

Summary
The Shaker family K+ channel protein, Kv1.3, is tyrosine phosphorylated by v-Src kinase at Tyr137 and Tyr449 to modulate current magnitude and kinetic properties. Despite two proline rich sequences and these phosphotyrosines contained in the carboxyl and amino terminals of the channel, v-Src kinase fails to co-immunoprecipitate with Kv1.3 as expressed in HEK 293 cells, indicating a lack of direct SH3- or SH2-mediated protein-protein interaction between the channel and the kinase. We show that the adaptor proteins, n-Shc and Grb10, are expressed in the olfactory bulb, a region of the brain where Kv1.3 is highly expressed. In HEK 293 cells, co-expression of Kv1.3 + Grb10, but not plus n-Shc, causes a decrease in Kv1.3 tyrosine phosphorylation and a reversal of v-Src-induced current suppression, v-Src-induced decrease in inactivation time constant (τinact), and v-Src-induced disruption of cumulative inactivation properties. These affects are dependent upon full v-Src kinase activity and are abolished when the severly impaired R385A v-Src kinase is substituted for Src kinase. Co-expression of Kv1.3 + n-Shc did not significantly alter v-Src-induced current suppression or decreased τinact, but right-shifted the voltage at half-activation (V1/2) by 10 mV. This significant shift in V1/2 was retained when Y220, Y221, and Y304 in the CH domain of n-Shc were mutated to F but was reversed back to control values when the family member Sck, which is not a substrate for Src kinase, was substituted for n-Shc. Thus the portion of the CH domain that excludes tyr220, 221, 304 that is not homologous to Sck may regulate a shift in voltage-dependence produced by n-Shc that is independent of tyrosine phosphorylation. Collectively these data indicate that Grb10 and n-Shc adaptor molecules differentially modulate the degree of Kv1.3 tyrosine phosphorylation, the channel's biophysical properties, and the physical complexes associated with Kv1.3 in the presence of Src kinase.
Introduction
Voltage-gated potassium (Kv) channels form highly potassium (K)-selective pores that are conformationally switched open or closed by changes in membrane voltage {1, 2, 3}. Kv channels may not function in isolation in the plasma membrane but rather be intricately involved in protein-protein interactions {4}. Such interactions build a molecular scaffold that may modulate the channel's subcellular distribution in the neuron, regulate the level of expression of the channel, introduce signaling interchange with particular proteins contained in specific transduction cascades, and ultimately affect the functional properties of the channel {4-10}. Protein-protein interactions occur between two or more proteins at discreet modules that may or may not require phosphorylation for recognition of the signaling motif.
Kv1.3, a mammalian homologue to the Shaker subfamily, is highly expressed in T-lymphocytes, the dentate gyrus of the hippocampus, the pyriform cortex, and the olfactory bulb (OB) {11}. In the olfactory bulb, Kv1.3 is localized to the mitral and granule neurons, where it is a substrate for protein phosphorylation by multiple tyrosine kinases that induce change in its function as assessed electrophysiologically {12,13}. Cellular Src kinase is among several of the tyrosine kinases that modulate Kv1.3 function through phosphorylation {12}. The transforming gene of the Rous sarcoma virus, viral Src (v-Src), encodes a protein with intrinsic tyrosine kinase activity and causes cell transformation into the cancerous state {14-18}. Ligand interactions with the src-homology 2 (SH2) or SH3 domains of Src kinases appear to destabilize the inactive conformation of the enzyme, allowing autophosphorylation at Src tyr416 and full activation of the kinase {14}. Binding at SH2 or SH3 domains of Src kinase thus propagates down-stream signaling and at times inappropriate signaling by phosphorylation that has been linked to several pathological conditions including asthma, autoimmune disease, oncogenesis, Alzheimer's, and allergies {19}.
Voltage and ligand-gated ion channels have been reported to interact with Src kinase SH3 domain by recognition of a proline-rich PXXP sequence motif in the channel {20} to cause biophysical changes in channel function due to the interaction or to tyrosine phosphorylation by Src. Kv1.3 contains two proline-rich stretches, one in the amino terminal (39-44 PLPPALP) and one in the carboxyl terminal (493-496 PXTP) of the channel protein that could interact with Src kinase SH3 domain. Kv1.3 also has a total of 16 tyrosine residues, 6 of which lie within good recognition motifs for tyrosine specific phosphorylation and could serve as potential interactive sites for Src kinase SH2 domain once phosphorylated by Src, which provides the free energy for binding (see Fig. 1). It has been previously shown, however, that while a related Kv family member, Kv1.5, co-immunoprecipitates with Src kinase at the Src SH3 domain, Kv1.3 fails to retain a strong interaction with Src kinase in HEK 293 cells by SDS-PAGE and Western blot analysis {7}. We thus hypothesized that adaptor molecules could form the physical complexes bringing together Kv1.3 and v-Src kinase to modulate the electrical activity by tyrosine phosphorylation. Thus the repertoire of adaptor proteins expressed in a given neuronal cell type and the specific capacity for regulating the proximity of kinase to its effector (the ion channel) may influence the electrical phenotype of a neuron to the same extent that expression of various voltage-gated channel species shape the overall macroscopic current properties of a cell.
Fig. 1. Diagrammatic representation of the tyrosine residues (Y) and potential protein-protein interactive modules (PTB, SH2, SH3, CH, and proline rich domains) between v-Src kinase, the Kv1.3 ion channel, and the adaptor proteins, n-shc and grb10.

For molecular details and abbreviations, please see text (INTRODUCTION).
Two adaptor proteins that have been shown to be substrates of Src kinase and which we found were well expressed in the olfactory bulb, are neuronal Src- and collagen-homology protein (n-Shc) and growth factor receptor binding protein 10 (Grb10) {21-29} (see Fig. 1). Adaptor proteins lack catalytic activity, but as their name implies, link other proteins together, often acting as platforms upon which enzymes act on their substrates {30}. Two members of the Shc family, neuronal Shc (n-Shc; ShcC) and Sck (ShcB), have been identified and shown to be neuronal-specific, however the latter member cannot be phosphorylated by Src kinase {31-34}. N-Shc contains SH2, phosphotyrosine binding (PTB), and Collagen Homology (CH) domains, and three key tyrosine residues, tyr 221, 222, and 304, that lie within phosphorylation-specific sequences {35, 36, 29}. Grb10 contains SH2, Pleckstrin homology (PH) and BPS interaction domains, as well as several proline-rich regions. These two adaptors could provide for both phosphorylation -dependent and -independent mechanisms, as predicted by their signaling motifs. The PTB domain of n-Shc is particularly interesting in that it is structurally similar to PH and PDZ domains, demonstrated to translocate proteins to membrane and sub-membrane cellular compartments and to link Kv channels {37-39}.
Herein we report that two adaptor proteins differentially modulate v-Src-induced Kv1.3 phosphorylation and modulation of channel function. N-Shc adaptor fails to alter v-Src-induced Kv1.3 phosphorylation, whereas Grb10 adaptor significantly reduces the Kv1.3 tyrosine phosphorylation produced by v-Src. The proline rich sequences contained in Grb10 adaptor protein may compete for the SH3 domain of Src, to decrease the ability for Src to phosphorylate Kv1.3 and suppress Kv1.3 current magnitude, alter the kinetics of Kv1.3 inactivation and deactivation, and disrupt cumulative inactivation during repetitive voltage stimulation. Through site-directed mutagenesis of the regulatory Tyr residues in the CH domain of n-Shc and comparison of the activity of n-Shc with a close family member Sck, data suggest that a portion of the CH domain that excludes tyr 221,222 and 304 and is not homologous to Sck, may regulate a shift in voltage-dependence produced by n-Shc that is independent of tyrosine phosphorylation.
Experimental Procedures
Solutions and Reagents
Human embryonic kidney cell (HEK 293) patch pipette solution contained (in mM): 30 KCl, 120 NaCl, 10 HEPES, and 2 CaCl2 (pH 7.4). HEK 293 cell recording bath solution contained (in mM): 150 KCl, 10 HEPES, 1 EGTA, and 0.5 MgCl2 (pH 7.4). Cell lysis buffer with protease and phosphatase inhibitors (PPI solution) contained (in mM): 25 Tris (hydroxymethyl) aminomethane (pH 7.5), 250 NaCl, 5 EDTA, 1% Triton X-100, 1 sodium orthovanadate, 1:100 dilution of Sigma Phosphatase Inhibitor Cocktail II (Catalog # P-5726), 1:500 dilution of Sigma Protease Inhibitor Cocktail (Catalog # P-8340). Wash buffer contained (in mM): 25 Tris (pH 7.5), 250 NaCl, 5 EDTA, and 0.1% Triton X-100. Homogenization buffer contained (in mM): 320 sucrose, 10 Tris base, 50 KCl, and 1 EDTA (pH 7.8). All salts were purchased from Sigma Chemical Company or Fisher Scientific. Tissue culture and transfection reagents were purchased from Gibco BRL.
