

Abstract
Trophoblast expression of immunomodulatory proteins in the human placenta is among the mechanisms that are critical for ensuring lymphocyte tolerance to the semi-allogeneic fetus. High levels of B7-H1 on trophoblast cells together with the known role of this protein in establishment of peripheral tolerance suggest that B7-H1 mediates immunological protection of the placenta during gestation. In this study we investigated the molecular mechanisms of regulation of B7-H1 in trophoblast cells by epidermal growth factor (EGF), a key regulator of trophoblast cell differentiation. EGF increased B7-H1 protein levels within 24 hours and mRNA levels within 4 hours of the initiation of treatment; by 24 hours B7-H1 mRNA levels were similar between control and EGF-treated cells. Analysis of two different potential promoter regions revealed strong promoter activity in response to IFN-γ. In contrast, no promoter activity could be induced by EGF, suggesting that this cytokine regulates B7-H1 expression post-transcriptionally in trophoblast cells. EGF-induced B7-H1 protein expression was completely blocked in the presence of inhibitors of the PI3Kinase/Akt/mTOR pathway, a pathway known to regulate gene expression at the translational level. Finally, analysis of monosomal and polysomal mRNA fractions of untreated and EGF-treated term trophoblast cells revealed that EGF induces a shift towards the translatable fractions and away from the untranslated fractions. These results highlight a novel mechanism for regulation of B7 family proteins in the placenta.
Introduction
Both central and peripheral tolerance mechanisms account for the ability of lymphocytes to discriminate between self- and non-self antigens, thereby avoiding inappropriate immune responses and autoimmune disease. Similar mechanisms may be involved in regulation of the maternal immune system such that recognition of paternally-inherited fetal antigens does not elicit an anti-fetal maternal immune response. Key to induction of fetal tolerance are cell-contact-dependent and cell-contact-independent interactions between maternal immune cells and fetal trophoblast cells. Multiple trophoblast-expressed proteins are postulated to mediate these interactions, including the class Ib members of the major histocompatibility complex family, tumor necrosis factor superfamily, the tryptophan-catabolizing enzyme, indoleamine-2,3-deoxygenase, and members of the B7 family of surface-associated proteins [1-3].
Members of the newly extended B7 family have been recognized as both positive and negative regulators of the immune response, depending on interaction with their specific counter-receptors [4]. B7-H1 (also called CD274 or PD-L1) is a surface-bound ligand that binds to the lymphocyte-expressed receptor, PD-1; this interaction results in inhibition of antigen-specific activation and cytokine production by T cells [5]. B7-H1 in peripheral tissues appears to prevent activation of self-reactive T cells, and mice lacking either B7-H1 or PD-1 suffer autoimmune disease [6, 7]. B7-H1 expression on tumor cells appears to be a mechanism by which the cells avoid eliciting an immune response, and its expression in carcinoma is correlated with poor prognosis in humans [8, 9]. Recent studies have also shown that persistent expression of PD-1 on T cells contributes to chronic infections due to its inhibition of an immune response. Blockade of PD-1 signaling reverses inhibition of the virus-specific T cells, resulting in a heightened ability for the T cells to resolve the infection [10, 11]. Collectively, these studies offer strong evidence for a role of this pathway in tolerance to both self and foreign antigens.
B7-H1 protein expression in the human placenta occurs selectively on trophoblast cells, including villous and extravillous cytotrophoblast cells, and syncytiotrophoblast cells [12, 13]. In addition, in the decidua, T cells are induced to express PD-1 protein early in pregnancy and appears to modulate cytokine production by these cells [14]. In the mouse, others have found that blockade of maternally-derived B7-H1 was found to result in rejection of allogeneic fetuses [15]. The genetic disparity between mother and fetus, the high level display of B7-H1 in the placenta, and functional studies together suggest an important role for the B7-H1/PD-1 pathway in maternal-fetal tolerance.
The elevated presence of B7-H1 protein on trophoblast cells is somewhat unusual in that, although its mRNA is broadly expressed, macrophages, dendritic cells, and some microvascular endothelial cells seem to be the only other cell types that constitutively expresses this protein under physiological circumstances [8]. Under inflammatory conditions, interferon (IFN)-γ induces B7-H1 protein expression in many cell types, including trophoblast cells [13]. Interestingly, physiologically relevant concentrations of epidermal growth factor (EGF), a central regulator of cytotrophoblast differentiation [16] also boost B7-H1 protein expression in trophoblast cells, an effect that mimics the in vivo observation of stronger B7-H1 protein expression within the syncytiotrophoblast than on the underlying mononuclear precursor trophoblast cells [12]. Because of the apparent physiological role of EGF- and differentiation-enhanced B7-H1 protein expression, together with the modulation of lymphocyte activity conferred by the B7-H1/PD-1 pathway in pregnancy, we sought to further elucidate the molecular mechanisms of B7-H1 regulation in trophoblast cells.
Materials and Methods
Tissue collection and processing
Normal term placentas were collected following Caesarian sections from the University of Kansas Hospital in accordance with protocols approved by the institutional Human Subjects Committee, and tissue was processed within 30 minutes of delivery. For most cell culture studies, 30-40g villous tissue was dissected, rinsed, and subjected to enzymatic digestion as described previously [17]. For studies involving analysis of monosomal and polysomal RNA, 80-120g villous tissue was dissected and processed. After mincing, placental villi were dissociated in three 30-minute stages of a digestion solution containing trypsin and DNase. The resulting cell suspension was layered over a 5-70% stepwise Percoll (Sigma) gradient and centrifuged. The trophoblast fraction was subjected to immunopurification by negative selection over magnetic bead columns coated with anti-HLA class I antibody (W6/32; BioExpress, West Lebanon, NH). Cells were routinely more than 95% pure as assessed by immunocytochemical staining for cytokeratin-7 (DAKO Corp., Carpinteria, CA).
