ending at the xiphoid process. Important: Be careful not to extend the incision beyond xiphoid process as there a number of large vessels within the skin and muscle layers which can bleed extensively. Attach hemostats to either side of the incision and xiphoid process and pull the abdominal cavity open (Figure 1). Hemostats can be placed on stacks of gauze to raise the abdominal walls and allow for easier visualization.
Moisten two pieces of gauze and place on the right side of the incision (left side of mouse). Using two moistened cotton swabs, carefully externalize the intestines as gently as possible from the cavity and place them on pre-moistened (sterile 0.9% saline) gauze to expose the portal vein and associated structures. Cover the intestine with the second piece of gauze to prevent drying. Carefully separate the quadrate lobe from its attachment with the left lateral lobe with scissors to better reveal the portal triad (Portal vein, Hepatic artery and Bile duct) (Figure 1). It may be necessary to first lift the median and left lateral lobes against the diaphragm to identify the connection between the left lateral and quadrate lobe.
Now place an atraumatic clip across the portal vein, hepatic artery, and bile duct just above the branching to the right lateral lobe (Figure 1). The median and left lateral lobes (approximately 70% of the liver) should quickly show significant blanching ie they should quickly change from their normal reddish brown color to a pale brown or cream color. Important: If the lobes do not show significant blanching (Figure 1), remove the clip and reapply. Depth of clamp placement can be critical as the hepatic artery (which you cannot see) can be significantly deeper than the portal vein and require deeper clamp placement for occlusion. Important: the clamps are very fragile and can become stretched with repeated uses decreasing their clamping force over time resulting in variable degrees of ischemia. Extreme care is therefore necessary when applying and removing clamps and specific clamp holder should always be used.
Following clamp placement, replace the intestines into the abdominal cavity carefully, add 500 μl of 10 U/ml heparinized-saline directly into the peritoneal cavity via syringe and cover the incision with well moistened gauze. Place the animal under a heat lamp to maintain body temperature at 37°C and monitor closely during the ischemic period making sure the covering gauze remains moist with saline. During this time, additional anesthesia may be required by placing a small volume (50 ul) of anesthetic into the open abdomen.
Following the desired period of ischemia, remove the clamp carefully and administer 500 μL of sterile saline to the peritoneal cavity to replenish any fluid loss during surgery. Important: Verify reperfusion visually as the blanched color of the ischemic lobes should begin to show restoration of the normal reddish-brown color within a few seconds of clamp removal. No reflow can occur especially following extended periods of ischemia, potentially causing significant variations in results.
Close the abdomen by suturing the muscle layer and then the skin with 4-0 silk sutures and allow the animal to recover for the required reperfusion period.
Immediately following the reperfusion period, blood may be collected from the anesthetized mouse as described below for serum preparation.
When blood samples have been obtained the mouse is euthanized by cutting the diaphram.
Representative samples of the post-ischemic liver (and/or bypass lobes) are immediately frozen in liquid nitrogen and stored at -80°C.
Figure 1.

Drawing depicting a ventral view of the normal mouse liver in which the left lateral and median lobes have been reflected back to expose the portal vein (blue), hepatic artery (red) and common bile duct (green). Cross clamping the portal vein and hepatic artery induces ischemia to the left lateral and median lobes of the liver.
Serum Preparation, Quantification of Liver Transaminase Activity and Cytokine Levels
Serum levels of alanine aminotransferase (ALT) as well as other liver-specific transaminases are quantitative indices of liver damage and can be quantified in serum obtained from mice subjected to liver I/R. At varying times following ischemia and/or reperfusion, blood is removed from the inferior vena cava following 45 or 90 minutes of ischemia and different times of reperfusion using a 25 gauge syringe attached to a 1 ml syringe and placed in a serum separator tube (Becton Dickson, Franklin Lakes, NJ).
The samples are allowed to clot on ice for approximately 10 minutes after which they are centrifuged at 4,000 × g for 10 minutes. Serum is removed and ALT is measured using a kit from Sigma Chemical Corporation (St. Louis, MO). Data are presented as units per liter (U/L) at 37°C. Aliquots (50 or 100 ul) of serum may be frozen prior to measurement of ALT at -80°C. Repeated freeze-thaw cycles should be avoided as ALT may become inactivated.
Serum levels of TNF-α as well as other cytokines are quantified using an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s specifications (Quantikine, R&D Systems, Abingdon, UK). Data are presented as pg/ml of serum cytokines
Histopathology
Following euthanasia, representative pieces of ischemic, I/R or bypass lobes are quickly removed and fixed in ice cold 10% phosphate-buffered formalin for 24 hours at 4°C.
