

Summary
The antibacterial peptide microcin J25 (MccJ25) inhibits transcription by bacterial RNA polymerase (RNAP). Biochemical results indicate that inhibition of transcription occurs at the level of NTP uptake or NTP binding by RNAP. Genetic results indicate that inhibition of transcription requires an extensive determinant, comprising more than fifty amino acid residues, within the RNAP secondary channel (also known as the “NTP-uptake channel” or “pore”). Biophysical results indicate that inhibition of transcription involves binding of MccJ25 within the RNAP secondary channel. Molecular modelling indicates that binding of MccJ25 within the RNAP secondary channel obstructs the RNAP secondary channel. We conclude that MccJ25 inhibits transcription by binding within, and obstructing, the RNAP secondary channel--acting essentially as a “cork in a bottle.” Obstruction of the RNAP secondary channel represents an attractive target for drug discovery.
Introduction
Microcin J25 (MccJ25) is a 21 residue peptide antibiotic with an unusual, “lariat-protoknot” structure (Salomon and Farias, 1992; Bayro et al., 2003; Rosengren et al., 2003; Wilson et al., 2003). MccJ25 is produced by Escherichia coli strains that harbor a plasmid-borne antibiotic-synthesis and antibiotic-export cassette, consisting of a gene for MccJ25 precursor (a 58 residue linear peptide), two genes for factors that process MccJ25 precursor into MccJ25, and one gene for export of MccJ25 (Solbiati et al., 1999). MccJ25 exhibits bacteriocidal activity against a range of Gram-negative bacterial species, including E. coli.
Recently, it has been established that the functional target of MccJ25 is RNA polymerase (RNAP). Delgado et al. (2001) showed that MccJ25 inhibits transcription and identified a single-substitution MccJ25-resistant mutant of rpoC--the gene for the RNAP β′ subunit. Yuzenkova et al. (2002) confirmed that MccJ25 inhibits transcription and identified six additional single-substitution MccJ25-resistant mutants of rpoC (two affecting the same codon as in the mutant of Delgado et al., 2001; four affecting other codons).
Here, we define the mechanism by which MccJ25 inhibits transcription. Our results indicate that MccJ25 inhibits transcription by binding within, and obstructing, the RNAP secondary channel (also known as the “NTP-uptake channel” or “pore”). This represents a novel mechanism for inhibition of a nucleotide polymerase and an attractive target for antibacterial drug discovery.
Results
MccJ25 inhibits transcription at the level of NTP uptake or NTP binding by RNAP
MccJ25 does not inhibit open-complex formation
Transcription involves the following steps (Record et al. 1996): (i) RNAP binds to promoter DNA, to yield an RNAP-promoter closed complex; (ii) RNAP melts ∼14 bp of promoter DNA surrounding the transcription start site, to yield an RNAP-promoter open complex; (iii) RNAP begins synthesis of RNA, typically carrying out multiple rounds of abortive initiation (synthesis and release of RNA products <9-11 nt in length), as an RNAP-promoter initial transcribing complex; and (iv), upon synthesis of an RNA product of a critical threshold length of 9-11 nt, RNAP breaks its interactions with promoter DNA and begins to translocate along DNA, processively synthesizing RNA as an RNAP-DNA elongation complex. To determine whether MccJ25 inhibits steps in transcription up to and including formation of the RNAP-promoter open complex, we performed electrophoretic mobility-shift experiments. We incubated RNAP holoenzyme with a fluorochrome-labelled DNA fragment containing the lacUV5 promoter--in parallel in the absence and presence of MccJ25--and we analyzed products by non-denaturing PAGE followed by x/y fluorescence scanning (Fig. 1A). The results indicate that MccJ25 at a concentration of 1, 10, or 100 μM has no effect on formation of open complex (Fig. 1A). We conclude that MccJ25 does not inhibit steps in transcription up to and including formation of open complex.
Figure 1. MccJ25 inhibits transcription at the level of NTP uptake or NTP binding by RNAP.

(A) Results of electrophoretic mobility shift experiments assessing effects of MccJ25 on open-complex formation. RPo, RNAP-promoter open complex; P, promoter DNA.
(B) Results of transcription experiments assessing effects of MccJ25 on abortive initiation and elongation.
(C) Results of transcription experiments assessing effects of MccJ25 on abortive initiation and elongation with RNAP derivative bearing Thr931→Ile substitution in RNAP β′ subunit (see Delgado et al., 2001).
(D) Results of transcription experiments assessing NTP-concentration-dependence of effects of MccJ25 on abortive initiation and elongation.
(E) Double-reciprocal plot for inhibition of synthesis of 3-mer and 4-mer abortive products. Filled circles, no MccJ25; open circles, 1 μM MccJ25; filled triangles, 10 μM MccJ25; open triangles, 100 μM MccJ25. Data are from (D). Lines are fits to a partial-competitive model of inhibition (Ki = 1.4±0.2 μM; α = 15±3; r2 = 0.99). Within experimental error, Vmax is independent of MccJ25 (2.2±0.1 mol/mol template/min at 0 μM MccJ25; 1.9±0.4 mol/mol template/min at 1 μM MccJ25; 1.4±0.7 mol/mol template/min at 10 μM MccJ25).
(F) Double-reciprocal plot for inhibition of synthesis of 3-mer and 4-mer abortive products. Filled circles, no MccJ25; open circles, 1 μM MccJ25; filled triangles, 10 μM MccJ25; open triangles, 100 μM MccJ25. Data are from fluorescence-detected abortive initiation assays (see Experimental Procedures). Lines are fits to a partial-competitive model of inhibition (Ki = 1.2±0.3 μM; α = 8.7±2; r2 = 0.97). Within experimental error, Vmax is independent of MccJ25 (4.0±1.3 mol/mol template/min at 0 μM MccJ25; 5.3±2.8 mol/mol template/min at 1 μM MccJ25; 2.4±2.9 mol/mol template/min at 10 μM MccJ25).
MccJ25 inhibits abortive initiation and elongation
To determine whether MccJ25 inhibits steps in transcription subsequent to formation of the RNAP-promoter open complex, we performed standard transcription experiments. We pre-incubated RNAP holoenzyme with a DNA fragment containing the lacUV5 promoter to form the RNAP-promoter open complex; we then added radiolabelled NTPs and allowed RNA synthesis to proceed for 5 min at 37°C--in parallel in the absence and presence of MccJ25--and analyzed products by urea-PAGE followed by storage-phosphor imaging (Fig. 1B). The results indicate that MccJ25 at a concentration of 1, 10, or 100 μM inhibits both formation of abortive products (3 nt and 4 nt RNA species produced in large stoichiometric excess over the DNA template; 86% inhibition at 100 μM MccJ25) and formation of the full-length product (26 nt RNA species; produced in stoichiometric equivalence with DNA template; 94% inhibition at 100 μM MccJ25) (Fig. 1B). The inhibition is specific; thus, inhibition is overcome completely upon substitution of residue 931 of the RNAP β′ subunit--the substitution shown by Delgado et al. (2001) to confer resistance to MccJ25 in vivo (Fig. 1C). Parallel experiments starting with a halted elongation complex, rather than with open complex, yield equivalent results with respect to inhibition of formation of full-length product (Supplementary Fig. 1). We conclude that MccJ25 inhibits both abortive initiation and elongation. We infer that MccJ25 interferes with an elementary reaction step common to both abortive initiation and elongation--i.e., NTP uptake, NTP binding, phosphodiester-bond formation, or translocation.
High concentrations of NTPs overcome inhibition
To assess whether MccJ25 interferes with an NTP-concentration-dependent elementary reaction step, we performed transcription experiments at each of four NTP concentrations (12.5 μM, 25 μM, 50 μM, and 100 μM). The results indicate that high NTP concentrations can overcome inhibition by MccJ25 (Fig. 1D). As the NTP concentration increases, the extent of inhibition by MccJ25 decreases--both at the level of abortive products and at the level of the full-length product (Fig. 1D). We conclude that MccJ25 interferes with an NTP-concentration-dependent elementary reaction step.