Assessment of cytotrophoblast fusion
Trophoblast fusion was assessed similarly to previously described procedures [18]. Purified cells were seeded into 4-well glass chamber slides (Nalge-Nunc) at a density of 750 × 106 cells per well. Control and EGF-treated cells were cultured for 48 hours, at which time they were stained for desmplakin to evaluate cell fusion. Trophoblast cells were rinsed in PBS, and nonspecific binding sites were then blocked for 1 hour in a solution of 50% SuperBlock buffer (Peirce, Rockford, IL), 2% donkey serum and 0.02% triton X-100. Anti-desmosomal protein (clone ZK-31, Sigma, St. Louis, MO) was added to the cells at a dilution of 1:400, and slides were incubated overnight at 4°C. Lack of binding of IgG1 control antibody confirmed specificity of desmoplakin staining. After rinsing in PBS, alexafluor 488-conjugated donkey anti-mouse IgG (eBiosciences) was added at room temperature for 1 hour. Cells were coverslipped in mounting medium (ProLong Gold with DAPI; Invitrogen). Digital images were captured at random locations within each well on a Nikon C1 Plus confocal microscope using EasyC1 software. A minimum of 100 nuclei were counted manually by ImageJ software, and data are expressed as percentage of cells containing two or more nuclei.
Protein isolation and immunoblot analysis
To study the influence of EGF and IFN-γ on B7-H1 expression by trohpoblast cells, immunopurified cells were plated overnight at a density of 7.5 × 106 cells in 60 mm Petri dishes in Iscove’s modified DMEM containing 10% FBS and antibiotics. The following morning, cells were rinsed twice, treated with medium with or without 5 ng/ml EGF (Peprotech, Rocky Hill, NJ) or 100 U/ml IFN-γ (R&D Systems; Minneapolis, MN) and incubated in a 5% CO2/95% air atmosphere for up to 144 hours. Long-term cultures (>48 hours) were replenished with fresh culture medium with or without treatments every other day. For signal transduction pathway studies, biochemical inhibitors were dissolved in DMSO (vehicle) and controls included medium alone and vehicle treatments. Cells were pretreated with each inhibitor for 1 hour prior to initiation of the 48 hour culture. Inhibitors included 20 μM tyrphostin AG1478 (EGF receptor), 20 μM PD98059 (ERK1/2 inhibitor; Sigma) and 10μM UO126 (MEK1/2 inhibitor; Sigma), 10 μM wortmannin (PI3K inhibitor; Calbiochem), 10 μM SB203580 (p38 MAPK inhibitor; Sigma) and 50 ng/ml rapamycin (mTOR inhibitor; Sigma). Wortmannin treatment induced a loss of nuclear morphology that was consistent with apoptosis by approximately 25%; other treatments did not affect cell viability.
Protein from cultured cells was collected by lysis in RIPA buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing protease inhibitors (100 μg/ml phenylmethylsulfonic acid, 10 μg/ml aprotinin, 10 μg/ml leupeptin). Protein (10 or 20 μg) was electrophoresed under denaturing conditions and transferred to nitrocellulose membranes. Membranes were blocked in 3% nonfat milk/TBS and probed with goat anti-human B7-H1 antibody (100 ng/ml) (R&D Systems, Minneapolis, MN). To verify equal loading of samples, membranes were stripped and re-probed with rabbit anti-actin (Sigma cat. no. A-5060). After incubating with the corresponding horseradish peroxidase-labeled secondary antibody (Jackson ImmunoResearch, West Grove, PA), bound antibodies were detected using a chemiluminescent detection system (Pierce Biotechnology, Rockford, IL) and autoradiography. Normalization of B7-H1 expression to actin was performed by taking the ratio of densitometric intensities using Gel-Pro image analysis software (Silver Spring, MD).
RNA Extraction and RT-PCR
To study the effects of EGF on B7-H1 mRNA expression, 7.5 × 106 trophoblast cells were plated in 60 mm Petri dishes and treated the following day for up to 48 hours. Total cellular RNA was extracted by addition of Trizol reagent (Invitrogen, Carlsbad, CA), following the manufacturer’s instructions. Total RNA was pretreated with DNase I (Sigma) and quantified by spectrophotometry. One μg RNA was reverse transcribed using MMLV reverse transcriptase and oligo dT primers (Invitrogen). Real time PCR was performed using an ABI Prism 7900 System (Applied Biosystems, Foster City, CA). Primers used included B7-H1 (5’-tca atg ccc cat aca aca aa-3’ (Fwd) and 5’-cga agt cat ctg gac aag c-3’ (Rev), and the housekeeping gene β-actin (Applied Biosystems). Each sample was assayed in triplicate and was compared to arbitrary values assigned to a standard curve generated from serial dilutions of placental RNA to obtain relative abundance of amplified product. These values were then normalized to those of β-actin products.