Olfactory Bulb Membrane Solubilization
One set of olfactory bulbs (OB) from Sprague-Dawley rats at postnatal day 17 (P17) and P30 (adult) were quickly dissected using American Veterinary Medical Association (AVMA)- and National Institutes of Health (NIH)-approved methods. Harvested OB tissue was homogenized on ice using a Kontes tissue grinder (size 20; 50 strokes) and homogenization buffer containing 0.5% nonidet P-40 (NP-40) detergent and Sigma protease and phosphatase inhibitor cocktails (see Solutions and Reagents). The mixture was vortexed every 15 minutes for 4 hours, centrifuged at 14,000 × g for 10 minutes at 4°C, and the supernatant (lysate) was stored at -80 °C until subsequent use. Protein concentration was determined by Bradford protein assay; 10 or 40 mg of lysate was separated by 10% SDS-PAGE, proteins were electrotransferred to nitrocellulose, and Western blots were probed with antisera for Src, Shc, and Grb (see below).
cDNA Constructs and Antibodies
All channel, kinase, and adaptor protein coding regions were downstream from a cytomegalovirus (CMV) promoter. The v-Src cDNA construct was a generous gift from Dr. Richard Huganir (Johns Hopkins University, Baltimore, MD) and was located in a modified PRK5 vector. The R385A v-Src cDNA construct (v-Src with severely impaired tyrosine kinase activity) was a generous gift from Dr. M. Senften (Friedrich Miescher-Institute, Basel Switzerland; {40}). The entire R385A v-Src coding region was inserted into a modified PRK5 vector {41}. Neuronal Shc (n-Shc), Sck, and n-Shc mutant cDNA (Y to F point mutations at tyr220, tyr221, tyr304) were generous gifts from Dr. T. Nakamura (Sumitomo Electric Industries, Yokohama, Japan) and were expressed in the vector pCMV1 {32}. The n-shc mutant will be referred to as the “triple”mutant. Grb10 cDNA was a gift from Dr. R. Roth (Stanford University) and was expressed in pBlueScript SK (-) vector (Stratagene).
AU13, a rabbit polyclonal Kv1.3 antiserum, was designed against the 46-amino acid sequence (478MVIEEGGMNHSAFPQTPFKTGNSTATCTTNNNPNDCVNIKKIFTDV523) representing the unique coding region of Kv1.3 between the carboxyl terminus and transmembrane domain 1. The purified peptide was produced by Genmed Synthesis (San Francisco, CA), and the antiserum was generated by Cocalico Biologicals (Reamstown, PA). This antibody was used for immunoprecipitation (1:1000) and Western blot detection (1:1500) of Kv1.3 as in Tucker and Fadool (submitted). The monoclonal antibody 4G10 (Upstate Biochemical) recognized phosphorylated tyrosine residues and was used at 3.5 μg/ml for immunoprecipitation and 1:1000 for Western analysis. Src monoclonal antibody, directed against the Src SH3 domain (Oncogene Ab-1), was used at 10 μg/ml for immunoprecipitation and 1:40-1:100 for Western analysis. Polyclonal antisera for Shc (H-108) and Grb10 (K-20) were from Santa Cruz; each was used at 20 μg/ml for immunoprecipitation and between 1:200-1000 for Western blot detection.
Maintenance of HEK 293 Cells and Transfection
HEK 293 cells were maintained in Minimum Essential Medium (MEM), 2% penicillin/streptomycin, and 10% FBS (Gibco BRL). Before transfection, cells were grown to 100% confluency (7 days), dissociated with trypsin-EDTA (Sigma) and mechanical trituration, diluted in MEM to a concentration of 600 cells/ml, and replated on Corning dishes (Catalog # 25000, Fisher Scientific). cDNA was introduced into HEK 293 cells with a lipofectamine reagent (Gibco BRL) 3-5 days after passage as previously described {12,13}. Briefly, cells were transfected for 4-5 hours with 0.5-0.75 μg of each cDNA construct per 35-mm dish for electrophysiology or 3.5-5.0 μg of each cDNA construct per 60-mm dish for biochemistry. Plasmid DNA with no coding insert (control vector) served as the control to equalize total μg of cDNA added to each dish. Cells were either harvested for biochemical analysis or used for electrophysiological recordings approximately 30-40 hours after transfection.
For patch-clamp experiments, transfection efficiency was monitored by co-transfecting with pHook (Invitrogen) as a means of rapidly selecting cells expressing Kv1.3 channels. pHook encodes a transmembrane domain from the platelet-derived growth factor receptor (PDGF-R), which is then anchored on the extracellular side of the plasma membrane. Before patch recording, a brief incubation with an appropriate antibody linked to a 5-μm polystyrene bead allowed recognition of transfected cells. Efficiency of co-transfection (greater than 95%) with our transfection method has been tested using double, sequential labeling and confocal microscopic visualization, as reported previously{13}.
Electrophysiology
Patch electrodes were fabricated from Jencons glass (Jencons Limited, Befordshire, UK), fire-polished to approximately 1 μm, and coated near the tip with beeswax to reduce the electrode capacitance. Pipette resistances were between 9 and 14 MΩ. Hoffman modulation contrast optics was used to visualize cells at 40× magnification (Axiovert 135, Carl Zeiss). Macroscopic currents in cell-attached membrane patches were recorded using an Axopatch-200B amplifier (Axon Instruments), filtered at 2 kHz, digitized at 2-5 kHz, and stored for later analysis. All voltage signals were generated and data were acquired with the use of either a Microstar DAP 800/2 board with in house written software (DapClamp; Microstar Lab, Bellevue, WA) or an Axon Digidata 1200 board with pClamp software (Axon Instruments). Data were analyzed using Microcal Origin software.
Outward macroscopic currents were recorded in the cell-attached rather than whole-cell configuration; Kv1.3 channel expression is so robust in the HEK 293 expression system that it is not routinely possible to record whole-cell currents without saturating the amplifier {41-43}. Patches were held routinely at a holding potential of -90 mV and stepped in 20 mV depolarizing potentials using a pulse duration of 1000 milliseconds. Pulses were generally delivered at intervals of 60 seconds or longer to prevent cumulative inactivation of the Kv1.3 channel {44}. Kv1.3 peak current amplitude, channel inactivation (τinact) and deactivation (τdeact) kinetics, voltage at one-half maximal activation (V1/2), and slope of voltage dependence (κ) were measured in the presence of Src kinase and the presence or absence of an adaptor protein (N-Shc or Grb10). Each biophysical property was analyzed in the form of non-normalized data by one-way ANOVA with a Student Newman Keuls (snk) follow-up test at the 95% confidence level to determine any statistical difference in Kv1.3 channel function in the presence of kinase or adaptor proteins. For graphical representations of peak current amplitudes among different transfection conditions, measurements were normalized to the transfection condition of Kv1.3 alone within a single recording session and respective transfection set. Kinetic properties of Kv1.3 have been reported to be independent of current magnitude {44}, thus peak current amplitude but not kinetic data were normalized to adjust for differences in channel expression between transfections.
Fitting parameters for inactivation and deactivation kinetics were as previously described {13}. Briefly, the inactivation of the macroscopic current, during a 1000 ms voltage step from -90 to +40 mV, was fit to the sum of two exponentials by minimizing the sums of squares using a bi-exponential function (y = y0 + A1e-(x-x0)/τ1 + A2e-(x-x0)/τ2). The two inactivation time constants (τ1 and τ2) were combined by multiplying each by its weight (A) and summing. The deactivation of the macroscopic current was fit similarly but to a single exponential (y = y0 + Ae-(x-x0)/τ). Tail current amplitudes were plotted in a current-voltage relationship and fit to a Boltzmann sigmoidal curve (Y = [(A1 + A2)/(1 + e(x-x0)/dx)] + A2) to calculate the slope of voltage dependence (κ) and voltage at half-activation (V1/2) for Kv1.3. To study cumulative inactivation of Kv1.3 under the control condition and in the presence of Src and/or an adaptor protein, patches were held at -90 mV and stepped to +40 mV twelve times using a similar pulse duration as above but recording using different interpulse intervals, including 60, 30, 10, 2, 1, and 0.5 seconds. Peak current amplitudes were normalized to the initial trace in each series for a given interpulse interval. Cumulative inactivation was always present with interpulse intervals less than 60 seconds in the control (Kv1.3 alone) transfection condition, and as much as 50% of channels were caught in the inactivated state with repeated stimulation less than 2 seconds.