Inhibition is partial competitive with respect to NTPs
Quantitative analysis of the NTP-concentration-dependence data indicates that mode of inhibition by MccJ25 is partial competitive--i.e., that MccJ25 binds to a site on RNA polymerase distinct from the NTP binding site and increases KM for NTPs (Fig. 1E). [In conventional, or full, competitive inhibition, binding of the inhibitor and binding of the substrate are mutually exclusive (i.e., the inhibitor increases the apparent KM for substrate to infinity) (Segel, 1973). In contrast, in partial competitive inhibition, binding of the inhibitor and binding of the substrate are mutually inhibitory, but not mutually exclusive (i.e., the inhibitor increases the apparent KM for substrate, but does not increase apparent KM for substrate to infinity) (Segel, 1973). Both full and partial competitive inhibition yield double-reciprocal plots that intersect at the y-axis; however, full and partial competitive inhibition yield double-reciprocal plots with different dependences of slope on inhibitor concentration and thus readily can be distinguished. Partial competitive inhibition implies that the inhibitor site and substrate site on the enzyme must be distinct, in part or in whole.] Ki, the reciprocal of the equilibrium binding constant for MccJ25-RNAP interaction, is estimated to be 1.4±0.2 μM; α, the factor by which MccJ25 increases KM for NTPs, is estimated to be 15±3 (Fig. 1E). Fluorescence-detected abortive initiation assays assessing iterative tri- and tetranucleotide synthesis--assays for which the initial-velocity assumption is rigorously valid--yield equivalent results: i.e., partial-competitive inhibition, with Ki = 1.2±0.3 μM, and α = 8.7±2 (Fig. 1F). We conclude that MccJ25 inhibits transcription by binding to a site on RNAP distinct from the NTP binding site and interfering with NTP uptake or NTP binding.
MccJ25 requires an extensive determinant within the RNAP secondary channel
MccJ25 requires residues that line the RNAP secondary channel
The MccJ25-resistant rpoC mutant of Delgado et al., 2001 results in substitution of residue 931 of RNAP β′ subunit (Thr931→Ile). In the primary structure of β′, residue 931 maps to conserved region G (Fig. 2A). In the three-dimensional structure of bacterial RNAP, residue 931 of β′ maps to the RNAP secondary channel (also referred to as the “NTP-uptake channel” or “pore”; Fig. 2B). The MccJ25-resistant rpoC mutants of Yuzenkova et al., 2002 also map to the RNAP secondary channel (Fig. 2B). The RNAP secondary channel is an ∼30 Å long, ∼10-15 Å wide, tunnel that connects the exterior surface of RNAP with the RNAP active center (Zhang et al., 1999; Cramer et al., 2000). According to generally accepted models, NTPs must pass through the RNAP secondary channel in order to access the RNAP active-center and NTP binding site (Zhang et al., 1999; Cramer et al., 2000; Korzheva et al., 2000; Gnatt et al., 2001; see however, Nedialkov et al., 2003). Thus, the location of the substitutions, in conjunction with our finding that MccJ25 inhibits transcription by interfering with NTP uptake, immediately suggests a possible mechanism of inhibition: i.e., MccJ25 may inhibit transcription by binding within, and obstructing, the RNAP secondary channel.
Figure 2. MccJ25 requires an extensive determinant within the RNAP secondary channel.
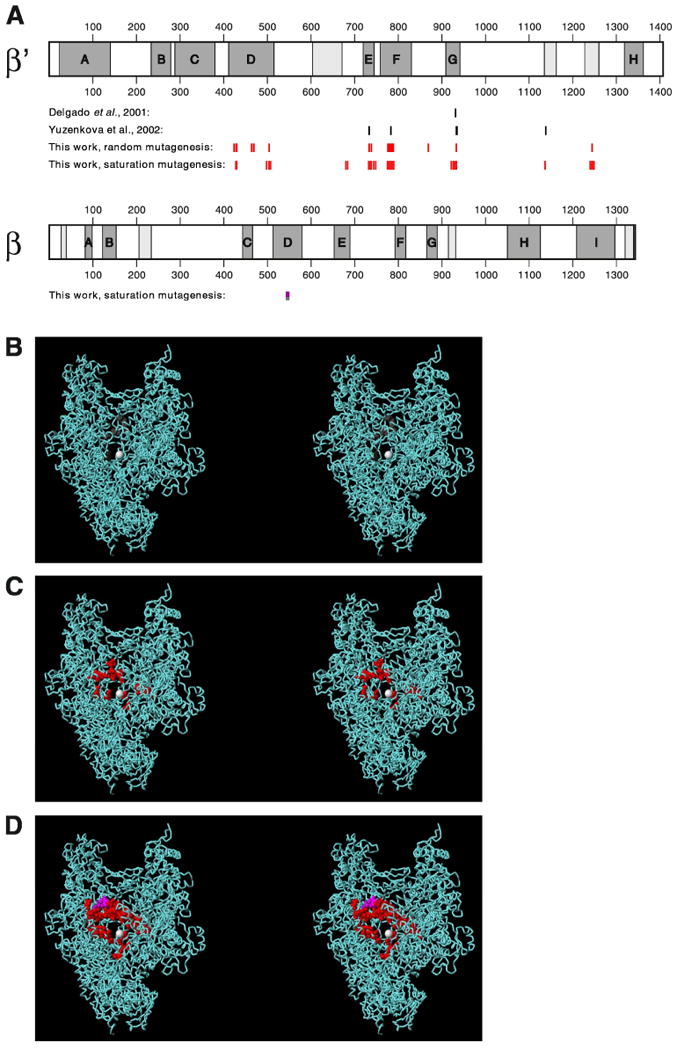
(A) Schematic maps of RNAP β′ and β subunits showing locations of conserved regions (conserved regions A-H of β′ and A-I of β lettered and shaded; additional conserved regions lightly shaded) and locations of single-residue substitutions that confer MccJ25-resistance (substitutions of Delgado et al., 2001 and Yuzenkova et al., 2003 in black; substitutions from this work in red and pink).
(B)-(D) Three-dimensional structure of RNAP showing locations of single-residue substitutions that confer MccJ25-resistance [substitutions of Delgado et al., 2001 and Yuzenkova et al., 2003 in red in (B); substitutions in from random mutagenesis in red and pink in (C); and substitutions from random and saturation mutagenesis in red and pink in (D)]. Each panel presents a stereodiagram, with a view directly into the RNAP secondary channel, toward the active-center Mg++ (white sphere at center). Atomic coordinates are based on the crystal structure of Thermus thermophilus RNAP at 2.6 Å resolution (Vassyleyev et al., 2002; PDB accession 1IW7; σ subunit omitted for clarity). Correspondences between residues of E. coli RNAP β′ and β and T. thermophilus RNAP β′ and β are based on comprehensive sequence alignments of bacterial, archaeal, and eukaryotic RNAP β′ and β homologs (R.H.E., unpublished).
To define determinants of β′ specifically required for transcription inhibition by MccJ25--and thereby to test the hypothesis that determinants for binding of MccJ25 are located within the RNAP NTP-uptake channel--we performed random mutagenesis of the entire gene encoding β′ and isolated and characterized MccJ25-resistant mutants. (RNAP β′ subunit comprises ∼50% of all residues of RNAP and comprises ∼90% of all residues of the RNAP secondary channel.) We performed mutagenesis using error-prone PCR (Zhou et al., 1991) of five DNA segments spanning the length of a plasmid-borne rpoC gene (Supplementary Table 1). Overall, we performed 20 mutagenesis reactions, analyzed ∼100,000 candidates, and isolated 22 independent plasmid-linked MccJ25-resistant mutants (Supplementary Table 1). Minimum-bacteriocidal-concentration (MBC) assays indicate that all 22 MccJ25-resistant mutants exhibit ≥5-fold increases in MBC, and 18 of 22 MccJ25-resistant mutants exhibit ≥50-fold increases in MBC (Table 1, column 5). Complementation assays indicate that all 22 MccJ25-resistant mutants can complement an rpoCts mutant for growth at the non-permissive temperature, confirming that each encodes a β′ derivative functional in transcription--indeed sufficiently functional in transcription to support viability (Table 1, column 4).