Analysis of fractionated RNA was performed by culturing 180-240 × 106 trophoblast cells divided equally between 100 mm Petri dishes (45-60 × 106/dish) and treated with either medium alone or 5ng/ml EGF. After 36 hours, cells were lysed as described [19] with several modifications. Briefly, cells were incubated (10 min) in media containing 10ug/mL cycloheximide, scraped and resuspended in 1mL PBS containing 100ug/ml cycloheximide and 1mM PMSF. A 50μl aliquot was placed in Trizol reagent for extraction of total RNA. The remaining cell suspension was pelleted and then lysed in low-salt buffer (LSB) (20mM Tris, 10mM NaCl, 3mM MgCl2) with 100U/ml RNAse Inhibitor (Invitrogen) on ice. Detergent buffer (1.2% Triton N-101 in LSB) (250μl) was added and the cells were homogenized on ice using a glass dounce homogenizer. The lysate was centrifuged (4°C, 10,000×g, 1 min) and the supernatant transferred to a tube containing 100ul of LSB with 10mg heparin/ml and 1.5M NaCl. The suspension was loaded directly onto a 11.5-ml continuous sucrose gradient (15-50% w:v in LSB with 25U/ml RNAse Inhibitor), and centrifuged at 36,000 × g for 4h. Each sucrose/RNA gradient was run through a UA6 UV detector/chart recorder (Teledyne Isco) and a running trace (250nm) was recorded as 250ul fractions were collected using the Foxy Jr. Fraction Collector (Teledyne Isco). RNA fractions corresponding to the 40S, 60S, monosomal, and individual polysomal (disomic, trisomic, etc.) were then combined to generate 11 fractions, and RNA was isolated and examined by real time RT-PCR as described above. Total RNA was examined for quantity and quality on a Dual Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Plasmid construction and luciferase assays
Two putative B7-H1 promoters corresponding to 1.069 kb upstream of exon 1 (UTR), and 1.2 kb upstream of the translation start codon (Int1) were analyzed for transcription factor binding sites using Transcription Factor Search online software (http://www.cbrc.jp/research/db/TFSEARCH.html) [20]. Human genomic DNA (Applied Biosystems) was amplified by PCR using primers to these regions containing cloning sites (underlined). Primers were: UTR-Fwd, 5’ – gca act cga ggt ggt caa gct cat – 3’; UTR-Rev: 5’ – cag aga tct tgc cct aat tat g – 3’; Int1-Fwd: 5’ – tga ctc gag gtg gct caa gcc tgt a – 3’, and (Int1-Rev: 5’ – ctc tcg agc cca aag aaa ggg tgt a – 3’. These regions were cloned into the promoterless luciferase expression vector pGL3 (Promega) using XhoI/BglII (to create B7H1.UTR) and XhoI (B7H1.Int1).
The human choriocarcinoma cell lines JEG-3, Jar or BeWo (ATCC, Manassas, VA) were grown to 50% confluence in 12-well tissue culture plates. pGL3-basic (negative control), pGL3-B7H1(UTR) or PGL3-B7H1(Int1) were transfected using Lipofectamine reagent (Invitrogen) with some modifications [21]. Following transfection, the cells were treated with medium alone, EGF (5 ng/ml) or IFN-γ (100 U/ml) for 8 hours. Luciferase assays were performed using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Transfection studies were conducted in triplicate in 3-4 separate experiments, and data are expressed as luciferase activity (relative light units [RLU]) of each promoter region, and normalized to that of Renilla.
Statistical analysis
Experimental data were analyzed by two-way analysis of variance at α = 0.05, using the Tukey post-hoc test where differences were found. For time-course and polysomal mRNA analyses, two-way ANOVA was performed using repeated measures so that treatment effect from all time points or fractions, respectively, could be analyzed simultaneously. A significant (P < 0.05) interaction indicated that the effects of the treatment on mRNA levels was dependent on the time point or fraction analyzed. Data are expressed as means ± SEM, and asterisks denote statistically significant differences (P < 0.05) from the indicated control.
Results
Time-course analysis of EGF-stimulated B7-H1 expression in trophoblast cells
We previously showed that the syncytiotrophoblast abundantly expresses B7-H1 protein in vivo, and that trophoblast cells syncytialized in vitro with EGF express higher levels of this protein than undifferentiated control cells [12]. To gain insight into the mechanism of EGF-induced B7-H1 protein induction, we performed a time-course analysis of B7-H1 mRNA and protein. Unstimulated levels of B7-H1 protein remained constant over the course of a 6-day culture period (P > 0.05), whereas EGF-induced protein doubled by 24 hours and further increased to nearly 3-fold by 48 hours. EGF-induced protein levels remained two- to three-fold above untreated control cells throughout the culture period high throughout the 6-day culture period (Figure 1A and B).
Figure 1. Time course analyses of B7-H1 expression.