Immunoprecipitation and Electrophoretic Separation
Transfected cells were harvested by lysis 2 days post-transfection in ice-cold PPI solution (see Solutions and Reagents). The lysates were clarified by centrifugation at 14,000 × g for 10 minutes at 4°C and incubated for 1 hour (h) with 0.2-0.3 mg/ml protein A-sepharose (Amersham-Pharmacia), followed by re-centrifugation to remove the protein A-sepharose. To test the phosphorylation state of Kv1.3 in the presence of v-Src kinase plus or minus an adaptor protein, tyrosine-phosphorylated proteins were immunoprecipitated from the clarified lysate by overnight incubation at 4°C with 3-4 μg/ml 4G10 antibody (Upstate Biochemical). Immunoprecipitated proteins were harvested by a 2-h incubation with protein A-sepharose and centrifugation as above. The immunoprecipitates (IPs) were washed 3 times with ice-cold wash buffer. Lysates and washed IPs were diluted in sodium dodecyl sulfate (SDS) gel loading buffer {45} containing 1 mM Na3VO4. Protein concentration was determined by Bradford protein assay. Proteins were separated on 10% acrylamide SDS gels and electrophoretically transferred to nitrocellulose for Western blot analysis. Nitrocellulose membranes were blocked with 5% nonfat milk and incubated overnight at 4°C in primary antibody against Kv1.3, v-Src, Shc, or Grb10. Membranes were then exposed to species-specific peroxidase-conjugated secondary (Amersham-Pharmacia or Sigma) for 90 minutes at room temperature. Enhanced chemiluminescence (ECL; Amersham-Pharmacia) exposure of Fuji RX film (Fisher) was used to visualize labeled protein.
Resultant bands were quantified by densitometry using a Hewlett-Packard Photosmart Scanner in conjunction with Quantiscan software (Biosoft, Cambridge, UK). Pixel densities for each band were normalized to Kv1.3 within the same cell passage, transfection, and autoradiograph. Mean pixel densities were then calculated across sets of normalized data contained in single autoradiographs. This type of quantification was designed to reduce inherent variability in cell culture, transient transfection efficiencies, and ECL exposure times. Statistical significance was set at the 95% confidence level for immunodensitometry data that were analyzed by one-way ANOVA with snk follow-up test.
Results
In this study we have found that the modulation of the voltage-gated potassium channel, Kv1.3, by the cellular tyrosine kinase, v-Src, is differentially affected by the presence of two adaptor proteins, n-Shc and Grb10. A schematic of the protein interaction modules and tyrosine phosphorylation recognition motifs contained in Kv1.3, n-Shc, Grb10, and Src is presented in Figure 1.
Signaling Proteins are Expressed in the Olfactory Bulb
Kv1.3 becomes robustly phosphorylated at tyr137 and tyr449 when co-expressed with v-Src kinase in HEK293 cells to induce current suppression and decreased kinetics of inactivation {41}. Likewise, patch pipette dialysis of c-Srcpp60 plus MgATP induces similar biophysical changes in the outward whole-cell current of OB neurons, that is carried largely by Kv1.3 {12}. Although Src kinase is ubiquitously expressed throughout the CNS, it was important to confirm the expression of Src in the OB in addition to its capacity for modulation of Kv1.3. Ten or forty μg of OB lysate was separated by 10% SDS-PAGE and electrotransferred to nitrocellulose membranes. Membranes were probed with a monoclonal antibody for viral Src (v-Src, 1:40-1:100) and Src expression was detected in the OB lysates at the expected molecular weight of 60 kDa (Fig. 2).
Fig. 2. Expression of Src kinase and related adaptor proteins in the rat olfactory bulb.

Western blot analysis of olfactory bulb (OB) lysates collected from adult (postnatal day 30) rats and homogenized in a NP40-based buffer. 10 μg (left) or 40 μg (right) of protein was loaded per set, separated by SDS-PAGE, electrotransferred to nitrocellulose, and probed with antisera against v-Src (1:100), Shc (1:800), or Grb10 (1:800).
In HEK 293 cells, a direct protein-protein interaction has been observed between v-Src kinase and Kv1.5 and this protein-protein interaction causes current suppression of Kv1.5 {7}. Although Src kinase modulates channel properties of Kv1.3 by tyrosine phosphorylation, the channel fails to co-immunoprecipitate with Src kinase in HEK 293 cells {7}. We conjectured that perhaps adaptor protein modules might facilitate channel-kinase interactions in native neurons expressing Src and Kv1.3. We thus probed the SDS-PAGE separated OB lysates with antisera for Shc (H-108) and Grb10 (K-20). Fig. 2 demonstrates the observed protein bands of the predicted molecular weight (46/52/66 kDa and 60 kDa, respectively) for these two adaptor proteins. Shc adaptor protein is not expressed in brain regions, however, a neuronal-specific member of the Shc family, n-Shc, is highly expressed in the brain and the OB, as shown by in situ hybridization{29}. Since H-108 antibody detects all Shc isoforms as well as n-Shc, it is highly likely that we are detecting predominantly the n-Shc family member in the OB.
v-Src kinase phosphorylation of Kv1.3 is altered by adaptor proteins, Grb10 and n-Shc
Deletion of the c-Src activation loop produces the viral form of Src, v-Src, which has unlimited, constitutive activity in the absence of other cellular signaling {46}. The rest of the experiments of this study were performed by transient co-transfection of v-Src plus Kv1.3 in an heterologous expression system (HEK 293 cells). This model facilitated the understanding of the potential mechanisms by which Src kinase could exert its effects on Kv1.3 in native OB neurons by allowing the experimenter to control the expression of putative protein-protein interacting partners and by having the ability to activate the kinase without induction of the endogenous upstream signaling cascade. Additionally, human embryonic kidney (HEK 293) cells are known to express high levels of cloned proteins {47}, which facilitates good protein yields for immunoprecipitation. Untransfected HEK 293 cells also show no C-type inactivating K currents in response to voltage stimulation {41}.
HEK 293 cells were co-transfected with cDNA for Kv1.3 + v-Src (see Experimental Procedures) plus or minus cDNA coding n-Shc or Grb10 adaptor proteins. Cells were given approximately 40 hours to express proteins prior to cell harvest by lysis. Kv1.3 transfected cells always served as the control and total cDNA across various transfection conditions was balanced by non-coding pcDNA3 vector. Precleared cell lysate was incubated overnight with the Kv1.3 directed antibody, α-AU13. Collected and washed immunoprecipitates (IPs) were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with a phosphotyrosine-specific antibody (see Experimental Procedures; Upstate 4G10). A complementary approach yielding similar results was also used, in which phosphorylated proteins were precipitated from lysates with the phosphotyrosine-specific antibody (4G10), and separated proteins were probed for Kv1.3 (data not shown). Kv1.3 plus v-Src co-transfected HEK 293 cells demonstrated that v-Src kinase significantly increased tyrosine phosphorylation of Kv1.3 14-fold over that found in Kv1.3-alone transfected cells (n = 10) (Fig. 3). Kv1.3 exhibited low levels of basal tyrosine phosphorylation, but as also observed by Holmes et al. {43}, its tyrosine phosphorylation increased dramatically when co-expressed with v-Src. The basal value of Kv1.3 tyrosine phosphorylation was assigned an arbitrary value of 1.0 (Fig. 3B). This value was used as a normalization factor for all other transfection conditions. The predicted molecular weight (Mr) of the Kv1.3 channel (58 kDa) shows a decreased electrophoretic mobility, i.e. upward band shift, on SDS-PAGE under protein tyrosine phosphorylation conditions {48, 49, 43}. The bracketed region (Fig. 3A, upper panel) from 55-72 kDa spanning all Kv1.3 immunoreactivity was thus quantified in assessing Kv1.3 tyrosine phosphorylation in this and all subsequent experiments. The expression of Kv1.3 protein in each condition, for this, and all remaining experiments, was verified by separating 5 μg of cell lysate for each transfection condition and probing the lysates with α-Kv1.3 antibody (Fig. 3A, lower panel).
Fig. 3. Effect of adaptor proteins on the phosphorylation of Kv1.3 by v-Src kinase.

Human embryonic kidney (HEK 293) cells were transiently transfected with Kv1.3 cDNA plus or minus v-Src kinase and plus or minus adaptor protein (grb10 or n-shc). A, (top panel), Kv1.3 protein was immunoprecipitated from Triton X-100-soluable cell lysates with α-Kv1.3 antiserum (IP:αKv1.3), separated by SDS-PAGE, electrotransferred to nitrocellulose, and probed with α-phosphotyrosine antibody (IB:αPY) that recognizes phosphorylated tyrosine residues. Upper arrow indicates Mr of Kv1.3 and lower arrow indicates the heavy chain of IgG (IgG). The box defines the area that was integrated (55-72 kDa) to generate the quantitative immunodensity data in B. A, (lower panel), Cell lysates used in the above immunoprecipitation experiment were probed with α-Kv1.3 antiserum to insure equal protein expression of the channel under varying transfection conditions. B, Histogram plot of the tyrosine phosphorylation of Kv1.3 (Kv1.3), as measured by quantitative immunodensitometry, indicates an increase in phosphorylation in the presence of v-src (+v-src) or v-src plus n-shc (+v-src + n-shc). This increase in v-src induced tyrosine phosphorylation of Kv1.3 is inhibited in the presence of grb10 (+ v-src + grb10). Pixel counts for each band were normalized to the value of the Kv1.3-only band within single gels for graphical representation. Non-normalized values from each transfection condition were compared statistically with one-way randomized design ANOVA, α = 0.05. * = significantly different from control (Kv1.3) by snk follow-up test.