The tissue is then partially dehydrated with ethanol and embedded in JB4 plastic mounting media using standard histological methods.
Five micrometer sections cut and stained with hematoxylin and eosin. Following staining, the sections are scored in a blinded fashion as described below.
NFkB Activation and Nuclear Translocation
Nuclear Extract Preparation
Place 5mls of cold Solution A (see below) into pre-chilled Dounce homogenizer.
Add frozen liver (one median lobe) and homogenize w/ ~ 10 strokes.
Decant homogenized liver into a cold 15ml conical centrifuge tube and centrifuge for 30sec @ 2000rpm (4°C).
Decant the supernatant into new cold 15 ml conical tube.
Incubate for 5min on ice.
Centrifuge for 5min @ 5000rpm (4°C).
Decant supernatant and keep nuclear pellet.
Resuspend the nuclear pellet in 400ul Solution B (see below) thoroughly as it may be difficult suspend.
Transfer the nuclear suspension to a cold 1.5ml microfuge tube.
Extract the protein by incubating suspension for 20 min on rotating platform at (4°C).
Centrifuge the extract for 15 sec @ 12,000 rpm.
Decant the supernatant into new cold microfuge tube.
Remove 50ul aliquots and place into cold 0.5ml microfuge tubes and flash freeze in liquid nitrogen.
Store extracts @ -80°C.
Determine protein concentration with Pierce BCA reagent.
| Solution A | Solution B |
| 0.6% NP-40 | 25% glycerol |
| 150mM NaCl | 20mM Hepes (pH 7.9) |
| 10mM Hepes (pH 7.9) | 420mM NaCL |
| 1mM EDTA | 1.2mM MgCl |
| 0.5mM PMSF (add Fresh!) | 0.2mM EDTA |
| 0.5mM DTT (add Fresh) | |
| 0.5mM PMSF (add Fresh) | |
| protein inhibitor cocktail (add fresh according to manufacturer) |
Oligonucleotide Prepartion and Eletrophoretic Mobility Shift Assay (EMSA)
The NF-κB consensus oligonucleotide 5’-AGTTGAGGGGACTTTCCCAGGC is end labeled with [γ-32P]ATP using T4 polynucleotide kinase according to the manufacturer’s instructions (Promega). Labeled oligonucleotide (35 fmol) is incubated with 20 μg nuclear extracts for 10 min on ice in binding buffer [1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris · HCl (pH 7.5), 0.05 μg/μl poly(dI-dC) · poly(dI-dC), and 4% glycerol] in a total volume of 35 μl. A 50-fold molar excess of nonlabeled consensus or mutated NF-κB consensus oligonucleotide (Santa Cruz Biotechnology, Santa Cruz, CA) is included in respective reactions.
Samples are incubated for 30 min at 25°C. For supershift studies, antibody specific for either the p50 (Santa Cruz Biotechnologies) or p65 (Rockland, Gilbertsville, PA) subunits are added after the initial incubation on ice. Protein-DNA complexes are resolved on 4% nondenaturing polyacrylamide gels by electrophoresis in 0.5× Tris-borate-EDTA. Gels were dried and visualized by a PhosphorImager (Molecular Dynamics, Hercules, CA). Activation of NF-κB (relative to non-TNF-treated controls) is determined by performing densitometric analysis (ImageQuant software; Molecular Dynamics, Sunnyvale, CA) on shifted bands from scanned autoradiograms.
Cytokine Message Determinations
Liver cytokine message expression is quantified using real time polymerase chain reaction in a BioRad iCycler. Total RNA is extracted from approximately 100mg of liver tissue using the Trizol method according to the manufacturer’s instructions.
One microgram of total RNA is subjected to DNAse treatment according to the manufacturer’s instruction. The DNAse treated RNA is then reverse transcribed using MuLV reverse trascriptase in a two step procedure: 30 minutes at 42°C followed by 5 minutes at 95°C. 200ng of cDNA is then amplified by real time polymerase chain reaction with SYBR Green as the fluorescent indicator (which binds to double stranded DNA) with the designated primer pairs.
The resultant data is analyzed using the BioRad MyIQ software package with samples being normalized against their sham vehicle controls.