Table 1. MccJ25r isolates from random mutagenesis and selection.
| Amino acid substitution | Codon substitution | Number of independent isolates | Complementation of rpoCts | MBC* (mg/ml) |
|---|---|---|---|---|
| none | none | - | ++ | 0.01 |
| 424 Asn→Ser | AAC→AGC | 1** | + | 0.1 |
| 428 Thr→Ile | ACT→ATT | 2 | ++ | 0.5 |
| 430 His→Leu | CAC→CTC | 3 | + | 0.5 |
| 464 Asp→Gly | GAC→GCC | 1 | ++ | 0.05 |
| 469 His→Arg | CAC→CGC | 1 | ++ | 0.05 |
| 504 Gln→Arg | CAG→CGG | 1 | ++ | 0.5 |
| 733 Ser→Phe | TCT→TTC | 1 | + | 0.5 |
| 738 Arg→Leu | CGT→CTT | 1 | ++ | 0.05 |
| 776 Thr→Ile | ACC→ATC | 1 | ++ | 0.5 |
| 779 Ala→Thr | GCT→ACT | 1 | ++ | 1 |
| 780 Arg→Cys | CGT→TGT | 1 | ++ | 1 |
| 782 Gly→Ala | GGT→GCT | 1 | ++ | 1 |
| 785 Asp→Gly | GAT→GGT | 1 | ++ | 0.5 |
| 786 Thr→Ile | ACC→ATC | 1 | ++ | 0.5 |
| 789 Lys→Arg | AAA→AGA | 1 | ++ | 0.5 |
| 789 Lys→Gln | AAA→CAA | 1 | ++ | 1 |
| 869 Cys→Arg | TGT→CGT | 1 | + | 1 |
| 933 Arg→Cys | CGT→TGT | 1 | ++ | 1 |
| 1244 Gln→Leu | CAG→CTG | 1 | ++ | 0.5 |
Minimum bacteriocidal concentration; defined as the lowest concentration of MccJ25 that yields a viable cell count of ∼0 after incubation 2 h at 37°C (see Experimental Procedures).
Isolated as double mutant 354 Val→Ile; 424 Asn→Ser; complementation and MBC data are for a single mutant constructed using site-directed mutagenesis.
For each of the 22 MccJ25-resistant mutants, the DNA-nucleotide sequence of the relevant segment of the rpoC gene was determined, and the amino acid sequence of the substituted β′ derivative was inferred (Table 1). Nineteen different substitutions, involving eighteen different sites within β′, were obtained (Table 1).
In the primary structure of β′, the sites at which substitutions were obtained map to conserved region D, conserved region E, conserved region F, the segment between conserved regions F and G, conserved region G, and the segment between conserved regions G and H (referred to as conserved region G′ by Zakharova et al., 1998) (Fig. 2A). In the three-dimensional structure of RNAP, the locations of the substitutions are tightly clustered--and are centered on the RNAP secondary channel (Fig. 2C). The substitutions, without exception, map to residues that line the RNAP secondary channel, or to residues that make direct contact with residues that line the RNAP secondary channel (Fig. 2C). The substitutions map to the floor, the roof, and the walls of RNAP secondary channel (Fig. 2C).
We conclude that the RNAP secondary channel contains a multi-residue determinant for function of MccJ25. Based on the fact that substitutions conferring MccJ25-resistance were obtained at none of the >1000 residues of β′ outside the immediate vicinity of the secondary channel, we tentatively conclude that no part of β′ outside the immediate vicinity of the secondary channel contains a determinant for function of MccJ25.
MccJ25 requires more than fifty residues that line the RNAP secondary channel
To define systematically the MccJ25 determinant within the RNAP secondary channel, we performed saturation mutagenesis of the rpoC gene, encoding β′, and the rpoB gene, encoding β, targeting all codons for residues that line the RNAP secondary channel. We performed saturation mutagenesis using a set of eleven “doped” oligodeoxyribonucleotide primers, designed to introduce all possible nucleotide substitutions at all positions of all codons for residues that line the RNAP secondary channel (sequences in Supplementary Table 2; methods as in Ner et al., 1988; Hermes et al., 1989). In total, we performed 25 mutagenesis reactions, analyzed ∼40,000 candidates, and isolated and characterized 114 independent plasmid-linked MccJ25-resistant mutants (Supplementary Table 3).
Sequencing indicates that 106 of the 114 MccJ25-resistant mutants are single-substitution mutants (Table 2). The single-substitution mutants from saturation mutagenesis comprise 75 different substitutions, involving 43 different sites within β′ and 4 different sites within β (Table 2). Taken together, the single-substitution mutants from random mutagenesis and saturation mutagenesis comprise 80 different substitutions, involving 47 different sites within β′ and 4 different sites within β (Tables 1,2).
Table 2. MccJ25r isolates from saturation mutagenesis and selection.
| Amino acid substitution | Number of independent isolates | MBC* (mg/ml) |
|---|---|---|
| rpoC | ||
| single-substitution mutants | ||
| 428 Thr→Ile | 3 | 0.5 |
| 428 Thr→Asn | 2 | 0.5 |
| 429 Leu→Gln | 2 | 0.5 |
| 430 His→Tyr | 1 | 0.5 |
| 498 Pro→Leu | 1 | 1 |
| 498 Pro→Gln | 1 | 1 |
| 503 Ser→Pro | 1 | 0.5 |
| 503 Ser→Tyr | 1 | 0.5 |
| 504 Gln→Arg | 1 | 0.5 |
| 504 Gln→Glu | 2 | 0.5 |
| 508 Leu→Val | 1 | 0.05 |
| 680 Asn→Lys | 2 | 0.5 |
| 684 Asp→Ala | 1 | 1 |
| 684 Asp→Tyr | 1 | 0.5 |
| 684 Asp→Glu | 1 | 0.5 |
| 684 Asp→Val | 1 | 0.5 |
| 732 Gly→Asp | 2 | 0.1 |
| 733 Ser→Phe | 1 | 0.1 |
| 733 Ser→Val | 1 | 0.5 |
| 733 Ser→Tyr | 1 | 0.1 |
| 735 Ala→Δ | 2 | 1 |
| 736 Gln→Pro | 1 | 0.5 |
| 738 Arg→Leu | 1 | 0.05 |
| 744 Arg→Pro | 1 | 0.05 |
| 744 Arg→His | 1 | 0.05 |
| 748 Ala→Pro | 3 | 0.1 |
| 775 Ser→Cys | 1 | 0.5 |
| 776 Thr→Ile | 3 | 0.5 |
| 777 His→Tyr | 1 | 1 |
| 777 His→Pro | 2 | 0.5 |
| 779 Ala→Gly | 1 | 0.5 |
| 779 Ala→Thr | 1 | 1 |
| 779 Ala→Pro | 2 | 0.5 |
| 780 Arg→Cys | 1 | 1 |
| 782 Gly→Ala | 1 | 1 |
| 783 Leu→Pro | 1 | 0.1 |
| 784 Ala→Glu | 1 | 0.5 |
| 785 Asp→Gly | 2 | 0.5 |
| 786 Thr→Ile | 1 | 0.5 |
| 786 Thr→Ala | 2 | 0.5 |
| 788 Leu→Met | 1 | 1 |
| 789 Lys→Asn | 2 | 1 |
| 789 Lys→Gln | 1 | 1 |
| 789 Lys→Arg | 1 | 0.5 |
| 790 Thr→Ala | 2 | 1 |
| 790 Thr→Ile | 2 | 1 |
| 790 Thr→Ser | 2 | 0.5 |
| 790 Thr→Asn | 1 | 0.5 |
| 922 Cys→Tyr | 1 | 0.5 |
| 926 Pro→Ser | 1 | 0.5 |
| 927 Gly→Ser | 2 | 1 |
| 927 Gly→Cys | 1 | 1 |
| 930 Leu→Met | 1 | 1 |
| 931 Thr→Ile | 2 | 1 |
| 931 Thr→Ala | 2 | 1 |
| 932 Met→Ile | 2 | 1 |
| 933 Arg→Cys | 1 | 1 |
| 1136 Gly→Cys | 1 | 1 |
| 1136 Gly→Ala | 1 | 0.5 |
| 1137 Gly→Ala | 1 | 0.5 |
| 1137 Gly→Arg | 1 | 1 |
| 1240 Val→Glu | 1 | 0.5 |
| 1241 Tyr→Ser | 2 | 0.1 |
| 1241 Tyr→His | 2 | 0.1 |
| 1241 Tyr→Cys | 1 | 0.1 |
| 1244 Gln→Pro | 2 | 0.1 |
| 1244 Gln→Leu | 1 | 0.5 |
| 1244 Gln→Glu | 2 | 0.1 |
| 1247 Lys→Glu | 2 | 0.1 |
| 1247 Lys→Gln | 1 | 0.1 |
| 1248 Ile→Ser | 1 | 0.1 |
| multiple-substitution mutants | ||
| 493 Pro→Thr; 498 Pro→Thr | 1 | 0.1 |
| 732 Gly→Asp;733 Ser→Ala | 1 | 0.1 |
| 732 Gly→Asp;735 Gly→Thr | 1 | 0.5 |
| 733 Ser→Val;734 Ala→Gly | 1 | 0.1 |
| 777 His→Ser;778 Gly→Ala | 1 | 0.5 |
| rpoB | ||
| single-substitution mutants | ||
| 544 Gly→Asp | 1 | 0.05 |
| 545 Phe→Ser | 2 | 0.05 |
| 548 Arg→Gln | 2 | 0.05 |
| 549 Asp→His | 1 | 0.05 |
| multiple-substitution mutants | ||
| 543 Ala→Pro; 546 Glu→Gly | 1 | 0.05 |
| 543 Ala→Leu; 549 Asp→Tyr | 1 | 0.05 |
| 545 Phe→Leu; 546 Glu→Asp | 1 | 0.05 |
Minimum bacteriocidal concentration; defined as the lowest concentration of MccJ25 that yields a viable cell count of ∼0 after incubation 2 h at 37°C (see Experimental Procedures).