Both Grb10 and n-Shc adaptor proteins have been shown to be substrates of Src kinase {51, 51, 27}, and each contain several signaling motifs for protein-protein interactions (Fig. 1). We tested the effect of each adaptor protein on v-Src-induced Kv1.3 phosphorylation using immunoprecipitation and Western analysis as described above. Addition of Grb10 cDNA to v-Src plus Kv1.3 co-transfected HEK 293 cells caused a significant decrease in Kv1.3 tyrosine phosphorylation, which approximated that of basal levels (Kv1.3 in the absence of v-Src kinase) (ANOVA, snk, α = 0.05) (Fig. 3B). Addition of n-Shc cDNA, however, slightly increased Kv1.3 tyrosine phosphorylation but not significantly greater than that found in Kv1.3 plus v-Src co-transfected cells (ANOVA, snk, α = 0.05) (Fig. 3B).
v-Src-induced modulation of Kv1.3 is altered by adaptor proteins, Grb10 and n-Shc
v-Src-induced current suppression of Kv1.3 {41} was first confirmed by comparing the biophysical properties of Kv1.3-transfected cells with those of Kv1.3-plus-v-Src-co-transfected cells. We then tested whether changes in v-Src-induced Kv1.3 tyrosine phosphorylation caused by co-expression with Grb10 or n-Shc also altered Kv1.3 physiology. For these patch-clamp experiments, HEK 293 cells were transfected with Kv1.3 alone or co-transfected with Kv1.3 plus v-Src, plus or minus an adaptor protein. Approximately 40 hours post-transfection, macroscopic outward currents were recorded in the cell-attached configuration. Cells were voltage-clamped at -90 mV and stepped to a depolarizing potential of +40 mV for 1 second to activate Kv1.3. Peak current amplitude was 1691.36 +/- 199.64 pA (n = 69) in the Kv1.3-only control transfection condition and 391.38 +/- 84.85 pA (n = 47) in the Kv1.3 plus v-Src condition, confirming a statistically significant 77% suppression of Kv1.3 current in the presence of the kinase (Student's t-test, α = 0.05) (Fig. 4A-B). This change in current magnitude appears to be independent of a shift in Kv1.3 voltage dependence. This is reflected in the lack of change of voltage at half-maximum activation (V1/2) in the presence of v-Src as calculated from a Boltzman fit of the tail currents when cells were held at -90 mV and stepped to +20 mV in increments of 5 mV using a pulse duration of 50 ms (Table 1 and 2). v-Src significantly decreased the τInact of Kv1.3 from 985 ms (n = 64) to 616 ms (n = 33), as shown in the examples of Figure 4C-D. v-Src also increased the τDeact of Kv1.3 from 34 ms (n = 63) to 43 ms (n = 40), although this did not represent a statistically significant difference (Student's t-test, α = 0.05) (Fig. 4E-F).
Fig. 4. Effect of adaptor proteins on the modulation of Kv1.3 by v-Src kinase.
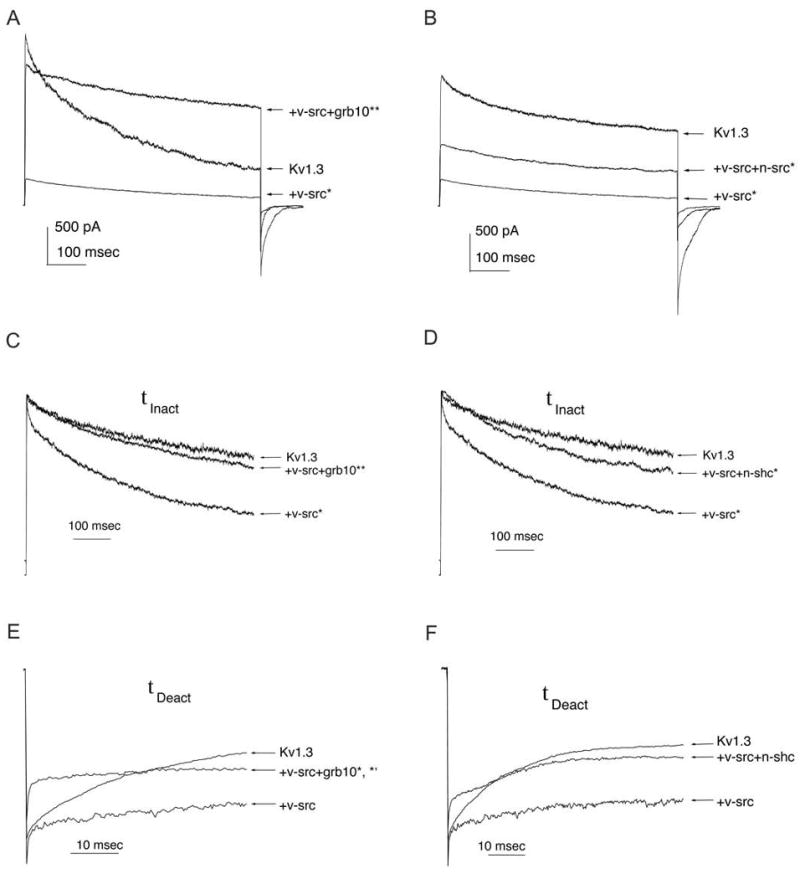
Human embryonic kidney (HEK 293) cells were transiently transfected with Kv1.3 cDNA +/- v-src +/- grb10 or n-shc, as in Fig. 3. A-B, Representative cell-attached patch-clamp recordings from HEK 293 cells that were held at -90 mV (Vh) and stepped to +40 mV (Vc) for 1000 msec. Three different HEK 293 cells are shown in each, demonstrating Kv1.3 alone (Kv1.3), Kv1.3 + v-src (+ v-src) and Kv1.3 + v-src + adaptor protein (+ v-src + grb10) or (+v-src + n-shc) transfection conditions. C-D, Same transfection and recording conditions as in A-B, respectively, but traces are normalized to peak current value to visualize rate of inactivation (τinact). E-F, Same transfection conditions as in A-B, respectively, but traces are normalized to peak tail current to visualize rate of deactivation (τdeact). τinact and τdeact were determined by fits to bi-exponential or single exponential functions, respectively, as described in Experimental Procedures. A-F, Values from transfection conditions were compared statistically by one-way completely randomized design ANOVA, snk follow-up test, α = 0.05. * = significantly different from control (Kv1.3). ** = significantly different from Kv1.3 + v-src co-transfection.
Table 1. The Affect of Grb10 Adaptor Protein on v-Src-Induced Kv1.3 Current Properties.
| Transfection Condition |
Normalized Peak Current |
τinact | τdeact | V1/2 | κ |
|---|---|---|---|---|---|
| Kv1.3 | 1.00(21) | 985+/-230(64) | 34+/-4(63) | -52+/-7(54) | 3+/-0(53) |
| Kv1.3 + v-Src | *0.14(18) | *616+/-115(33) | 43+/-6(40) | -49+/-4(24) | *13+/-5(24) |
| Kv1.3 + R385A | 0.77(19) | 819+/-76(18) | 29+/-4(17) | -51+/-3(13) | 5+/-1(13) |
| Kv1.3 + v-Src + Grb10 | 0.88(19) | 793+/-147(18) | *18+/-3(15) | -45+/-4(10) | 4+/-0(9) |
| Kv1.3 + R385A + Grb10 | *4.20(13) | *508+/-119(13) | 43+/-10(12) | -55+/-2(10) | 4+/-1(10) |
HEK 293 cells were transfected with cDNAs encoding Kv1.3 +/- v-Src or R385A Src kinase +/- Grb10 adaptor protein. Several voltage-stimulating paradigms were completed (see text) to collect the tabled biophysical properties by cell-attached patch-clamp recording. Peak current amplitudes were normalized to the control condition (Kv1.3) for each cell passage prior to averaging. Mean values (+/- s.e.m.) for inactivation time constant (τinact), deactivation time constant (τdeact), voltage at half-activation (V1/2), and slope (κ) of the voltage-dependence were compared across various transfection conditions. Data were analyzed by completely randomized one-way ANOVA with Student Newman Keuls (snk) followup test as described in the text. * = Significantly different from Kv1.3 (control).