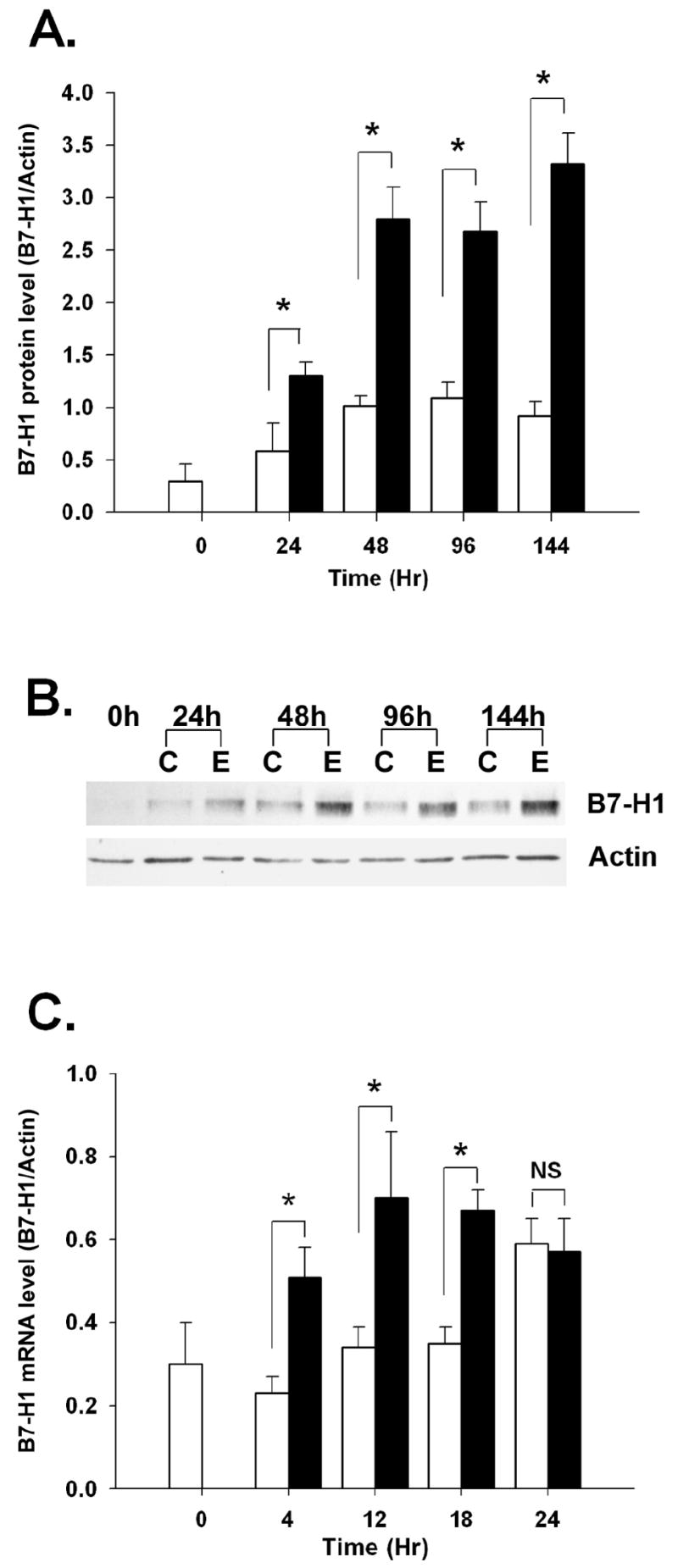
(A) Trophoblast cells from three different placentas were treated with medium alone (open bars) or 5ng/ml EGF (filled bars) for up to 6 days, at which time protein was extracted and subjected to western blot analysis using anti-B7-H1 or anti-actin (control). Densitometric intensities of specific B7-H1 and actin bands were digitally obtained and expressed as a ratio. (B) Representative western blot from three experiments. (C) Trophoblast cells three different placentas were treated similarly to the experiment in A, but cultured for up to 24 hours. RNA was extracted and subjected to qRT-PCR using primers against B7-H1 or actin. Data are expressed as mean ratios of Ct values for B7-H1 and β-actin ± SEM, and asterisks denote significant differences (P < 0.05) of EGF-treated cells from their respective controls. NS, P > 0.05. C, control; E, EGF.
B7-H1 can be regulated at the level of transcription by cytokines such as IFN-γ [22]. To determine whether increased transcription of the B7-H1 gene might also account for EGF-induced elevation of protein levels in trophoblast cells, a short-term time course was performed in which trophoblast cells were treated with EGF for 0-24 hours and analyzed by real-time RT-PCR. EGF significantly upregulated B7-H1 mRNA expression as early as 4 hours after the onset of treatment, and message levels remained high at levels for the duration of the 24 hour culture (Figure 1C). In untreated control cells, B7-H1 mRNA levels did not increase before 24 hours of culture (P < 0.05). Thus, EGF treatment resulted in an accelerated increase in B7-H1 mRNA expression in comparison to untreated control cells.
B7-H1 upstream regions do not respond to EGF
Given the rapid induction of mRNA, we next tested the hypothesis that this growth factor could regulate B7-H1 at the transcriptional level. Two potential promoter regions of the B7-H1 gene were each cloned into a promoterless vector such that promoter activity of the cloned regions would drive luciferase expression. These two regions, depicted in Figure 2A and termed B7H1.UTR and B7H1.Int1, were each tested for their responsiveness to EGF. JEG-3 cells, which, like primary trophoblast cells, express B7-H1 constitutively, and Jar cells, which lack B7-H1 even when treated with proinflammatory cytokines, were transiently transfected with vectors containing these regions. IFN-γ, a positive control, induced B7H1.UTR promoter activity in both JEG-3 and Jar cells, and moderately induced B7-H1.Int1 promoter activity in JEG-3, but not Jar cells (Figure 2C-F). In contrast, EGF failed to stimulate activity of either of the two promoters, regardless of whether JEG-3 or Jar cells were used, despite its ability to upregulate the protein in JEG-3 cells (Figure 2B). Similar results were obtained with BeWo cells; these cells exhibited a 3.6-fold increase (P = 0.002) and 2.5-fold increase (P = 0.001) in luciferase activity in response to IFN-γ over untreated controls when transfected with B7H1.UTR or B7H1.Int1, respectively, whereas they did not support EGF-stimulated activity of either putative B7-H1 promoters (P > 0.05).
Figure 2. Promoter activity of the B7-H1 gene in JEG-3 and Jar cells.

(A) Schematic diagram of the B7-H1 genetic regions from which the B7H1.UTR and B7H1.Int1 putative promoters were cloned. The adenosine of the ATG start codon is designated +1, and the locations of other primers are expressed relative to this base. Exon sequences are shaded, and intronic sequences unfilled; arrows represent directions of primer sequences. (B) JEG-3 cells were treated with medium alone (Contr) or EGF (5 ng/ml) in two separate experiments (Exp) for 48 hours, and subjected to western blot analysis. (C) JEG-3 cells were transfected with the B7H1.UTR or (D) B7H1.Int1 promoter, and treated for 6 hours with medium alone (Control), EGF (5 ng/ml), or IFN-γ (100 U/ml). (E) Jar cells were similarly transfected with B7H1.UTR or (F) B7H1.Int1 and treated. The transfection experiments (C-F) were repeated three separate times. Luciferase activity of each of the promoters (RLU) was normalized to Renilla activity and expressed as mean ± SEM. NS, P > 0.05.