In the three-dimensional structure of RNAP, the sites at which single substitutions were obtained define a nearly continuous surface, comprising most of the interior lining and part of the rim of the RNAP secondary channel (Fig. 2D). The sites span nearly the full circumference of the RNAP secondary channel (Fig. 2D). The side chains of the majority of implicated residues are solvent-accessible--directed into the lumen of the RNAP secondary channel or toward the exterior of RNAP--and make no obvious interactions important for RNAP structure or function.
We conclude that the RNAP secondary channel contains an extensive determinant for function of MccJ25. Based on the size of the determinant (more than fifty residues; Tables 1 and 2), the architecture of the determinant (interior of hollow cylinder), and the solvent-accessibility of the determinant, we propose that the determinant corresponds to the binding site on RNAP for MccJ25. We propose that the sites of substitutions that confer MccJ25-resistance map the binding site on RNAP for MccJ25 and, in effect, serve as a genetic footprint of the binding site.
Thirteen of fifteen sites associated with the highest level of MccJ25-resistance (MBC = 1 mg/ml; Tables 1 and 2) cluster in an ∼20 Å × ∼20 Å × ∼20 Å sub-region of the RNAP secondary channel, bounded by the α-helix containing residue 684 (D/E helix), the α-helix containing residue 735 (E helix), the α-helix containing residues 777-790 (F helix), the α-helix and loop containing residues 927-933 (G helix and loop), and loop containing residues 1136-1137 (G′ loop) (sub-region above and to left of the active-center Mg++ in view in Fig. 2D). We infer that this sub-region is the most important part of the determinant.
MccJ25 binds within the RNAP secondary channel
MccJ25 binds to RNAP
To determine whether MccJ25 binds to RNAP, we performed fluorescence resonance energy transfer (FRET) binding experiments (Selvin, 2000). We prepared a fluorescent-probe-labelled MccJ25 derivative, Cy3-MccJ25, and verified that Cy3-MccJ25 was functional in transcription inhibition. We then titrated a fluorescent-probe-labelled RNAP derivative, DAC-σ70 RNAP holoenzyme, with Cy3-MccJ25 in the concentration range 0-20 μM and monitored FRET. The results reveal a large, saturable increase in FRET, indicating that Cy3-MccJ25 binds to RNAP (Fig. 3A, filled circles). Analysis of the concentration dependence of the increase in FRET indicates that the equilibrium dissociation constant, Kd, for interaction of Cy3-MccJ25 with RNAP is 1.4 μM (Fig. 3A). Stopped-flow analysis of the kinetics of interaction of Cy3-MccJ25 with RNAP indicates that the association rate constant, kon, is 7 × 105 M-1 s-1, and the dissociation rate constant, koff, is 0.7 s-1 (Supplementary Fig. 2). The value of Kd estimated from stopped-flow analysis is 1 μM (Kd = koff/kon), which, within experimental error, agrees with the value of Kd estimated from direct titration.
Figure 3. MccJ25 binds within the RNAP secondary channel.

(A) Binding of Cy3-MccJ25 to DAC-σ70 RNAP holoenzyme (filled circles) or DAC-σ70 [Ile931]β′ RNAP holoenzyme (open circles).
(B) Competition by MccJ25 for binding of Cy3-MccJ25 to DAC-σ70 RNAP holoenzyme.
(C) Competition by GreB for binding of Cy3-MccJ25 to DAC-σ70 RNAP holoenzyme.
(D) Results of systematic FRET measurements defining distances between Cy3 and DAC chromophores in complexes of Cy3-MccJ25 and DAC-labelled RNAP holoenzyme derivatives (ten with DAC at sites within σ70, two with DAC at sites within ω). E, FRET efficiency; Ro, Förster parameter; R, distance.
(E) Results of systematic FRET measurements and distance-restrained docking defining possible positions of Cy3 chromophore in complexes of Cy3-MccJ25 and DAC-labelled RNAP holoenzyme derivatives (view as in Fig. 2). Green spheres, possible positions of Cy3 chromophore (125 highest-ranked solutions; see Supplementary Information: Distance-restrained docking); yellow spheres, DAC sites within σ70; orange spheres, DAC sites within ω; red and pink van der Waals surfaces, sites of substitutions in β′ and β that confer MccJ25-resistance; white sphere, active-center Mg++; cyan ribbons, β′, β, α, and ω; yellow ribbon, σ70.
MccJ25 binds to RNAP with Kd comparable to Ki
To determine the equilibrium dissociation constant, Kd, for interaction of native, unlabelled MccJ25 with RNAP, we performed FRET competition experiments. We started with complexes of Cy3-MccJ25 and DAC-σ70 RNAP holoenzyme, challenged complexes with unlabelled MccJ25 in the concentration range 0-20 μM, and monitored FRET. The results reveal a concentration-dependent decrease in FRET, indicating that unlabelled MccJ25 competes with Cy3-MccJ25 for binding to RNAP (Fig. 3B). Analysis of the concentration dependence of the decrease in FRET and application of the Cheng-Prusoff equation (Cheng and Prusoff, 1973), indicates that the equilibrium dissociation constant, Kd, for interaction of unlabelled MccJ25 with RNAP is 0.5 μM (Fig. 3B). Within experimental error, Kd for interaction of unlabelled MccJ25 with RNAP is equal to Ki for inhibition of transcription (1.2±0.3 μM; Fig. 1E,F). We infer that the same binding process is monitored by the FRET experiments as is monitored in the transcription-inhibition experiments.
Binding requires the genetically defined determinant within the RNAP secondary channel
To determine whether binding of MccJ25 requires the genetically defined determinant within the RNAP secondary channel (Fig. 2B-E), we performed FRET binding experiments with Cy3-MccJ25 and a fluorescent-probe-labelled RNAP derivative having a single-amino-acid substitution within the determinant (Thr931→Ile; see Fig. 1C and Table 2; see also Delgado et al., 2001; Yuzenkova et al., 2002). Cy3-MccJ25 exhibited no detectable interaction with the substituted RNAP derivative in the concentration range analyzed (Fig. 3A, open circles). We conclude that binding of MccJ25 requires the genetically defined determinant within the RNAP secondary channel. We infer that the genetically defined determinant represents a binding determinant for MccJ25 (as opposed to a conformational determinant required for MccJ25 function but not for MccJ25 binding).
Binding occurs within the RNAP secondary channel: competition with GreB
To test the hypothesis that Cy3-MccJ25 binds within the RNAP secondary channel, we performed FRET competition experiments with Cy3-MccJ25 and the transcript-cleavage factor GreB, which has been shown to bind within the RNAP secondary channel (Opalka et al., 2003; Laptenko et al., 2003; Sosunova et al., 2003). We started with complexes of Cy3-MccJ25 and DAC-σ70 RNAP holoenzyme, challenged complexes with GreB in the concentration range 0-1 μM, and monitored FRET. The results show that GreB competes with Cy3-MccJ25 for binding to RNAP (Fig. 3C). The calculated value of Kd for interaction of GreB with RNAP is 0.05 μM, which, within experimental error, agrees with previously reported values (Koulich et al., 1997) (Fig. 3C). We conclude that binding of Cy3-MccJ25 to RNAP and binding of GreB to RNAP are mutually exclusive. We infer that the binding location for MccJ25 overlaps the binding location for GreB: i.e., that MccJ25 binds within, or immediately adjacent to, the RNAP secondary channel.
Binding occurs within the RNAP secondary channel: systematic FRET
To test further the hypothesis that Cy3-MccJ25 binds within the RNAP secondary channel, and to define the location of Cy3-MccJ25 relative to RNAP within the Cy3-MccJ25-RNAP complex by a direct, physical approach, we performed systematic FRET measurements of distances and distance-restrained docking (methods essentially as in Mekler et al., 2002). We used FRET to measure the distances between the Cy3 probe of Cy3-MccJ25 and DAC probes incorporated into RNAP holoenzyme at each of ten sites in σ70 (Fig. 3D, left) and each of two sites in ω (Fig. 3D, right). We then used automated distance-restrained docking to define locations of the Cy3 probe of Cy3-MccJ25 relative to RNAP holoenzyme consistent with the measured distances (green spheres in Fig. 3E). The results indicate that the Cy3 probe of MccJ25 is located in the RNAP secondary channel, at the external mouth of the RNAP secondary channel (the mouth distal to the RNAP active center; green spheres in Fig. 3E). The Cy3 probe of MccJ25 is located immediately adjacent to the genetically defined determinant within the RNAP secondary channel (green spheres and red van der Waals surfaces in Fig 3E). We conclude that MccJ25 binds within, or immediately adjacent to, the RNAP secondary channel.