Table 2. The Affect of n-Shc or Sck Adaptor Protein on v-Src-Induced Kv1.3 Current Properties.
| Transfection Condition |
Normalized Peak Current |
τinact | τdeact | V1/2 | κ |
|---|---|---|---|---|---|
| Kv1.3 | 1.00(42) | 985+/-230(64) | 34+/-4(63) | -52+/-7(54) | 3+/-0(53) |
| Kv1.3 + v-Src | *0.25(32) | *616+/-115(33) | 43+/-6(40) | -49+/-4(24) | *13+/-5(24) |
| Kv1.3 + R385A | 0.77(19) | 819+/-76(18) | 29+/-4(17) | -51+/-3(13) | 5+/-1(13) |
| Kv1.3 + v-Src + n-Shc | 0.59(38) | *593+/-152(27) | 34+/-5(35) | *-39+/-5(11) | *18+/-6(14) |
| Kv1.3 + R385A + n-Shc | 0.49(8) | *693+/-145(7) | 29+/-8(8) | -50+/-3(8) | 4+/-2(8) |
| Kv1.3 + v-Src + Sck | 0.56(21) | 898+/-352(20) | 27+/-4(20) | -49+/-5(15) | 7+/-2(15) |
| Kv1.3 + v-Src + triple | 0.45(7) | *610+/-180(5) | 21+/-5(5) | *-37+/-11(5) | 5+/-3(5) |
Same experimental protocol, electrophysiological recording, statistical analysis, and notation as in Table 1 but for n-Shc and related (Sck) or mutated (nShc triple mutant) family members.
Upon co-transfection of Grb10 cDNA with Kv1.3 plus v-Src, Grb10 adaptor protein relieved v-Src-induced current suppression of Kv1.3 to return the peak current amplitude to 88% of the control level (Kv1.3-only transfection) (n = 19) (Fig. 4A, Table 1). Grb10 adaptor protein relieved the v-Src-induced decrease in τinact to values near those of the Kv1.3-only control condition, but Grb10 shifted τdeact to values significantly lower than either the Kv1.3 or Kv1.3 plus v-Src transfection conditions (ANOVA, snk, α = 0.05) (Fig. 4C,E). In contrast, co-transfection of n-Shc cDNA only partially relieved v-Src-induced Kv1.3 current suppression (Fig. 4B, Table 2, n = 38). Kv1.3 current in the presence of v-Src and n-Shc was 59% of the Kv1.3-only control condition (Table 2). n-Shc also did not significantly relieve the v-Src-induced decrease in τinact but returned τdeact to values similar to those of the Kv1.3-only transfection condition (ANOVA, snk, α = 0.05) (Fig. 4D,F).
Taken together, the data from our combined SDS-PAGE-immunoprecipitation and patch-clamp electrophysiological experiments indicate that the adaptor protein Grb10 inhibits a large proportion of the v-Src-induced tyrosine phosphorylation of Kv1.3 and that this decreased phosphorylation correlates with a decrease in Src kinase modulation of Kv1.3 current amplitude and kinetics. The adaptor molecule n-Shc does not significantly relieve v-Src-induced modulation of Kv1.3 current amplitude, the kinetics of inactivation, or the corresponding total tyrosine phosphorylation of the channel that is retained or increased in the presence of the adaptor protein. Although we do not know if different tyrosine residues in Kv1.3 are targeted by v-Src in the presence of n-Shc, clearly the structural differences between n-Shc and Grb10 could provide differential interaction with Kv1.3 to account for the differences in Kv1.3 physiology in the presence of Src kinase and n-Shc or Grb10. The fact that Grb10 inhibits most of v-Src-induced phosphorylation and functional modulation could suggest that Grb10 blocks access of v-Src kinase to Kv1.3 in the cell. Since both Grb10 and Kv1.3 contain proline-rich sequences specific for interaction with the SH3 domain of v-Src kinase, and the channel cannot tightly co-immunoprecipitate with Src at this domain {7}, Grb10 may have a stronger affinity for this domain and physically block the kinase access to the channel.
v-Src kinase disrupts Kv1.3 cumulative inactivation, an effect that is restored by Grb10 and accentuated by n-Shc
Kv1.3 ion channel inactivates slowly and remains in the inactivated state for up to 60 seconds {44, 52}. When the interpulse interval is less than 60 seconds, Kv1.3 exhibits a cumulative inactivation upon repeated stimulation due to its long dwell time in the inactivated state. Kv1.3 cumulative inactivation has not been studied under a kinase-induced phosphorylated state. To test whether v-Src kinase affected Kv1.3 cumulative inactivation, cells were held at -90 mV and stepped to +40 mV 12 times for 1 second each at varying interpulse intervals. Peak current amplitudes were normalized to that of the first trace in each series of 12 traces and plotted against pulse number. Sample size for each interpulse interval varied slightly due to the technical difficulty of performing all needed recordings on a cell without losing the voltage-clamp. Kv1.3 showed the expected cumulative inactivation when expressed alone: no inactivation at 60 second interpulse intervals, slight cumulative inactivation was initiated at 30 second interpulse intervals, and marked cumulative inactivation was apparent at 10, 2, 1, and 0.5 s interpulse intervals (n = 22-26) (Fig. 5A). With an interpulse interval permitting only a 2 second recovery from inactivation, as much as 50% of the Kv1.3 channels were caught in the inactivated state. In the condition of v-Src co-transfection, Kv1.3 cumulative inactivation was not initiated until the interpulse interval was shortened to 2 seconds. A maximum of 20-25% of Kv1.3 channels were caught in the inactivated state, even when recovery from inactivation was only 500 milliseconds, compared with Kv1.3 alone transfections in which typically 75% of channels remain inactivated with such short recovery intervals (n = 19-23) (Fig. 5B). The severe decrease of Kv1.3 cumulative inactivation in the presence of the kinase was not due to overall kinase-induced current suppression because the current amplitudes were normalized to that of the first trace in each series and thus percent decreases could be calculated, if present. These data indicate that v-Src kinase could contribute a permissive function in the excitability of cells by increasing the number of Kv1.3 channels available for activation during rapid repetitive stimulation. The Src-induced disruption of Kv1.3 cumulative inactivation would be consistent with either Kv1.3 failing to enter the inactivated state or Kv1.3 exiting the inactivated state more quickly in the presence of the kinase. These alternatives could be best deciphered at the unitary current level in future studies.
Fig. 5. Cumulative inactivation of Kv1.3 is interrupted by v-Src Kinase and re-established in the presence of grb10 adaptor protein.

HEK 293 cells were transfected as in Fig. 3. Cells were voltage-clamped in the cell-attached configuration with Vh = -90 mV and Vc = +40 mV for 1000 msec with different interpulse intervals including, 30, 10, 2, 1, 0.5 seconds. Peak current amplitudes were normalized to that of the first voltage stimulation (In/I1) and this normalized current was plotted against pulse number (n). Error bar represents s.e.m. for 19-26 cells recorded at a given interpulse interval.
Co-transfection of v-Src plus Kv1.3 cDNAs in the presence of either Grb10 or n-Shc adaptor proteins differentially affected the disruption of cumulative inactivation of Kv1.3 by v-Src kinase (Fig. 5C-D). Addition of n-Shc adaptor protein failed to restore cumulative inactivation of Kv1.3 in Kv1.3 plus v-Src co-transfected cells (Fig. 5D). When recovery from inactivation was shortened to only 500 milliseconds, nearly 90% of Kv1.3 channels could still be available for reactivation upon voltage stimulation. Addition of Grb10 adaptor protein, however, re-established cumulative inactivation properties of Kv1.3 in the presence of v-Src kinase (Fig. 5C).
The effects of Grb10 and n-Shc on the phosphorylation and modulation of Kv1.3 by v-Src are phosphorylation-dependent
To determine the importance of v-Src kinase activity and to eliminate the possibility that this kinase exerts its effects on Kv1.3 only through its own non-catalytic interaction domains, we substituted a mutant Src kinase for v-Src in our transfection scheme described above. Mutation of arginine 385 of Src to alanine produces a severely impaired Src kinase (R385A Src) {40}. To confirm that the mutant Src kinase is in fact significantly impaired, we tested its ability to phosphorylate Kv1.3, and we show that this mutant only phosphorylates Kv1.3 to 4-fold above its basal level as opposed to the 14-fold increase by wild-type v-Src (n = 5) (Fig. 6B). The outward current of Kv1.3 in the presence of R385A Src kinase is 76% that of the controls, showing little suppression (n = 19) (Fig. 6A, Tables 1 and 2). The R385A Src kinase does not change the inactivation, deactivation, or cumulative inactivation of Kv1.3 in comparison to the control condition of Kv1.3 alone (n = 8) (Tables 1 and 2, Fig. 6A). When co-transfected with Kv1.3 plus R385A Src kinase, Grb10 decreases what little phosphorylation of Kv1.3 remains to less than that of the Kv1.3-only transfection condition (n = 3) (Fig. 6B). Physiologically, Kv1.3 peak current amplitude is increased 4-fold when R385A Src is substituted for Src kinase in the presence of Grb10, so there is no current suppression in this condition (n = 13) (Fig. 6A, Table 1). Grb10 fails to reverse the decreased τinact of the Kv1.3 plus v-Src co-transfection condition when R385A Src kinase is substituted for Src kinase (Table 1). Kv1.3 exhibits normal cumulative inactivation in the presence of R385A Src and Grb10 adaptor protein (n = 4-6) (Fig. 7B). These results demonstrate that the effects of Grb10 on v-Src-induced phosphorylation and subsequent modulation of Kv1.3 are dependent on the kinase activity of Src.