EGF regulates B7-H1 through the PI3 Kinase/mTOR pathway
To gain a better understanding of the mechanisms of EGF-induced B7-H1 protein expression, we next analyzed the role of components of intracellular signaling cascades. As shown in Figure 3A, the EGF receptor tyrosine kinase inhibitor AG1478 completely blocked B7-H1 protein stimulation by EGF, confirming the requirement for tyrosine phosphorylation of the EGF receptor.
Figure 3. Effects of metabolic inhibitors of signal transduction pathways.

Primary trophoblast cells were pretreated for 1 hour with (A) AG1478 (AG; EGFR inhibitor); (B) rapamycin (Rap; mTOR inhibitor); or (C) wortmannin (Wort; PI3K inhibitor). (D) and (E), representative western blots. Control wells were treated with vehicle ((Veh; DMSO) alone. After removal of the inhibitors, cells were treated with medium or EGF (5 ng/ml) for 48 hours, and subjected to western blot analysis. Data represent means ± SEM of four experiments, each from separate placentas, and letters denote statistically significant differences.
mTOR, which can be regulated by several different signal transduction pathways, is a key mediator of translation. To examine a potential role for mTOR, we tested the effect of rapamycin on EGF-induced B7-H1 protein expression. Figure 3B shows that rapamycin blocked the ability of EGF to upregulate B7-H1 in trophoblast cells (P < 0.05). To determine the likely regulator of EGF-induced mTOR activity, we next tested the ERK1/2 inhibitor PD98059 and the PI3K inhibitor wortmannin on EGF-treated trophoblast cells (Figure 3B and C). Only wortmannin prevented the rise in B7-H1 protein, indicating that the PI3K-Akt but not the ERK1/2-MEK1/2 pathway controls mTOR activation in this EGF-induced response.
EGF is well-known to induce differentiation trophoblast cells as measured by syncytialization. EGF-mediated syncytialization has previously been shown to be dependent upon the p38 MAPK pathway [23]. To evaluate whether the B7-H1-inducing effect of EGF is direct or secondary to differentiation, we measured the degree of syncytialization of trophoblast cells in the presence an absence of rapamycin and SB203580, an inhibitor of p38 kinase. Treatment with EGF for 48 hours induced a morphological transformation of the cells, from small aggregates of 5-15 cells to larger aggregates of cells (Figure 4A). In addition, a modest but consistent increase in syncytialization was observed as a result of EGF treatment, from approximately 15% to about 25% in EGF-treated wells, although this level did not reach statistical significance (P = 0.134; Figure 4B). Treatment of cells with the p38 MAPK inhibitor, SB203580 neither prevented this modest increase in syncytialization, nor did it interfere with EGF-mediated induction of B7-H1; two experiments using trophoblast cells from different placentas revealed 1.6- and 3.7-fold increases in B7-H1 expression following EGF treatment. In contrast, rapamycin, which inhibited B7-H1 expression (Figure 3B), appeared to promote EGF-induced syncytialization.
Figure 4. EGF-induced syncytialization of trophoblast cells.
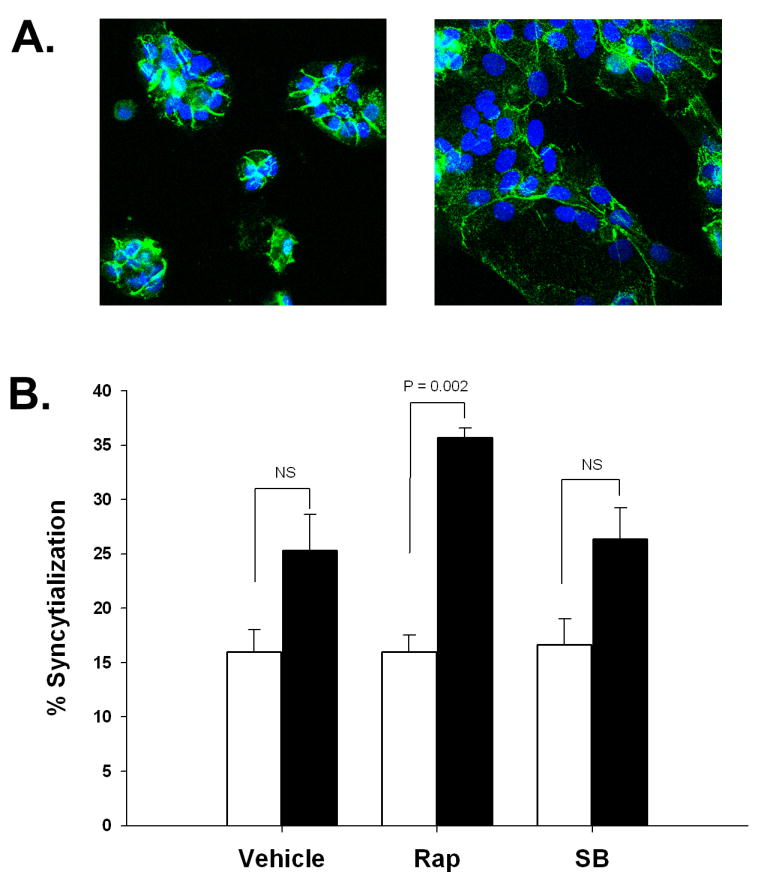
(A) Illustration of syncytialization. Cytotrophoblast cells were cultured for 48 hours, fixed and stained for desmoplakin, followed by DAPI counterstaining. Cells were treated with medium alone (left panel) or EGF (right panel). Images are representative of 3 separate experiments using trophoblast cells from different placentas. (B) Quantification of syncytialization in the presence and absence of biochemical pathway inhbitors. Open bars represent data from control cells; filled bars represent data from EGF-treated cells. NS, P > 0.05.