Evaluating the roles of superoxide or nitric oxide in liver I/R
To assess specifically the role of superoxide we have utilized genetically engineered polycationic chimeric form of manganese SOD consisting of fusion protein of the mature human Mn-SOD (SOD2) sequence followed by the 26 C-terminal residues of human extracellular SOD (SOD3) (18). This chimeric SOD2/3 fusion proteins binds to the microvascular endothelium as well as the extracellular matrix (25). Mice are treated with SOD2/3 (1000U/kg; i.v.) 15 mins prior ischemia.
The role of eNOS-derived NO (or any other NOS or enzyme) in I/R injury may be evaluated using the genetically engineered eNOS-deficient mouse. In addition, the NOS inhibitor L-nitroarginine methyl ester (L-NAME) (Sigma-Aldrich, St. Louis, MO) may be adminstrered at a dose of 4 mg/kg (iv) or vehicle (0.9% sodium chloride) in a volume of 100 μl 15 minutes prior to ischemia (23;24;26).
Statistical Analysis
All values are presented as mean ± SEM (standard error of mean). Data are analyzed using Students t test or analysis of variance where significance was set at p<0.05.
Results and Discussion
Depriving 70% of the liver of blood flow for 90 minutes followed by varying times of reperfusion induces dramatic and time-dependent increases in liver injury as assessed by increases in serum ALT levels (Figure 2). Important: We have found that addition of heparin during the surgical preparation reduces animal-to-animal variation by maintaining more consistent reperfusion while limiting the “no reflow” problems associated with coagulation. Histopathological evaluation of the livers confirms liver damage showing necrosis in the absence of significant PMN infiltration. These data are consistent with the acute phase of liver injury as described above (Figure 3). One of the earliest molecular events that occurs following liver I/R is the activation and nuclear translocation of the transcription factor NFkB. It has been demonstrated by several different laboratories that I/R-induced activation of NFkB may represent an important pathophysiological event in liver damage (5;10;16)(17). Figure 4 demonstrates that as early as 1 hour following reperfusion, a time when liver injury is minimal, the p50/p65 active heterodimeric transcription factor has translocated into the nuclei of the liver cells. Important: Since the nuclear preparation is obtained from whole liver the vast majority of the nuclei are derived from hepatocytes. This is an important consideration if one wishes to assess the role of NFkB activation in other cell types such as SECs or Kupffer cells (KCs) both of which represent a small population of the total cells in the liver. We as well as others have reported that I/R-induced NFkB activation may induce hepatocellular injury in the acute, PMN-independent phase by promoting the expression of injurious cytokines and mediators. One such cytokine is the TNF-α (TNF).
Figure 2.

Serum alanine aminotransferase (ALT) levels following 90 minutes of ischemia and varying periods of reperfusion. Sham operated animals underwent identical surgical manipulations without clamp placement. * p< 0.05 for I/R (black bar) versus time-matched sham-operated controls (white bar). Data derived from reference 18.
Figure 3.

Histopathology of livers subjected to sham surgery or 90 minutes of ischemia and 6 hours of reperfusion. A. Sham operated control liver. B. Liver subjected to 90 minutes of ischemia and 6 hours of reperfusion. Note the absence of PMN infiltration but the presence of hepatocellular necrosis, vacuolization and pyknotic nuclei.
Figure 4.

Electromobility shift assay (EMSA) demonstrating activation and nuclear localization of the p50/p65 heterodimeric transcription factor NFkB following 90 minutes of ischemia and varying times of reperfusion.
Figure 5 shows the time dependent increase in both message and serum protein levels of TNF following 90 min of ischemia and varying times of reperfusion. As expected, TNF message level increases rapidly preceding enhanced protein expression by 1 hour following reperfusion. Serum protein levels of TNF increase in a time dependent fashion with maximum production at 6 hrs following reperfusion. Because a number of different gene products are up-regulated in response to I/R, a systematic effort to indentify which of the many cytokines and mediators are important in the pathophysiology of post-ischemic tissue damage should be undertaken. Figure 6 demonstrates that pretreatment of mice with a single injection of a TNF monoclonal antibody (mAb) 15 minutes prior to ischemia attenuates I/R-induced liver injury following 90 of ischemia and 6 hours of reperfusion. It is interesting to note the pleiotropic properties of TNF as immuno-neutralization of TNF actually enhances post-ischemic injury at 1 hour of reperfusion. The reasons for these interesting but perplexing activities of TNF are not known at the present time.
Figure 5.

Liver message and serum TNF-α levels following 90 minutes of ischemia and varying periods of reperfusion. * p< 0.05 vs. sham operated control.
Figure 6.