Fig. 6. Phosphorylation-dependence of adaptor protein affecting v-src-induced phosphorylation and modulation of Kv1.3.

HEK 293 cells were transiently transfected with cDNAs for Kv1.3 +/- v-src or R385A v-src +/- grb10, n-shc, sck, or triple n-shc mutant as in Fig. 3. A, Normalized peak current amplitude for cells recorded with the same voltage paradigm and patch configuration as Fig. 4. Amplitudes for each transfection condition was normalized to that of control (Kv1.3) for each experiment to control for variation in cell passage and tranfection efficiency. Normalized values were then averaged across experimental days. Error bar represents s.e.m., number represents sample size for recordings. B, (top panel), Western-blot demonstrating the degree of tyrosine phosphosphorylated Kv1.3 under various transfection conditions. Protocol and abbreviations as in Fig. 3. B, (lower panel), Histogram plot of the tyrosine phosphorylation of Kv1.3 (Kv1.3), as measured by quantitative immunodensitometry for several experiments as in B, (top panel). Error bar represents s.e.m. with number representing sample size of Western blots.
Fig. 7. Phosphorylation-dependence of v-src induced interruption of Kv1.3 cumulative inactivation and the participation of the CH domain of n-shc.

HEK 293 cells were transfected as in Fig. 6 and patch recorded using the voltage-stimulation paradigm described in Fig. 5. Peak current amplitudes were normalized to that of the first voltage stimulation (In/Ii) and this normalized current was plotted against pulse number (n). Error bar represents s.e.m. for 4-11 cells recorded at a given interpulse interval. R385A = mutation of v-Src kinase at arginine 385; severely impaired v-Src kinase. Triple = n-shc triple mutant; Y to F mutation of n-shc at tyr220, 221, 304 in the CH domain. Sck = shc family member with intact CH domain but not a substrate for v-Src kinase.
Transfection of n-Shc with Kv1.3 plus R385A Src results in no increase in phosphorylation over that of Kv1.3 + R385A Src (n = 3) compared with the 18-fold increase in channel tyrosine phosphorylation in the presence of the adaptor and live v-Src kinase (Fig. 6B). As with transfection of n-Shc with Kv1.3 + v-Src, substitution with R385A Src yielded an intermediate relief in v-Src-induced current suppression (Fig. 6A, Table 2) compared with control Kv1.3 alone conditions. Substitution of R385A Src kinase for v-Src also does not significantly reverse the v-Src-induced decrease in Kv1.3 τinact (Table 2). Kv1.3 exhibits normal cumulative inactivation in the presence of R385A Src and n-Shc adaptor protein (n = 2-3) (Fig. 7C). These results show that the lack of n-Shc effect on v-Src-induced Kv1.3 phosphorylation, Kv1.3 current suppression, decreased τinact, and cumulative inactivation are independent of an intact kinase domain of Src, whereas the n-Shc reversal effect on v-Src-induced 10 mV shift in V1/2 and increase slope of voltage dependence (κ) is dependent on Src kinase activity. Taken together, the data from experiments using R385A Src suggest that Grb10 and n-Shc adaptor proteins each differently regulate the properties of Kv1.3 channel in the presence of v-Src kinase, and do so by a mechanism that requires Src kinase activity.
The CH domain is responsible for n-Shc regulation of v-Src-induced phosphorylation and current suppression of Kv1.3
Members of the Shc family of adaptor proteins contain highly homologous SH2 and PTB domains, responsible for recognizing specific sequences of other proteins containing phosphotyrosines, and CH domains that are not highly conserved and contain different varieties of proline-rich regions and tyrosine phosphorylation-specific sequences{29}. Within the CH domain, the Shc protein is known to be phosphorylated on three specific tyrosine residues, tyr239 and tyr240, and tyr317 {35, 36, 51, 53, 54} that correspond to tyr220, tyr221, and tyr304 of n-Shc, respectively {29}. Sck is a member of the Shc family, which is expressed predominantly in the peripheral nervous system, and differs from n-Shc in the structure of its CH domain. Sck is also NOT a substrate for Src kinase{29}. Sck none the less contains homologous tyrosines in its CH domain. Dr. Takeshi Nakamura (Sumitomo Electric Industries) generously provided our laboratory with cDNA constructs encoding n-Shc, Sck, and a n-Shc mutant in which these three key tyrosines were conservatively mutated to phenylalanine (Y220F, Y221F, and Y304F). The cDNA construct in which all three tyrosine residues in the CH domain were collectively altered from tyrosine to phenylalanine as a cassette, we have termed “n-Shc triple tyrosine mutant.” We used this n-Shc triple tyrosine mutant and the Sck member of the Shc family to determine the contribution of the CH domain toward the n-Shc effect on v-Src-induced changes in Kv1.3 function and tyrosine phosphorylation. n-Shc triple tyrosine mutant lacks the tyrosine residues that would normally be phosphorylated by Src kinase to provide phosphotyrosine residues for interaction with SH2-containing proteins, whereas Sck contains the tyrosine residues, but these tyrosines are not normally substrates for Src kinase. We performed transfection of HEK 293 cells similar to that described for n-Shc above, substituting either Sck or the n-Shc triple tyrosine mutant for n-Shc.
When co-transfected with Kv1.3 and v-Src, Sck adaptor protein maintained an increased Kv1.3 phosphorylation by v-Src (n = 4) (Fig. 6B). Similar to n-Shc, Sck partially relieved v-Src-induced Kv1.3 current suppression to 56% of the control level (n = 21) (Fig. 6A, Table 2). Unlike n-Shc, however, Sck reversed the decreased τinact found in Kv1.3 plus v-Src co-transfected cells to an inactivation rate approaching Kv1.3 alone (Table 2). The increased τdeact induced by v-Src kinase was decreased close to Kv1.3 control τdeact when co-transfected with either n-Shc or Sck. Lastly, Sck, but not n-Shc, restored the cumulative inactivation of Kv1.3 in Kv1.3 plus v-Src co-transfected conditions (n = 10-11) (Fig. 7D). Results from these Sck experiments suggest that regions of the n-Shc CH domain that differ from those of the Sck CH domain are responsible for reversing the decreased τinact, the increased slope (κ) of voltage dependence, the right-shifted voltage at half-activation (V1/2) and reestablishing the cumulative inactivation that becomes disrupted in v-Src + Kv1.3 co-transfected cells. The common inability to relieve current suppression and common ability to increase τdeact back toward Kv1.3 control values compared with Kv1.3 + v-Src transfected conditions suggests that these properties are governed by CH domain residues in common across n-Shc and Sck.
When n-Shc triple tyrosine mutant was substituted for n-Shc in the transfection scheme of Kv1.3 plus v-Src kinase plus adaptor protein, phosphorylation of the ion channel was 8-fold over that of the control, slightly less than that of Kv1.3 plus v-Src kinase (n = 5) (Fig. 6B). Similar to that of wild-type n-Shc, the n-Shc triple tyrosine mutant relieved current suppression to 45% of that of the control (n = 7) (Fig. 6A, Table 2). In a manner similar to that of n-Shc, the n-Shc triple tyrosine mutant had no effect on τinact of Kv1.3 plus v-Src co-transfected cells but further decreased τdeact to rates less than that of Kv1.3 alone. Similar to n-Shc, but not Sck, the n-Shc triple tyrosine mutant caused a 10 mV right shift in V1/2 compared with Kv1.3 plus v-Src co-transfected cells lacking an adaptor molecule (Table 2). In contrast to n-Shc, but similar to Sck, the n-Shc triple tyrosine mutant restored cumulative inactivation of Kv1.3 to similar levels as that observed for Kv1.3 alone (n = 2) (Fig. 7E). The data from experiments performed with the n-Shc triple tyrosine mutant suggest that tyr 220, tyr221, tyr304 are not important for increasing Kv1.3 phosphorylation by v-Src and are apparently also not necessary for the partial recovery of Src-induced Kv1.3 current suppression observed in the presence of n-Shc. Tyr220, tyr221, and/or tyr304 in n-Shc, however, must participate in the loss of cumulative inactivation of Kv1.3 as disrupted by v-Src kinase, because removal of these residues (v-Src triple tyrosine mutant) or their capacity to be phosphorylated by Src (Skc) reverses Src disruption of Kv1.3 cumulative inactivation.