MccJ25 obstructs the RNAP secondary channel
MccJ25 is a 21-residue “lariat protoknot,” consisting of an 8-residue cyclic segment followed by a 13-residue linear segment that loops back and threads through the cyclic segment (Fig. 4A; Bayro et al., 2003; Rosengren et al., 2003; Wilson et al., 2003). MccJ25 has dimensions of ∼27 Å × ∼14 Å and is roughly comma-shaped, with the cycle and threaded segment (residues 1-8 and 18-21) corresponding to the head of the comma, and with the connector segment (residues 9-17) corresponding to the tail of the comma (Fig. 4A).
Figure 4. MccJ25 obstructs the RNAP secondary channel.

(A) Structure of MccJ25 (Bayro et al., 2003; PDB accession 1PP5; see also Rosengren et al., 2003; Wilson et al., 2003).
(B,C) Model for the structure of the MccJ25-RNAP complex (view orientation as in Figs. 2,3). Green van der Waals surface, MccJ25 (oriented as in panel A); red and pink van der Waals surfaces, sites of substitutions in β′ and β that confer MccJ25-resistance; yellow labels, sites of substitutions in β′ that confer highest-level MccJ25-resistance (MBC = 1 mg/ml).
MccJ25 exhibits structural complementarity, in both size and shape, to the RNAP secondary channel. In cross-section, the RNAP secondary channel has dimensions of ∼28 Å × ∼14 Å and is roughly comma-shaped, with the secondary-channel sub-region bounded by the β′ D/E helix, E helix, F-helix, and G-helix and loop corresponding to the head of the comma, and with the secondary-channel sub-region bounded by β′ residues 498-504, 732-733, 922-926, and 1244-1248 corresponding to the tail of the comma.
We have used this structural complementarity to dock the structure of MccJ25 into the structure of RNAP in order to construct a model for the structure of the MccJ25-RNAP complex (Fig. 4B,C). In the resulting model, the cycle and threaded segment of MccJ25 (the head of the comma) is placed in the secondary-channel sub-region bounded by the β′ D/E helix, E helix, F-helix, and G-helix and G-loop; and the connector segment of MccJ25 (the tail of the comma) is placed in the secondary-channel sub-region bounded by β′ residues 498-504, 732-733, 922-926, and 1244-1248 (Fig. 4B,C). The resulting model is supported by four observations. First, the model is sterically acceptable (Fig. 4B,C). Second, the model accounts for genetic data defining critical determinants of RNAP (Fig. 2; Tables 1,2)--placing MccJ25 in contact with, or adjacent to, the majority of sites of substitutions conferring MccJ25-resistance (red and pink residues in Fig. 4B,C), and, in particular, placing the cycle and threaded segment of MccJ25 in contact with, or adjacent to, the majority of sites of substitutions conferring highest-level MccJ25-resistance (labelled residues in Fig. 4C). Third, the model accounts for genetic and biochemical data defining critical determinants of MccJ25 (J.M., E.S., and R.H.E, unpublished data)--placing essential residues of MccJ25 on the face of MccJ25 directed toward RNAP, and placing non-essential residues of MccJ25 on the face of MccJ25 directed toward solvent. Fourth, the model accounts for systematic-FRET/distance-restrained-docking data defining possible locations of the Cy3 probe of Cy3-MccJ25 (Fig. 3E)--placing the attachment site of Cy3 on the face of MccJ25 directed toward the external mouth of the secondary channel, at a position of MccJ25 close to the external mouth of the secondary channel.
The most striking feature of the model is that it suggests that MccJ25 essentially completely seals the RNAP secondary channel--like a “cork in a bottle” (Fig. 4B,C). The model suggests that binding of MccJ25 would block passage of molecules the size of NTPs and possibly also would block passage of smaller molecules. It has been proposed that NTPs pass through the RNAP secondary channel to enter the RNAP active center (Zhang et al., 1999; Cramer et al., 2000; Korzheva et al., 2000; Gnatt et al., 2001; see however, Nedialkov et al., 2003), and that abortive RNA fragments, edited RNA fragments, and backtracked RNA segments pass through the secondary channel to exit the active center (Cramer et al., 2000; Korzheva et al., 2000; Gnatt et al., 2001). The model suggests that binding of MccJ25 would block these transactions. It also has been proposed that pyrophosphate may pass through the RNAP secondary channel to exit the RNAP active center. The model suggests that binding of MccJ25 might block this transaction. Finally, it has been shown that the transcript-cleavage factors GreA, GreB, and TFIIS must enter the secondary channel to access the active center (Opalka et al., 2003; Laptenko et al., 2003; Sosunova et al., 2003; Kettenberger et al., 2003). The model suggests that binding of MccJ25 would block function of transcript-cleavage factors, consistent with the observed binding competition between Cy3-MccJ25 and GreB (Fig. 3C).
Discussion
Our results establish that MccJ25 inhibits the abortive-initiation and elongation phases of transcription, that inhibition involves interference with NTP uptake or NTP binding, and that inhibition is partial-competitive with respect to NTPs (i.e., involves a site distinct from the RNAP NTP binding site). Our results further establish that inhibition involves an extensive determinant within the RNAP secondary channel, comprising nearly the entire lining of the RNAP secondary channel (more than fifty sites for substitutions conferring MccJ25-resistance). Our results further establish that MccJ25 binds within the RNAP secondary channel and suggest that binding of MccJ25 within the RNAP secondary channel obstructs the RNAP secondary channel.
The RNAP secondary channel (also referred to as the “NTP-uptake channel” or “pore”) is an ∼30 Å long, ∼10-15 Å wide, fully enclosed tunnel that connects the exterior surface of RNAP with the RNAP active center (Zhang et al., 1999; Cramer et al., 2000). In structural models of the transcription complexes responsible for abortive initiation and elongation, the RNAP secondary channel is the sole evident solvent-accessible path between the exterior surface of RNAP and the RNAP active center (Zhang et al., 1999; Cramer et al., 2000; Korzheva et al., 2000; Gnatt et al., 2001). Therefore, it has been proposed that NTPs must pass through the RNAP secondary channel in order to access the RNAP active-center and NTP binding site (Zhang et al., 1999; Cramer et al., 2000; Korzheva et al., 2000; Gnatt et al., 2001). Based on the results presented here, we conclude that MccJ25 inhibits transcription by interfering with NTP uptake by binding within, and obstructing, the RNAP secondary channel--acting essentially as a “cork in a bottle.” Severinov and co-workers, using different experimental approaches, have reached the same conclusion (personal communication).
Obstruction of the RNAP secondary channel represents a novel mechanism of inhibition of RNAP. Rifampicin inhibits bacterial RNAP by sterically preventing synthesis of an RNA product longer than ∼4 nt (Campbell et al., 2001). Streptolydigin, arylhydroxamidines, and pyrazoles are proposed to inhibit bacterial RNAP through interference with active-center conformational changes associated with phosphodiester-bond formation and/or translocation (Epshtein et al., 2002; Artsimovitch et al., 2003). α-Amanitin is proposed to inhibit eukaryotic RNAP II through interference with active-center conformational changes associated with phosphodiester-bond formation and/or translocation (Bushnell et al., 2002).
Several arguments suggest that obstruction of the RNAP secondary channel represents an exceptionally attractive target for development of novel antibacterial agents. First, the RNAP secondary channel is eminently “druggable,” presenting an extended, encircling surface complementary to a range of molecules--like MccJ25--that have molecular weights of 500-2,500 Da. Second, the RNAP secondary channel exhibits distinct patterns of sequence conservation in bacterial RNAP and eukaryotic RNAP, permitting identification of agents--like MccJ25--that inhibit bacterial RNAP but do not inhibit eukaryotic RNAP. Third, the RNAP secondary channel is distinct from the binding site of the RNAP inhibitor in current use in antibacterial therapy, rifampicin, permitting identification of agents--like MccJ25 (E.S. and R.H.E., unpublished data)--that do not exhibit cross-resistance with rifampicin.