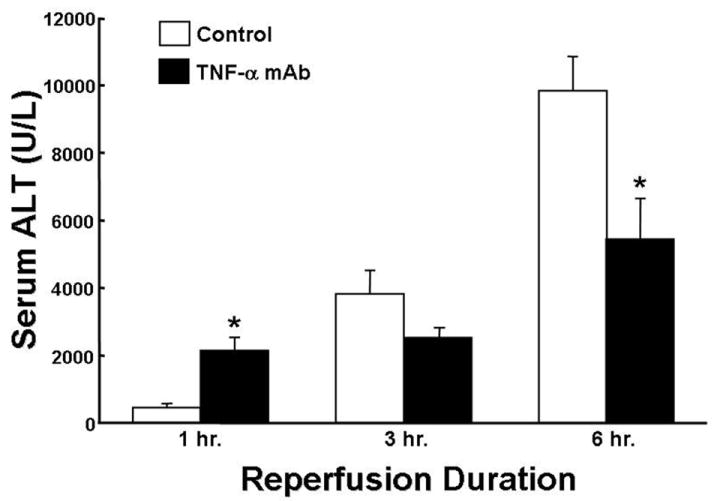
Effect of a monoclonal antibody to mouse TNF-α (200 ug/mouse) on serum ALT levels following 90 minutes of ischemia and varying periods of reperfusion. * p< 0.05 for antibody treated versus vehicle-treated control. Data derived from reference 18.
Numerous investigations have identified ROS as important mediators of reperfusion injury in the liver (5;7-11). In order to assess specifically the role that O2- plays in post-ischemic liver damage following ischemia, we have tested the effectiveness of a genetically engineered fusion protein consisting of the mature human manganese superoxide dismutase (MnSOD; SOD2) sequence followed by the polycationic 26 C-terminal residues of human extracellular SOD (SOD3). This fusion protein (SOD2/3) is membrane impermeable but capable of binding to the surface of sinusoidal and microvascular endothelial cells (estimated half-life of 30 hours) (18;25). We have found that SOD2/SOD3 administration is very effective at attenuating I/R-induced liver injury (Figure 7). Indeed, we have shown, using mice deficient in KCs or NADPH oxidase that KC-derived O2- plays a major role in the pathophysiology of postischemic liver injury (18). The protective effect of SOD2/3 correlates well with its ability to significantly reduce serum levels of TNF-α (Figure 8). The mechanisms by which superoxide may directly or indirectly mediates post-ischemic tissue are not known however we and others have suggested that it may act to rapidly inactivate the important cytoprotective molecule NO. Indeed, if we subject eNOS ko mice to liver I/R we observe a dramatic exacerbation of liver injury as shown in Figure 9 (24;26). Very similar effects are observed when wild type mice are pretreated with the NOS inhibitor L-NAME (data not shown)(24;26). Important: It should be noted that all NOS experiments have to be performed using only 45 min ischemia as longer periods increase dramatically the mortality of the mice. In order to test the hypothesis that superoxide indirectly mediates I/R injury by inactivating NO, eNOS deficient mice were treated with SOD2/3 prior to I/R. We reasoned that SOD2/3 administration should provide no protection to the enhanced liver injury observed eNOS deficient mice if our hypothesis were correct. Indeed, SOD2/3 did not provide any protection to the eNOS ko animals (23). Taken together, these data suggest that eNOS-derived NO acts to limit post-ischemic tissue injury and that the overproduction of superoxide indirectly mediates this injury by rapidly inactivating the cytoprotective free radical NO.
Figure 7.

Effect of polycationic SOD2/3 on serum ALT levels following 90 minutes of ischemia and varying times of reperfusion. * p<0.05 versus sham operated control. Data derived from reference 18.
Figure 8.

Effect of polycationic SOD2/3 on serum TNF-α protein expression following 90 minutes of ischemia and 6 hours of reperfusion. * p<0.05 versus sham operated control. Data derived from reference 18.
Figure 9.

Exacerbation of liver I/R injury in eNOS deficient (eNOS-/-) mice following 45 minutes of ischemia and varying times of reperfusion. *p<0.05 vs. sham; #p<0.05 vs. wild type. Data derived from reference 26.