Substituting two proteins very similar to n-Shc with differences only in their CH domains has provided results suggesting that other residues than tyr220, tyr221, and tyr304 of the n-Shc CH domain that are similar in the Sck CH domain are required for retaining or increasing v-Src-induced phosphorylation of Kv1.3. Differing regions of the CH domain, excluding the three key tyrosines, are important for reversing the decreased τinact observed in the Kv1.3 plus v-Src co-transfected condition. n-Shc, Sck, and the triple n-Shc mutant all show an intermediate, non-significant relief of v-Src-induced-Kv1.3 current suppression and all return τdeact to rates less than that of the Kv1.3-only controls. Therefore, the ability for n-Shc to modulate current magnitude and τdeact is likely to lie within its SH2 and/or PTB domains and not with residues or signaling structures contained in its CH domain.
Discussion
We have begun to investigate the mechanisms by which signaling modules could regulate ion channel physiology and subsequent sensory processing in the OB by using cloned signaling components in a controlled heterologous expression system. In determining the excitability and plasticity of single nerve cells, it will be important to consider not only the diversity of voltage-gated ion channels expressed in a neuron, but also the array of protein modules that could interact with any given channel at discrete phosphorylation-dependent and -independent targets. There are several good examples of how adaptor proteins may play central roles in directing tyrosine kinase signaling. For example, the novel ZIP1 and ZIP2 adaptor proteins link PKC activity to an auxiliary potassium channel subunit, Kvβ2, thereby linking kinase activity to potassium channel octomers {55}. PDZ-domain containing postsynaptic density protein 95 (PSD-95) is required for Src modulation of NMDA-sensitive glutamate receptors, and PSD-95 has also been shown to bind NMDA receptor subunits, the inward rectifiers Kir2.1 and Kir2.3, and the Shaker homolog Kv1.4 {8, 56-58}. The human homolog of the PDZ-containing Drosophila discs large protein (Hdlg) is able to bind both the Src-family member Lck and Kv1.3 {59}. PSD-95 forms the bridge in a complex between Src kinase and Kv1.3 in cultured microglia {60}. Finally, Nitabach et al. {61} demonstrate that Kv1.5 channels may act as adaptor molecules themselves by forming heteromultimeric complexes with Kv1.4 subunits otherwise insensitive to interaction with Src kinase by providing the necessary proline rich regions for Src kinase SH3 domain interaction through the Kv1.5 subunit.
The degree of excitability of OB neurons is dependent upon the activity of voltage-gated ion channels that may greatly shape the coding of olfactory information. Modulation of voltage-gated potassium (Kv) channels, in particular, by tyrosine phosphorylation is a dynamic means of changing neuronal excitability, and thus, the ability of the involved brain area to encode information {62}. We have found that while Src kinase is expressed in the OB and modulates Kv1.3 current magnitude, kinetics of inactivation, and cumulative inactivation, adaptor proteins further shape the nature of the kinase-induced modulation. Regulation of ion channel modulation by adaptor proteins is most likely via protein-protein interactions across a channel-kinase-adaptor complex to alter tyrosine phosphorylation of the channel.
The differential degree of Kv1.3 tyrosine phosphorylation and modulation of Kv1.3 current in v-Src + Kv1.3 co-transfected cells in the presence of Grb10 and n-Shc may be attributed to the inherent uniqueness in signaling modules contained in these two adaptor proteins (Fig. 8). Unlike n-Shc, Grb10 decreases or eliminates v-Src-induced Kv1.3 tyrosine phosphorylation, current suppression, decreased τdeact, and disrupted cumulative inactivation properties. As described in the introduction, the Src SH3 domain recognizes and binds to proline-containing sequences in other proteins capable of forming a polyproline type II helix {63}, minimally recognizing PXXP and most favorably recognizing sequences that conform to the canonical Src SH3 domain binding site RPLPXXP {64}. Kv1.3 (amino acids 39-44 (PLPPALP) and 493-496 (PQTP)) and Grb10 (amino acids 30-36 (PAGPGLP), 136-139 (PAIP), and 148-154 (PGSPPVL)) both contain two or more such proline-rich regions. Thus it is conceivable that the Kv1.3 and Grb10 proline-rich regions compete for interaction with the Src SH3 domain such that the Src SH3 domain preferentially binds to a Grb10 proline-rich target, allowing Src access to the Kv1.3 ion channel only in the absence of Grb10 expression or correct subcellular localization (Fig. 8A). Grb10 adaptor protein may therefore regulate v-Src modulation of Kv1.3 by blocking the access of the kinase to the ion channel. Because v-Src induced Kv1.3 tyrosine phosphorylation is dramatically reduced in the presence of Grb10, tyr137 and tyr449, the normal molecular targets for Kv1.3 phosphorylation by v-Src, are unlikely interaction modules for either the Src SH2 or Grb10 SH2 domain. It is highly probable that Grb10 is phosphorylated as a result of interaction with v-Src (Fig. 8A). Our data substituting R385A Src kinase for v-Src show that the mechanism for Grb10 alteration of v-Src-induced channel phosphorylation and modulation is phosphorylation-dependent. This idea is further supported by evidence that Grb10 is phosphorylated by v-Src on its own tyr67 {27}.
Fig. 8. Putative model of n-Shc and Grb10 modulation of v-src-induced changes in Kv1.3 channel properties and upregulation in phosphorylation.

Src kinase is known to phosphorylate (P) Kv1.3 at tyr137 and tyr449 (noted by a *). A, Kv1.3 and Grb10 proline-rich regions may compete for interaction with the Src SH3 domain such that the Src SH3 domain preferentially binds to a Grb10 proline-rich target, allowing Src access to the Kv1.3 ion channel only in the absence of Grb10 expression or correct subcellular localization. Grb10 adaptor protein may therefore regulate v-Src modulation of Kv1.3 by blocking the access of the kinase to the ion channel. B, A potential sequence of events leading to n-Shc regulation of v-Src-induced modulation of Kv1.3 is as follows: 1) the v-Src SH3 domain binds to n-Shc via proline-rich regions in the CH domain, 2) v-Src phosphorylates n-Shc at tyr220, tyr221, and/or tyr 304, 3) n-Shc translocates itself and v-Src to membrane regions in which Kv1.3 is located using its PTB domain, 4) n-Shc binds to Kv1.3 at v-Src phosphorylated tyr137 and tyr449 using its SH2 domain or at non-phosphorylated tyrosines with its PTB domain, and 5) n-Shc uses its PTB domain to bind to and shield non-phosphorylated key tyrosines from being phosphorylated by v-Src. N-Shc could possibly retain some degree of phosphorylation at tyr137 and tyr449 and direct Src kinase to add phosphate groups to other tyrosine sites unimportant for channel current suppression, thereby bringing about an increase in phosphorylation but not increasing the degree of current suppression. Alternatively, n-Shc could perhaps bind to basally phosphorylated tyrosines (possibly including tyr137 and/or tyr449) via its SH2 domain and then redirect further phosphorylation onto other tyrosines not important for current suppression.
The specific mechanism by which n-Shc may regulate the phosphorylation and subsequent modulation of Kv1.3 by v-Src in HEK 293 cells is somewhat complex. In addition to the SH3 domain, Src kinase also contains a SH2 domain, which recognizes and binds to phosphorylated tyrosines of other proteins. Kv1.3 has at least six potential phosphotyrosine sites that are accessible to a SH2 domain from the cytoplasmic face of the channel: tyr 111, 112, 113, tyr137, tyr449, and tyr479. N-Shc contains three key tyrosines in its central CH domain: tyr220, tyr221, and tyr304. N-Shc also contains a SH2 domain at its C-terminus, as well as a phosphotyrosine binding (PTB) domain at its N-terminus. As mentioned above, Kv1.3 and n-Shc both contain numerous proline-rich sequences. Because of the many possible protein-protein interactions among Kv1.3, v-Src, and n-Shc due to their interaction motifs, we attempted to simplify our study by substituting two members of the Shc family for n-Shc: Sck, which differs from n-Shc at its CH domain, and the n-Shc triple mutant, which lacks the three key tyrosines. Our data from these experiments and those using R385A Src indicate that interactions that enable n-Shc to right shift the V1/2 of Kv1.3 by 10 mV and restore Kv1.3 cumulative inactivation disrupted by v-Src are phosphorylation-dependent and require the three key tyrosines of the CH domain of n-Shc to be intact. Our data which demonstrate that Sck, but not n-Shc, reverses the v-Src-induced decrease in τinact of Kv1.3 suggests that release of the three key tyrosines of the CH domain from being substrates for Src phosphorylation permits an inactivation rate for the channel that approximates that of Kv1.3 alone. N-Shc returns τDeact to a value similar to that of Kv1.3 alone, and the causative interaction is phosphorylation-independent and most likely occurs via the PTB domain of n-Shc. The PTB domain has been shown to bring about phosphorylation-independent interactions {65-67}, making this domain an interesting possibility for this interaction. Finally, the ability of n-Shc to partially relieve Kv1.3 current suppression brought about by v-Src results from a phosphorylation-dependent interaction by either its SH2 or PTB domain. Taken together, our results suggest that n-Shc could exerts its effects in this system by using proline-rich regions and the three key tyrosines of its CH domain, its PTB domain, and possibly its SH2 domain. A potential sequence of events leading to n-Shc regulation of v-Src-induced modulation of Kv1.3 is as follows (Fig. 8B): 1) the v-Src SH3 domain binds to n-Shc via proline-rich regions in the CH domain, 2) v-Src phosphorylates n-Shc at tyr220, tyr221, and/or tyr 304, 3) n-Shc translocates itself and v-Src to membrane regions in which Kv1.3 is located using its PTB domain, 4) n-Shc binds to Kv1.3 at v-Src phosphorylated tyr137 and tyr449 using its SH2 domain or at non-phosphorylated tyrosines with its PTB domain, and 5) n-Shc uses its PTB domain to bind to and shield non-phosphorylated key tyrosines from being phosphorylated by v-Src. N-Shc could possibly retain some degree of phosphorylation at tyr137 and tyr449 and direct Src kinase to add phosphate groups to other tyrosine sites unimportant for channel current suppression, thereby bringing about an increase in phosphorylation but not increasing the degree of current suppression. Alternatively, n-Shc could perhaps bind to basally phosphorylated tyrosines (possibly including tyr137 and/or tyr449) via its SH2 domain and then redirect further phosphorylation onto other tyrosines not important for current suppression.