We note that a FRET competition assay employing fluorochrome-labelled MccJ25 and fluorochrome-labelled RNAP provides an effective, high-throughput-screening-compatible, means to identify agents that bind within the RNAP secondary channel (Fig. 3 B,C). In unpublished work, we have used this assay to identify additional agents that bind within, and obstruct, the RNAP secondary channel (J.M. and R.H.E., unpublished data).
Key priorities for future work include: (i) determination of the three-dimensional structure of the MccJ25-RNAP complex, (ii) construction of “minimized” analogs of MccJ25 (analogs trimmed to the shortest length consistent with function), and (iii) isolation of novel secondary-channel-directed inhibitors. The results will provide information about RNAP structure and function, research tools for dissection of specific steps in transcription, and lead compounds for antibacterial therapy.
Experimental Procedures
Plasmids
Plasmids used in this work are described in Supplementary Information: Plasmids.
MccJ25
MccJ25 was prepared essentially as in Blond et al. 1999 (details in Supplementary Information: MccJ25).
Cy3-MccJ25
Cy3-MccJ25 was prepared using Lys-specific chemical modification. Reaction mixtures contained (200 μl): 1 mM [Lys13]MccJ25 derivative (prepared as for MccJ25, but using plasmid pTUC202-13K), 2 mM Cy3 NHS-ester (Amersham-Pharmacia Biotech), and 0.5 % (v/v) triethylamine in dimethylsulfoxide. Following 15 h at 4°C, products were purified by reversed-phase HPLC on a C18, 5 μm, 300 Å column (Rainin), with solvent A = 0.1% trifluoroacetic acid, solvent B = 90% acetonitrile and 0.1% trifluoroacetic acid, and gradient = 25-40% solvent B in solvent A in 30 min, and flow rate = 1 ml/min. Fractions containing Cy3-MccJ25 (retention time ∼29 min; detected by UV absorbance at 276 nm) were pooled, lyophilized, re-dissolved in 500 μl 30% methanol, and stored in aliquots at -20°C.
RNAP
RNAP core and holoenzyme were prepared as in Niu et al., 1996. [Ile931]β′ RNAP core and holoenzyme were prepared from strain 397c [rpoCts397 argG thi lac (λcI857h80St68dlac+); Christie et al., 1996] transformed with a pRL663 derivative encoding [Ile931]β′-RNAP using analogous procedures. Yields typically were 6 mg. Purities typically were >95%.
DAC-σ70 RNAP
DAC-σ70 was prepared using Cys-specific chemical modification. Reaction mixtures contained (1 ml): 20 μM single-Cys σ70 derivative [prepared as in Mukhopadhyay et al., 2001; subjected to solid-phase reduction on Reduce-Imm (Pierce) per manufacturer's instructions immediately before use], 200 μM N-(7-dimethylamino-4- methylcoumarin-3-yl)maleimide (Molecular Probes), 100 mM sodium phosphate (pH 8.0), and 1 mM EDTA. Following 1 h at 4°C, products were purified by gel-filtration chromatography on Bio-Gel P6DG (Bio-Rad), and stored in 20 mM Tris-HCl (pH 7.9), 100 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT, and 50% glycerol at -20°C. Efficiencies of labelling, determined by measurement of UV-absorbance, were ∼90%; site-specificities of labelling, determined by comparison to products of control reactions with wild-type σ70, were ∼90%.
DAC-σ70 RNAP holoenzyme and DAC-σ70 [Ile931]β′ RNAP holoenzyme were prepared by incubation of 5 pmol unlabelled RNAP core and [Ile931]β′ RNAP core, respectively, with 4 pmol DAC-σ70 in 20 μl TB for 20 min at 25°C.
DAC-ω RNAP
DAC-ω was prepared using Cys-specific chemical modification. Reaction mixtures contained (1 ml): 20 μM single-Cys ω derivative [prepared essentially as described for ω in Mekler et al., 2002; subjected to solid-phase reduction on Reduce-Imm (Pierce) per manufacturer's instructions immediately before use], 200 μM N-(7-dimethylamino-4- methylcoumarin-3-yl)maleimide (Molecular Probes), 100 mM sodium phosphate (pH 8.0), 1 mM EDTA, and 6 M guanidine hydrochloride. Following 1 h at 25°C, products were purified by gel-filtration chromatography on Bio-Gel P6DG (Bio-Rad,). Site-specificities of labelling, determined by comparison to products of control reactions with wild-type ω, were ∼90%. DAC-ω RNAP holoenzyme and DAC-ω [Ile931]β′ RNAP were prepared by reconstitution as described for unlabelled RNAP holoenzyme in Naryshkin et al., 2001, using 500 μg β′ or [Ile931]β′, 300 μg β, 30 μg hexahistidine-tagged α, and 250 μg σ70, including 100 μg DAC-ωn, and including a final incubation for 45 min at 30°C. Products were purified using metal-ion-affinity chromatography on Ni:NTA-agarose and ion-exchange chromatography on Mono-Q (methods essentially as in Naryshkin et al., 2001 and Niu et al., 1996); concentrated (methods essentially as in Naryshkin et al., 2001); and stored in 20 mM Tris-HCl (pH) 7.9, 100 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT, and 50% glycerol at -20°C.
Electrophoretic mobility shift assays
Reaction mixtures contained (20 μl): 100 nM RNAP holoenzyme, 20 nM DNA fragment lacUV5-12(Cy5,+26) (Mukhopadhyay et al., 2001), and 0-100 μM MccJ25 in TB [50 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μg/ml bovine serum albumin, and 5% glycerol]. Following 15 min at 37°C, 0.5 μl 1 mg/ml heparin was added (to disrupt non-specific complexes), and, following a further 2 min at 37°C, reaction mixtures were applied to 5% polyacrylamide slab gels (30:1 acrylamide/bisacrylamide; 6 × 9 × 0.1 cm) and electrophoresed in 90 mM Tris-borate (pH 8.0) and 0.2 mM EDTA (20 V/cm; 30 min at 37°C) and analyzed using a fluorescence scanner (Storm 860; Molecular Dynamics). Identities of tri- and tetranucleotide abortive products were defined as in Borowiec and Gralla (1985).
Transcription assays
Reaction mixtures contained (18 μl): 100 nM RNAP holoenzyme, 20 nM DNA fragment lacUV5-12(Cy5,+26) (Mukhopadhyay et al., 2001), and 0-100 μM MccJ25 in TB. Following 15 min at 37°C, 0.5 μl 1 mg/ml heparin was added, and, following a further 2 min at 37°C, RNA synthesis was initiated by addition of 2 μl 5 mM ApA and 125 μM (or 250 μM, 500 μM, and 1 mM) each of [α-32P]UTP (0.6 Bq/fmol), ATP, CTP, GTP. Following 5 min at 37°C, reactions were terminated by addition of 10 μl 80% formamide, 10 mM EDTA, 0.04% bromophenol blue, and 0.04% xylene cyanol. Products were heated 10 min at 90°C, resolved by urea-PAGE (Sambrook and Russel, 2001), and quantified using a storage-phosphor scanner (Storm 860; Molecular Dynamics). Data were fit to full-competitive, partial-competitive, full-noncompetitive, partial-noncompetitive, full-uncompetitive, partial-uncompetitive, full-mixed, and partial-mixed models of inhibition using the Fit-To-Model feature of the SigmaPlot Enzyme Kinetics Module (SPSS).
Fluorescence-detected abortive initiation assays
Reaction mixtures contained (46.5 μl): 100 nM RNAP holoenzyme, 20 nM DNA fragment lacUV5-12 (Mukhopadhyay et al., 2001), and 0-100 μM MccJ25 in TB. Following 15 min at 37°C, 0.5 μl 1 mg/ml heparin was added, and, following a further 2 min at 37°C, 1 μl of 0.25-5 mM (γ-AmNS)UTP (Molecular Probes) was added, and reaction mixtures were transferred to sub-micro fluorometer cuvettes (Starna Cells). Following 2 min at 37°C, RNA synthesis was initiated by addition of 2.5 μl 10 mM ApA, and fluorescence emission intensity was monitored 5 min at 37°C [excitation wavelength = 360 nm and emission wavelength = 500 nm; excitation and emission slit widths = 4 nm; QuantaMaster QM1 spectrofluorometer (PTI)]. The quantity of UMP incorporated into RNA was determined from the quantity of (γ-AmNS)UTP consumed, which, in turn, was calculated as follows (Schlageck et al. 1979):
| (1) |
where (γ-AmNS)UTP0 is the quantity of (γ-AmNS)UTP at time 0, F0 is the fluorescence emission intensity at time 0, and Ft is the fluorescence emission intensity at time t. Data were fit to full-competitive, partial-competitive, full-noncompetitive, partial-noncompetitive, full-uncompetitive, partial-uncompetitive, full-mixed, and partial-mixed models of inhibition as in the preceding section.