EGF induces an increase in the proportion of translatable B7-H1 mRNA
The identification of the mTOR pathway in EGF-induced B7-H1 expression prompted us to next evaluate whether EGF operates through a post-transcriptional gene regulatory mechanism to upregulate B7-H1. We therefore examined whether induction of B7-H1 protein expression by EGF is accompanied by a shift in the proportion of B7-H1 mRNA being actively translated. In 3 separate experiments, primary cytotrophoblast cells were treated with either medium alone or EGF, lysed, and subjected to density gradient fractionation and analysis of mRNA within each fraction. The majority of β-actin mRNA migrated with the heavier polysomal RNA fractions, regardless of treatment (Figure 5). There was no significant fraction × treatment interaction (P = 0.478), revealing that EGF had no effect on β-actin mRNA distribution among polysomal RNA. Conversely, B7-H1 mRNA exhibited a markedly different distributional pattern in response to EGF treatment. In untreated control cells B7-H1 mRNA was biphasically distributed within the monosomal fraction (fraction 5) and the heavier polysomal fractions (fractions 6-11; Figure 5). Upon treatment with EGF, however, the proportion of total B7-H1 mRNA present in the monosomal fraction shifted to the polysomal fractions (Figure 5). This was reflected by a significant fraction × treatment interaction (P = 0.043).
Figure 5. Monosomal and polysomal fractionation of B7-H1 mRNA.

Representative experiment of 3 in which primary cytotrophoblast cells from different placentas were treated with medium alone (closed symbols) or EGF (open symbols) for 36 hours, lysed, and subjected to size fractionation by sucrose gradient centrifugation. RNA was isolated from fractions and subjected to reverse transcription and real-time PCR for B7-H1 and actin. The standard errors of the means for the three experiments were 3.835 (B7-H1 mRNA) and 2.912 (Actin mRNA).
Discussion
Trophoblast cells express a unique array of immunomodulatory proteins that directly regulate maternal leukocytes during pregnancy, such that the leukocytes are directed toward physiologically favorable roles. B7-H1 is one such protein that appears to regulate T cells in pregnancy. In most tissues, the B7-H1 gene is transcribed, as evidenced by the broad distribution of its mRNA [5, 24, 25]. However, constitutive expression of the protein is confined to only a few cell types, and protein expression within peripheral tissues is rare. Even so, expression by these cells under physiologic, noninflammatory circumstances is likely important for maintaining tissue-specific immune tolerance, since mice lacking either B7-H1 or its receptor, PD-1, are unable to control attack by self reactive T cells, and are thus susceptible to autoimmune disease [6, 7, 26, 27].
In pregnancy, the B7-H1/PD-1 pathway regulates cytokine production by maternal T ccells in the decidua [14]. B7-H1 protein is temporally and spatially regulated in the human placenta such that it is displayed on cells that interface directly with maternal blood and tissues. For example, its induction by oxygen ensures that it is highly expressed during the second and third trimester, the stages in which the placenta is immersed in maternal blood [28]. Relevant to these studies, the spatial regulation of B7-H1 protein occurs coincidentally with trophoblast differentiation, resulting in strong expression at the microvillous surface of the syncytiotrophoblast, the point of contact with the maternal blood [12].
EGF is a key mediator of syncytialization in trophoblast cells, and reproduces many differentiation-associated events in vitro [16]. Here, we confirmed and expanded our previous findings of regulation of trophoblast B7-H1 expression by EGF. The rapid doubling of B7-H1 mRNA levels initially led us to hypothesize that EGF regulates this protein by means of inducing mRNA expression, and we tested this by asking whether either of two potential promoter regions of the B7-H1 gene are responsive to EGF (Figure 2A). B7H1.UTR was cloned from the 5’ untranslated region located upstream of exon 1; this region includes sequences previously described as responsive to IFN-γ [22]. B7H1.Int1, a second potential promoter region, was cloned from the region of the B7-H1 gene directly upstream of the ATG translation start site; this region lies within the first intron of B7-H1 and contains numerous putative regulatory sequences. Each was cloned into a promoterless Luciferase expression vector, transfected into JEG-3, BeWo and Jar cells, and assessed for responsiveness to EGF. Both JEG-3 and BeWo cells constitutively express B7-H1, while Jar cells do not express B7-H1 even when stimulated with IFN-γ. Surprisingly, neither promoter exhibited the ability to induce luciferase activity in response to EGF in any of the three cell lines tested, even though IFN-γ robustly induced the promoters, particularly B7H1.UTR. Interestingly, Jar cells were the only of the three lines that could not support activity of B7H1.Int1 in response to IFN-γ, even though they could support B7H1.UTR activity. These cells are also unable to respond to IFN-γ by upregulating B7-H1 mRNA [13]. Thus, even though the B7H1.UTR promoter appears stronger under both basal and IFN-γ-induced conditions, the B7H1.Int1 region might be critical in ensuring a full response of the gene to IFN-γ. More studies are needed to fully test this hypothesis.