Concluding Remarks
The mouse model of liver I/R has proven invaluable for our understanding of the role that reactive oxygen and nitrogen metabolites play in modulating and mediating tissue injury in vivo. This model should be useful to investigators who are interested in exploring the different molecular, cellular and/or physiological mechanisms associated with vascular pathobiology, free radical metabolism and inflammation in vivo.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81:22–29. [PubMed] [Google Scholar]
- 2.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–G26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 4.Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 5.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 6.Lefer AM, Lefer DJ. Nitric oxide. II. Nitric oxide protects in intestinal inflammation. Am J Physiol. 1999;276:G572–G575. doi: 10.1152/ajpgi.1999.276.3.G572. [DOI] [PubMed] [Google Scholar]
- 7.Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med. 1999;77:577–592. doi: 10.1007/s001099900029. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J Leukoc Biol. 1992;52:377–382. doi: 10.1002/jlb.52.4.377. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury. J Gastroenterol Hepatol. 2000;15:718–724. doi: 10.1046/j.1440-1746.2000.02207.x. [DOI] [PubMed] [Google Scholar]
- 10.Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, Oberley L, Engelhardt JF. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med. 1998;4:698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 11.Muller MJ, Vollmar B, Friedl HP, Menger MD. Xanthine oxidase and superoxide radicals in portal triad crossclamping-induced microvascular reperfusion injury of the liver. Free Radic Biol Med. 1996;21:189–197. doi: 10.1016/0891-5849(96)00028-7. [DOI] [PubMed] [Google Scholar]
- 12.Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, Messmer K. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5:34–40. doi: 10.1097/00024382-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Gaboury J, Woodman RC, Granger DN, Reinhardt P, Kubes P. Nitric oxide prevents leukocyte adherence: role of superoxide. Am J Physiol. 1993;265:H862–H867. doi: 10.1152/ajpheart.1993.265.3.H862. [DOI] [PubMed] [Google Scholar]
- 14.Granger DN, Kubes P. Nitric oxide as antiinflammatory agent. Methods Enzymol. 1996;269:434–442. doi: 10.1016/s0076-6879(96)69044-2. [DOI] [PubMed] [Google Scholar]
- 15.Kurose I, Wolf R, Grisham MB, Aw TY, Specian RD, Granger DN. Microvascular responses to inhibition of nitric oxide production. Role of active oxidants. Circ Res. 1995;76:30–39. doi: 10.1161/01.res.76.1.30. [DOI] [PubMed] [Google Scholar]
- 16.Shin T, Kuboki S, Lentsch AB. Roles of nuclear factor-kappaB in postischemic liver. Hepatol Res. 2007 doi: 10.1111/j.1872-034X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- 17.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 18.Hines IN, Hoffman JM, Scheerens H, Day BJ, Harada H, Pavlick KP, Bharwani S, Wolf R, Gao B, Flores S, et al. Regulation of postischemic liver injury following different durations of ischemia. Am J Physiol Gastrointest Liver Physiol. 2003;284:G536–G545. doi: 10.1152/ajpgi.00400.2002. [DOI] [PubMed] [Google Scholar]
- 19.Jaeschke H. Mechanisms of reperfusion injury after warm ischemia of the liver. J Hepatobiliary Pancreat Surg. 1998;5:402–408. doi: 10.1007/s005340050064. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia- reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 21.Dezfulian C, Raat N, Shiva S, Gladwin MT. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc Res. 2007;75:327–338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grisham MB, Granger DN, Lefer DJ. Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radic Biol Med. 1998;25:404–433. doi: 10.1016/s0891-5849(98)00094-x. [DOI] [PubMed] [Google Scholar]
- 23.Hines IN, Kawachi S, Harada H, Pavlick KP, Hoffman JM, Bharwani S, Wolf RE, Grisham MB. Role of nitric oxide in liver ischemia and reperfusion injury. Mol Cell Biochem. 2002;234-235:229–237. [PubMed] [Google Scholar]
- 24.Kawachi S, Hines IN, Laroux FS, Hoffman J, Bharwani S, Gray L, Leffer D, Grisham MB. Nitric oxide synthase and postischemic liver injury. Biochem Biophys Res Commun. 2000;276:851–854. doi: 10.1006/bbrc.2000.3559. [DOI] [PubMed] [Google Scholar]
- 25.Gao B, Flores SC, McCord JM. A site-directed mutant of Cu,Zn-superoxide dismutase modeled after native extracellular superoxide dismutase. Biol Trace Elem Res. 1995;47:95–100. doi: 10.1007/BF02790105. [DOI] [PubMed] [Google Scholar]
- 26.Hines IN, Harada H, Flores S, Gao B, McCord JM, Grisham MB. Endothelial nitric oxide synthase protects the post-ischemic liver: potential interactions with superoxide. Biomed Pharmacother. 2005;59:183–189. doi: 10.1016/j.biopha.2005.03.011. [DOI] [PubMed] [Google Scholar]