FRET binding assays
Assay mixtures (50 μl) contained 100 nM DAC-σ70 RNAP holoenzyme (labelled at residue 517 of σ70) and 0-20 μM Cy3-MccJ25 in TB at 25°C. Fluorescence emission intensities were measured before and 5 min after addition of 1 μl 2 mM unlabeled MccJ25 [excitation wavelength = 385 nm; emission wavelength = 465 nm; excitation and emission slit widths = 5 nm; QuantaMaster QM1 spectrofluorometer (PTI)]. Efficiencies of fluorescence resonance energy transfer (E) and fractional saturations (θ) were calculated as:
| (2) |
| (3) |
where F and Fx are emission intensities before and after addition of 40 μM unlabeled MccJ25, and where Emax is E at saturation. The equilibrium dissociation constant (Kd,Cy3-MccJ25) was extracted by non-linear regression using the equation:
| (4) |
where [Cy3-MccJ25] is the concentration of Cy3-MccJ25.
To assess kinetics of binding, using a stopped-flow instrument (SFM-4/QS; Bio-Logic), 100 μl 5 μM or 10 μM Cy3-MccJ25 in TB was mixed with 100 μl 100 nM DAC-σ70 RNAP in TB, and fluorescence emission intensity was monitored as a function of time following mixing [excitation wavelength = 400 nm; emission wavelength = 455 nm; QuantaMaster QM1 spectrofluorometer (PTI); data from four shots averaged]. The observed rate of complex formation, kobs, the association rate constant, kon, and the dissociation rate constant, koff, were calculated as:
| (5) |
| (6) |
| (7) |
where F is fluorescence emission intensity at time, t; Fo + a is the fluorescence emission intensity at t = 0; Fo is fluorescence emission intensity at t = ∞; and kobs,1 and kobs,2 are observed rates at [Cy3-MccJ25]1 and [Cy3-MccJ25]2.
FRET competition assays
Assay mixtures (50 μl) contained 100 nM DAC-σ70 RNAP holoenzyme (labelled at residue 517 of σ70), 5 μM Cy3-MccJ25, and 0-20 μM competitor [MccJ25 or GreB (prepared as in Koulich et al., 1997)] in TB at 25°C. Fluorescence emission intensities were measured before and 5 min after addition of 1 μl 2 mM unlabeled MccJ25, and E and θ were calculated as above. Equilibrium dissociation constants (Kd) were calculated as:
| (8) |
where IC50 is the concentration of competitor that yields half-maximal competition.
FRET distance measurements
Assay mixtures (50 μl) contained 100 nM DAC-labelled RNAP holoenzyme derivative or corresponding DAC-labelled [Ile931]β′ RNAP holoenzyme derivative in 50 mM Tris-HCl (pH 8.0), 800 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μg/ml bovine serum albumin, and 5% glycerol at 25°C. Fluorescence emission intensities were measured before and 5 min after addition of 2 μl 250 μM Cy3-MccJ25, and values of E were calculated as:
| (9) |
where Fo,R and FR are emission intensities before and after addition of Cy3-MccJ25 to DAC-labelled RNAP holoenzyme derivative, and Fo,R′ and FR′ are emission intensities before and after addition of Cy3-MccJ25 to the corresponding DAC-labelled [Ile931]β′ RNAP holoenzyme derivative.
Donor-acceptor distances (R) were calculated as:
| (10) |
where Ro is the Förster parameter (49.4 Å in this study, calculated essentially as in Mekler et al., 2002). Distance-restrained docking was performed essentially as in Mekler et al., 2002 (details in Supplementary Information: Distance-restrained docking).
Random mutagenesis
Random mutagenesis was performed by error-prone PCR amplification of the XbaI-SnaBI (codons 1-292), SnaBI-SphI (codons 292-546), SphI-SalI (codons 546-876), SalI-BspEI (codons 876-1213), and BspEI-XhoI (codons 1213-1408) rpoC segments of plasmid pRL663 (Wang et al., 1995)(methods as in Zhou et al. 1991). Mutagenized plasmid DNA was introduced by transformation into strain Stbl2 (Invitrogen), transformants (∼104 cells) were applied to LB-agar plates (Sambrook and Russel, 2001) containing 1 μg/ml MccJ25 and 200 μg/ml ampicillin, and plates were incubated 24 h at 37°C followed by 0-48 h at 25°C. For each MccJ25r clone (identified as a clone yielding a colony on the original selective plate and also yielding colonies when re-streaked to the same medium and incubated 16 h at 37°C), plasmid DNA was prepared, plasmid DNA was introduced by transformation into strain DH5α (Invitrogen), transformants (∼104 cells) were applied to LB-agar plates containing 1 μg/ml MccJ25 and 200 μg/ml ampicillin and, in parallel, to LB-agar plates containing 200 μg/ml ampicillin, and plates were incubated 16 h at 32°C. For each plasmid-linked MccJ25r clone (identified as a clone yielding comparable numbers of colonies on the plates with and without MccJ25), the nucleotide sequence of the mutagenized rpoC segment was determined by dideoxy nucleotide sequencing.
Saturation mutagenesis
A set of “doped” oligodeoxyribonucleotide primers corresponding to codons 425-437, 487-509, 592-608, 675-703, 723-743, 739-757, 772-793, 918-937, 1132-1141, and 1236-1251 of the rpoC gene of plasmid pRL663 (Wang et al., 1995), and codons 542-549 of the rpoB gene of plasmid pRL706 (Severinov et al., 1998), was synthesized on an AB392 automated synthesizer (Applied Biosystems) using solid-phase β-cyanoethylphosphoramidite chemistry (sequences in Supplementary Table 3). The level of “doping” (nucleotide misincorporation) was selected to yield an average of 0.4-1 substitution per molecule of oligodeoxyribonucleotide primer (equations in Hermes et al., 1989). Thus, the nucleotides corresponding to codons 429-433, 597-603, and 1136-1137 of rpoC were synthesized using phosphoramidite reservoirs containing 92% of the correct phosphoramidite and 8% of a 1:1:1:1 mix of dA, dC, dG, and dT phosphoramidites (i.e., 94% total correct phosphoramidite and 6% total incorrect phosphoramidite); the nucleotides corresponding to codons 492-504, 680-698, 726-740, 741-754, 775-790, 922-933, and 1239-1248 of rpoC, and codons 542-549 of rpoB, were synthesized using phosphoramidite reservoirs containing 98% of the correct phosphoramidite and 2% of a 1:1:1:1 mix of dA, dC, dG, and dT phosphoramidites (i.e., 98.5% total correct phosphoramidite and 1.5% total incorrect phosphoramidite); and all other nucleotides were synthesized using phosphoramidite reservoirs containing 100% of the correct phosphoramidite. Primer-extension mutagenesis reactions were performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), with a “doped” oligodeoxyribonucleotide primer, a complementary wild-type oligodeoxyribonucleotide primer, and pRL663 as template (primers at 75 nM; all other components at concentrations as specified by the manufacturer). Mutagenized plasmid DNA was introduced into cells, and plasmid-linked MccJ25r clones were identified and characterized, as in the preceding section.
Complementation assays
Strain 397c [rpoCts397 argG thi lac (λcI857h80St68dlac+); Christie et al., 1996] was transformed with pRL663 or a pRL663 derivative, transformants (∼104 cells) were applied to LB-agar plates containing 1 μg/ml MccJ25 and 200 μg/ml ampicillin, plates were incubated 16 h at 37°C, and bacterial growth was scored.
Minimum bacteriocidal concentration (MBC) assays
Strain DH5α (Invitrogen) was transformed with pRL663 or a pRL663 derivative, or pRL706 or a pRL706 derivative, transformants (106 cells) were incubated 2 h at 37°C in 1 ml LB (Sambrook and Russel, 2001) containing 0.01, 0.05, 0.1, or 1 mg/ml MccJ25; aliquots (10 μl) were applied to LB-agar plates containing 200 μg/ml ampicillin; plates were incubated 16 h at 37°C; and colonies were counted. The MBC was defined as the lowest concentration of MccJ25 that yielded a colony count of <5.
Supplementary Material
Acknowledgments
We thank Drs. S. Borukhov, S. Busby, T. Heyduk, F. Moreno, A. Ishihama, R. Landick, and K. Severinov for plasmids. This work was supported by NIH grants GM41376 to R.H.E. and GM64375 to R.L. and R.H.E and by a Howard Hughes Medical Institute Investigatorship to R.H.E.