From these results, we cannot rule out the possibility that the promoter regions used to transfect the cells were of insufficient length to encompass an EGF-responsive region responsible for its effects on B7-H1 mRNA; this could explain the early increase in B7-H1 mRNA after EGF treatment. A second explanation for the early EGF-induced rise in mRNA could be an increase in stability of the RNA, both as a result of EGF treatment and as a result of maintenance in culture, as observed by the increase in RNA of control cells at 24 hours. Nevertheless, other, non-transcriptional mechanisms could account for the EGF-associated increase in B7-H1 protein. In trophoblast cells, EGF promotes survival, invasiveness, syncytialization, and endocrine function through multiple signal transduction pathways, including MAPK, JAK/Stat, PI3K and SPHK-1 [29-32], and we reasoned that identification of the signal transduction cascade(s) responsible would yield further insight into the mechanisms regulating the B7-H1 gene in trophoblast cells. We first asked whether mTOR, which functions to enhance translation of 7-methylguanosine 5’-capped mRNAs, is involved in EGF-induced B7-H1 protein expression. Indeed, treatment of the cells with rapamycin indicated the dependence of B7-H1 protein expression on mTOR, since it completely blocked EGF-upregulated B7-H1. Activation of mTOR can be achieved through either the PI3K-Akt and MEK1/2-ERK1/2 pathways [33-35], and our results suggest the former, since wortmannin, but not PD98059, prevented the effect of EGF. In agreement with these observations, JEG-3 cell upregulation of B7-H1 by EGF was also sensitive to both rapamycin and wortmannin (data not shown). Collectively, these results suggest that the sequence of events leading to elevated B7-H1 protein involves tyrosine phosphorylation of the EGF receptor, activation of the PI3K-Akt pathway, and subsequent targeting of mTOR.
mTOR functions to permit the formation of the translation initiation complex by hyperphosphorylating and thus releasing the inhibitory 4E-binding proteins (4E-BPs) from the eukaryotic initiation factor 4F (eIF4F), which is followed by recruitment of ribosomes to the mRNA [36]. Untranslated or inefficiently translated mRNA is associated with ≤ 1 ribosome (unassociated or monosomal RNA), and highly efficiently translated mRNA is associated with ≥ 2 ribosomes (polysomal RNA). Density-based fractionation of RNA permits the visualization of ribosomal association of individual mRNAs, so we next examined the effect of EGF on the ribosomal association of B7-H1 message. Since neither mRNA nor protein levels of β-actin were altered by EGF, we chose to use this message as our control gene. In control cells, most of the β-actin mRNA sedimented within the polysomal RNA fractions, suggesting that virtually all of this mRNA species is constitutively translated. As predicted, EGF did not affect the distribution of β-actin mRNA among polysomal fractions. In contrast, B7-H1 mRNA was found within both monosomal and polysomal fractions in untreated control cells, suggesting that some, but not all of the B7-H1 mRNA is actively translated in these cells. This agrees with the observation that undifferentiated cytotrophoblast cells constitutively express moderate amounts of this protein. However, in EGF-treated cells, B7-H1 mRNA was predominantly distributed among the polysomal fractions, revealing that the EGF-induced increases in B7-H1 levels were associated with an increase in B7-H1 mRNA levels in polysomal fractions.
From these experiments, it is not directly clear whether the rise in B7-H1 protein due to EGF is a direct effect or is secondary to its effect on syncytialization. It is possible that the effect of EGF in due to syncytialization since the level of increase in syncytialization at 48 hours was similar to the level of induction of B7-H1 protein: both increased by 1.5- to 2-fold. On the other hand, this likely does not hold true for later times; whereas B7-H1 appeared to be maximally induced by 48 hours, syncytialization in response to EGF is known to progressively increase over time. These results also reveal that the p38 kinase pathway does not act in response to EGF to increase either syncytialization or B7-H1 protein in these shorter-term cultures. These results contrast with those previously reported about the involvement of this pathway in syncytialization possibly because of the longer time period analyzed in the latter study.
In conclusion, our results reveal that activation of the PI3K-Akt-mTOR pathway upon EGF-mediated trophoblast differentiation posttranslationally promotes syncytiotrophoblast expression of B7-H1 protein. Identification of the PI3K-Akt-mTOR pathway draws out further similarity between trophoblast cells and tumor cells, as neuroblastoma was recently shown to use this axis to induce B7-H1 protein, thus protecting the cells from lymphocyte-mediated cytotoxicity [37]. Thus there are thus at least two ways in which EGF might promote trophoblast cell survival through the PI3K-Akt pathway: directly, by preventing cytokine-induced apoptosis in trophoblast cells [29, 38], and indirectly, by promoting expression of B7-H1 and perhaps other immunomodulatory proteins, in turn modulating activation and production of toxic cytokines by lymphocytes [39]. Finally, B7-H1 has recently been suggested to deliver an antiapoptotic signal directly to cells via reverse signaling following ligation of its receptor, PD-1 [40]. Altogether, these results highlight a new mechanism of regulation of the immunomodulatory protein B7-H1 in trophoblast cells.
Acknowledgments
This work was supported by NIH grants R01 HD045611 (M.G.P.), P01 HD49480 (M.G.P., Project Director), HD051870 (L.C.K) and the Madison and Lila Self Fellowship (M.Z.C.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunt JS. Stranger in a strange land. Immunol Rev. 2006;213:36–47. doi: 10.1111/j.1600-065X.2006.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mellor AL, Jayabalan S, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 3.Petroff MG. Immune interactions at the maternal-fetal interface. J Reprod Immunol. 2005;68:1–13. doi: 10.1016/j.jri.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Greenwald R, Freeman G, Sharpe A. The B7 family revisited. Annu Rev Immunol. 2004;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 5.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 6.Dong H, Zhu G, Tamada K, Flies DB, van Deursen JMA, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8+ T lymphocytes. Immunity. 2004;20:327–336. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 7.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong H, Strome SE, DR S, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 9.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111:3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 11.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJR, Klenerman P, Ahmed R, Freeman GJ, Walker BD. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 12.Petroff MG, Chen L, Phillips TA, Azzola D, Sedlmayr P, Hunt JS. B7 family molecules are favorably positioned at the human maternal-fetal interface. Biol Reprod. 2003;68:1496–1504. doi: 10.1095/biolreprod.102.010058. [DOI] [PubMed] [Google Scholar]
- 13.Petroff MG, Chen L, Phillips TA, Hunt JS. B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta. 2002;23(Suppl A):S95–101. doi: 10.1053/plac.2002.0813. [DOI] [PubMed] [Google Scholar]
- 14.Taglauer ES, Trikhacheva AS, Slusser JG, Petroff MG. Expression and function of PDCD1 at the human maternal-fetal interface. 2008;79:562–569. doi: 10.1095/biolreprod.107.066324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guleria I, Khosroshahi A, Ansari MJ, Habicht A, Azuma M, Yagita H, Noelle RJ, Coyle A, Mellor AL, Khoury SJ, Sayegh MH. A critical role for the programmed death ligand 1 in fetomaternal tolerance. J Exp Med. 2005;202:231–237. doi: 10.1084/jem.20050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrish DW, Dakour J, Li H, Xiao J, Miller R, Sherburne R, Berdan RC, Guilbert LJ. In vitro cultured human term cytotrophoblast: a model for normal primary epithelial cells demonstrating a spontaneous differentiation programme that requires EGF for extensive development of syncytium. Placenta. 1997;18:577–585. doi: 10.1016/0143-4004(77)90013-3. [DOI] [PubMed] [Google Scholar]
- 17.Petroff MG, Phillips TA, Ka H, Pace JL, Hunt JS. Isolation and culture of term human trophoblast cells. Meth Mol Med. 2006;121:203–217. doi: 10.1385/1-59259-983-4:201. [DOI] [PubMed] [Google Scholar]
- 18.Douglas GC, King BF. Differentiation of human trophoblast cells in vitro as revealed by immunocytochemical staining of desmoplakin and nuclei. 1990;96:131–141. doi: 10.1242/jcs.96.1.131. [DOI] [PubMed] [Google Scholar]
- 19.Raun H, Brown CY, Morris DR. Analysis of ribosome loading onto mRNA species: implications for translation control. In: Richter JD, editor. mRNA Formation and Function. Academic Press; 1997. pp. 305–321. [Google Scholar]
- 20.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel EE, Kel OV, Ingnatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFACT, TRRD, and COMPEL. Nucleic Acids Res. 1998;26:364–370. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfe MW. Culture and transfection of human choriocarcinoma cells. Methods Mol Med. 2006;121:229–239. doi: 10.1385/1-59259-983-4:227. [DOI] [PubMed] [Google Scholar]
- 22.Lee S-J, Jang B-C, Lee S-W, Yang Y-I, Suh S-I, Park Y-M, Oh S, Shin J-G, Yao S, Chen L, Choi I-H. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-[gamma]-induced upregulation of B7-H1 (CD274) FEBS Letters. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 23.Johnstone ED, Sibley CP, Lowen B, Guilbert LJ. Epidermal growth factor stimulation of trophoblast differentiation requires MAPK11/14 (p38 MAP kinase) activation. 2005;73:1282–1288. doi: 10.1095/biolreprod.105.044206. [DOI] [PubMed] [Google Scholar]
- 24.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 25.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne C, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 27.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 28.Holets LM, Hunt JS, Petroff MG. Trophoblast CD274 (B7-H1) is differentially expressed across gestation: influence of oxygen concentration. Biol Reprod. 2006;74:352–358. doi: 10.1095/biolreprod.105.046581. [DOI] [PubMed] [Google Scholar]
- 29.Mackova M, Kilani RT, Davidge ST, Guilbert LJ. The effect of oxygen tension on intracellular survival signalling in primary villous trophoblasts. Placenta. 2003;24 doi: 10.1016/s0143-4004(03)00056-0. [DOI] [PubMed] [Google Scholar]
- 30.Johnstone M, Mackova M, Das S, Payne SG, Lowen B, Sibley CP, Chan G, Guilbert LJ. Multiple anti-apoptotic pathways stimulated by EGF in cytotrophoblasts. Placenta. 2005;26:548–555. doi: 10.1016/j.placenta.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Qiu Q, Yang M, Tsang BK, Gruslin A. Both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signalling are required in epidermal growth factor-induced human trophoblast migration. Mol Hum Reprod. 2004;10:677–684. doi: 10.1093/molehr/gah088. [DOI] [PubMed] [Google Scholar]
- 32.Kamei T, Jones SR, Chapman BM, KL MC, Dai G, Soares MJ. The phosphatidylinositol 3-kinase/Akt signaling pathway modulates the endocrine differentiation of trophoblast cells. Mol Endocrinol. 2002;16:1469–1481. doi: 10.1210/mend.16.7.0878. [DOI] [PubMed] [Google Scholar]
- 33.Naegele S, Morley SJ. Molecular cross-talk between MEK1/2 and mTOR signaling during recovery of 293 cells from hypertonic stress. J Biol Chem. 2004;279:46023–46034. doi: 10.1074/jbc.M404945200. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res. 2002;91:821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 35.Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–234. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]
- 36.Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 37.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 38.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 39.Taglauer ES, Trikhacheva AS, Slusser JG, Petroff MG. Expression and Function of PDCD1 at the Human Maternal-Fetal Interface. Biol Reprod. 2008 doi: 10.1095/biolreprod.107.066324. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous anti-apoptotic receptor on cancer cells. 2008 doi: 10.1182/blood-2007-11-123141. blood-2007-2011-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]