References
- Artsimovitch I, Chu C, Lynch A, Landick R. A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science. 2003;302:650–654. doi: 10.1126/science.1087526. [DOI] [PubMed] [Google Scholar]
- Bayro M, Mukhopadhyay J, Swapna GVT, Huang J, Ma LC, Sineva E, Dawson P, Montelione G, Ebright R. Structure of antibacterial peptide microcin J25: a 21-residue lariat protoknot. J Am Chem Soc. 2003;125:12382–12383. doi: 10.1021/ja036677e. [DOI] [PubMed] [Google Scholar]
- Blond A, Peduzzi J, Goulard C, Chiuchiolo M, Barthelemy M, Prigent Y, Salomon R, Farias R, Moreno F, Rebuffat S. The cyclic structure of microcin J25, a 21-residue peptide antibiotic from Escherichia coli. Eur J Biochem. 1999;259:747–755. doi: 10.1046/j.1432-1327.1999.00085.x. [DOI] [PubMed] [Google Scholar]
- Borowiec J, Gralla J. Supercoiling response of the lacps promoter in vitro. J Mol Biol. 1985;184:587–598. doi: 10.1016/0022-2836(85)90305-5. [DOI] [PubMed] [Google Scholar]
- Bushnell D, Cramer P, Kornberg R. Structural basis of transcription: α-amanitin-RNA polymerase II cocrystal at 2.8 Å resolution. Proc Natl Acad Sci USA. 2002;99:1218–1222. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst S. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Prusoff W. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Christie G, Cale S, Iraksson L, Jin D, Xu M, Sauer B, Calendar R. Escherichia coli rpoC397 encodes a temperature-sensitive C-terminal frameshift in the β′ subunit of RNA polymerase that blocks growth of bacteriophage P2. J Bacteriol. 1996;178:6991–6993. doi: 10.1128/jb.178.23.6991-6993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Bushnell D, Kornberg R. Structural basis of transcription: RNA polymerase II at 2.8 Å resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- Delgado M, Rintoul M, Farias R, Salomon R. Escherichia coli RNA polymerase is the target if the cylcopeptide antibiotic microcin J25. J Bacteriol. 2001;183:4543–4550. doi: 10.1128/JB.183.15.4543-4550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Mustaev A, Markovtsov V, Bereshchenko O, Nikiforov V, Goldfarb A. Swing-gate model of nucleotide entry into the RNA polymerase active center. Mol Cell. 2002;10:623–634. doi: 10.1016/s1097-2765(02)00640-8. [DOI] [PubMed] [Google Scholar]
- Gnatt A, Cramer P, Fu J, Bushnell D, Kornberg R. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- Hermes J, Parekh S, Blacklow S, Koster H, Knowles J. A reliable method for random mutagenesis: the generation of mutant libraries using spiked oligodeoxyribonucleotide primers. Gene. 1989;84:143–151. doi: 10.1016/0378-1119(89)90148-0. [DOI] [PubMed] [Google Scholar]
- Kettenberger H, Armache K, Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–357. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov V, Goldfarb A, Darst S. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- Koulich D, Orlova M, Malhotra A, Sali A, Darst S, Borukhov S. Domain organization of Escherichia coli transcript cleavage factors GreA and GreB. J Biol Chem. 1997;272:7201–7210. doi: 10.1074/jbc.272.11.7201. [DOI] [PubMed] [Google Scholar]
- Laptenko O, Lee J, Lomakin I, Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis A, Niu W, Ebright Y, Levy R, Ebright R. Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell. 2002;108:599–614. doi: 10.1016/s0092-8674(02)00667-0. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J, Kapanidis A, Mekler V, Kortkhonjia E, Ebright Y, Ebright R. Translocation of σ70 with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell. 2001;106:453–463. doi: 10.1016/s0092-8674(01)00464-0. [DOI] [PubMed] [Google Scholar]
- Naryshkin N, Kim Y, Dong Q, Ebright R. Site-specific protein-DNA photocrosslinking: analysis of bacterial transcription initiation complexes. Meths Mol Biol. 2001;148:337–361. doi: 10.1385/1-59259-208-2:337. [DOI] [PubMed] [Google Scholar]
- Nedialkov Y, Gong X, Hovde S, Yamaguchi Y, Handa H, Geiger J, Yan H, Burton Z. NTP-driven translocation by human RNA polymerase II. J Biol Chem. 2003;278:18303–18312. doi: 10.1074/jbc.M301103200. [DOI] [PubMed] [Google Scholar]
- Ner S, Goodin D, Smith M. A simple and efficient procedure for generating random point mutations and for codon replacements using mixed oligodeoxynucleotides. DNA. 1988;7:127–134. doi: 10.1089/dna.1988.7.127. [DOI] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, Ebright R. Transcription activation at Class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opalka N, Chlenov M, Chacon P, Rice W, Wriggers W, D S. Structure and function of the transcription elongation factor GreB bound to bacterial RNA polymerase. Cell. 2003;114:335–345. doi: 10.1016/s0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- Record MTJ, Reznikoff W, Craig M, McQuade K, Schlax P. Escherichia coli RNA polymerase (Eσ70), promoters, and the kinetics of the steps of transcription initiation. In: Neidhart FC, editor. Escherichia coli and Salmonella. Washington, D.C.: ASM Press; 1996. pp. 792–820. [Google Scholar]
- Rosengren K, Clark R, Daly N, Goransson U, Jones A, Craik D. Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J Am Chem Soc. 2003;125:12464–12474. doi: 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- Salomon R, Farias R. Microcin J25, a novel antimicrobial peptide produced by Escherichia coli. J Bacteriol. 1992;174:7428–7435. doi: 10.1128/jb.174.22.7428-7435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- Schlageck JG, Baughman M, Yarbrough LR. Spectroscopic techniques for study of phosphodiester bond formation by E. coli RNA polymerase. J Biol Chem. 1979;254:12074–12077. [PubMed] [Google Scholar]
- Segel I. Enzyme Kinetics. New York: Wiley; 1975. [Google Scholar]
- Selvin P. The renaissance of fluorescence resonance energy transfer. Nature Structl Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- Severinov K, Mooney R, Darst SA, Landick R. Tethering of the large subunits of Escherichia coli RNA polymerase. J Biol Chem. 1997;272:24137–24140. doi: 10.1074/jbc.272.39.24137. [DOI] [PubMed] [Google Scholar]
- Solbiati J, Ciaccio M, Farias R, Gonzalez-Pastor J, Moreno F, Salomon R. Sequence analysis of the four plasmid genes required to produce the circular peptide antibiotic microcin J25. J Bacteriol. 1999;181:2659–2662. doi: 10.1128/jb.181.8.2659-2662.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunova E, Sosunov V, Kozlov M, Nikiforov V, Goldfarb A, Mustaev A. Donation of catalytic residues to RNA polymerase active center by transcription factor Gre. Proc Natl Acad Sci USA. 2003;100:15469–15474. doi: 10.1073/pnas.2536698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev D, Sekine S, Laptenko O, Lee J, Vassylyeva M, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Wang D, Meier T, Chan C, Feng G, Lee D, Landick R. Discontinuous movements of DNA and RNA in RNA polymerase accompany formation of a paused transcription complex. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- Wilson K, Kalkum M, Ottesen J, Yuzenkova J, Chait B, Landick R, Muir T, Severinov K, Darst SA. Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J Am Chem Soc. 2003;125:12475–12483. doi: 10.1021/ja036756q. [DOI] [PubMed] [Google Scholar]
- Yuzenkova J, Delgado M, Nechaev S, Savalia D, Epshtein V, Artsimovitch I, Mooney R, Landick R, Farias R, Salomon R, Severinov K. Mutations of bacterial RNA polymerase leading to resistance to microcin J25. J Biol Chem. 2002;277:50867–50875. doi: 10.1074/jbc.M209425200. [DOI] [PubMed] [Google Scholar]
- Zakharova N, Bass I, Arsenieva E, Nikiforov V, Severinov K. Mutations in and monoclonal antibody binding to evolutionary hypervariable region of Escherichia coli RNA polymerase β′ subunit inhibit transcript cleavage and transcript elongation. J Biol Chem. 1998;273:24912–24920. doi: 10.1074/jbc.273.38.24912. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell E, Minakhin L, Richter C, Severinov K, Darst S. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang X, Ebright R. Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucl Acids Res. 1991;19:6052. doi: 10.1093/nar/19.21.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.