Phosphotyrosine signaling depends heavily upon protein-protein interactions mediated by adaptor proteins to carry out its function{68}. Although the assemblage of Kv1.3, v-Src kinase, n-Shc, and Grb10 is expressed in the olfactory bulb, it is not known how Src kinase might be activated in native neurons and modulated by adaptor molecules. We do know that receptor-lined kinases expressed in the bulb differentially modulate Kv1.3 dependent upon stage of development, the animal's degree of satiety, and odor-sensory experience ({13}; Tucker and Fadool, submitted). Growth factors, such as IGF and BDNF, have been demonstrated to drive up Src kinase activity which subsequently alters voltage- or ligand gated channels {69}. Given that production and release of growth factors from neurons are activity dependent {70,71} provides a venue for activation of Src kinase with increased electrical activity.
Future co-immunoprecipitation experiments in native olfactory bulb tissue combined with targeted-gene deletions in mice will be valuable in accessing the detailed function and contribution of adaptor molecules for encoding chemical communication.
Acknowledgments
We would like to thank Drs. Huganir, Senften, Roth, and Nakamura for generous gifts of cDNA constructs used in our study. We are grateful for the technical assistance of Ms. Kristal R. Tucker. This work was supported by NIH R29 DC03387 from the NIDCD and a State of Florida Neuroscience Fellowship to K.C.
References
- 1.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 2.Biggin PC, Toosild T, Choe S. Cur Opin Struct Biol. 2000;10:456–461. doi: 10.1016/s0959-440x(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 3.Yi BA, Jan LY. Neuron. 2000;27:423–425. doi: 10.1016/s0896-6273(00)00052-0. [DOI] [PubMed] [Google Scholar]
- 4.Scannevin RH, Trimmer JS. Biochem Biophys Res Com. 1997;232:585–589. doi: 10.1006/bbrc.1997.6333. [DOI] [PubMed] [Google Scholar]
- 5.Yu XM, Askalan R, Keil GJ, Salter MW. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 6.Tiffany AM, Manganas LN, Kim E, Hsueh YP, Sheng M, Trimmer JS. J Cell Biol. 2000;148:1–147. doi: 10.1083/jcb.148.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes TC, Fadool DA, Ren R, Levitan IB. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 8.Kornau HC, Seeburg PH, Kennedy MB. Cur Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- 9.Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 10.Arnold DB, Clapham DE. Neuron. 1999;23:149–157. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 11.Kues WA, Wunder F. Eur J Neurosci. 1992;4:1296–1308. doi: 10.1111/j.1460-9568.1992.tb00155.x. [DOI] [PubMed] [Google Scholar]
- 12.Fadool DA, Levitan IB. J Neurosci. 1998;18:6126–6137. doi: 10.1523/JNEUROSCI.18-16-06126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadool DA, Tucker K, Phillips JJ, Simmen JA. J Neurophysiol. 2000;83:2332–2348. doi: 10.1152/jn.2000.83.4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abram CL, Courtneidge SA. Exp Cell Res. 2000;254:1–13. doi: 10.1006/excr.1999.4732. [DOI] [PubMed] [Google Scholar]
- 15.Parker RC, Varmus HE, Bishop JM. Proceedings of the National Academy of Sciences USA. 1981;78:5842–5846. doi: 10.1073/pnas.78.9.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fung YK, Crittenden LB, Fadly AM, Kung HJ. Proceedings of the National Academy of Sciences USA. 1983;80:353–357. doi: 10.1073/pnas.80.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon MA, Kornberg TB, Bishop JM. Nature. 1983;302:837–839. doi: 10.1038/302837a0. [DOI] [PubMed] [Google Scholar]
- 18.Takeya T, Hanafusa H. Cell. 1983;32:881–890. doi: 10.1016/0092-8674(83)90073-9. [DOI] [PubMed] [Google Scholar]
- 19.Bradshaw JM, Mitaxov V, Waksman G. J Mol Biol. 1999;293:971–985. doi: 10.1006/jmbi.1999.3190. [DOI] [PubMed] [Google Scholar]
- 20.Cohen GB, Ren R, Baltimore D. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 21.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci PG. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- 22.Liu F, Roth RA. Proceedings of the National Academy of Sciences USA. 1995;92:10287–10291. doi: 10.1073/pnas.92.22.10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ooi J, Yajnik V, Immanuel D, Gordon M, Moskow JJ, Buchberg AM, Margolis B. Oncogene. 1995;10:1621–1630. [PubMed] [Google Scholar]
- 24.Hansen H, Svensson U, Zhu J, Laviola L, Giorgino F, Wolf G, Smith RJ, Riedel H. The Journal of Biological Chemistry. 1996;271:8882–8886. doi: 10.1074/jbc.271.15.8882. [DOI] [PubMed] [Google Scholar]
- 25.O'Neill TJ, Rose DW, Pillay TS, Hotta K, Olefsky JM, Gustafson TA. The Journal of Biological Chemistry. 1996;271:22506–22513. doi: 10.1074/jbc.271.37.22506. [DOI] [PubMed] [Google Scholar]
- 26.Dong LQ, Du H, Porter SG, Kolakowski LF, Jr, Lee AV, Mandarino J, Fan J, Yee D, Liu F. The Journal of Biological Chemistry. 1997;272:29104–29112. doi: 10.1074/jbc.272.46.29104. [DOI] [PubMed] [Google Scholar]
- 27.Langlais P, Dong LQ, Hu D, Liu F. Oncogene. 2000;19:2895–2903. doi: 10.1038/sj.onc.1203616. [DOI] [PubMed] [Google Scholar]
- 28.Frantz JD, Giorgetti-Peraldi S, Ottinger EA, Shoelson SE. The Journal of Biological Chemistry. 1997;272:2659–2667. doi: 10.1074/jbc.272.5.2659. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura T, Muraoka S, Sanokawa R, Mori N. The Journal of Biological Chemistry. 1998;273:6960–6967. doi: 10.1074/jbc.273.12.6960. [DOI] [PubMed] [Google Scholar]
- 30.Zuker CS, Ranganathan R. Science. 1999;283:650–651. doi: 10.1126/science.283.5402.650. [DOI] [PubMed] [Google Scholar]
- 31.Kavanaugh WM, Williams LT. Science. 1994;266:1862–1865. doi: 10.1126/science.7527937. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura T, Sanokawa R, Sasaki Y, Ayusawa D, Oishi M, Mori N. Oncogene. 1996;13:1121. [PubMed] [Google Scholar]
- 33.O'Bryan JP, Songyang Z, Cantley L, Der CJ, Pawson T. Proceedings of the National Academy of Sciences USA. 1996;93:2729–2734. doi: 10.1073/pnas.93.7.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelicci G, Dente L, De Giuseppe A, Verducci-Galletti B, Giuli S, Mele S, Vetriani C, Giorgio M, Pandolfi PP, Cesareni G, Pelicci PG. Oncogene. 1996;13:633–641. [PubMed] [Google Scholar]
- 35.van der Geer P, Wiley S, Gish GD, Pawson T. Current Biology. 1996;6:1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- 36.Ishihara H, Sasaoka T, Ishiki M, Takata Y, Imamura T, Usui I, Langlois J, Sawa T, Kobayashi M. The Journal of Biological Chemistry. 1997;272:9581–9586. doi: 10.1074/jbc.272.14.9581. [DOI] [PubMed] [Google Scholar]
- 37.van der Geer P, Pawson T. Trends in Biological Science. 1995;20:277–280. doi: 10.1016/s0968-0004(00)89043-x. [DOI] [PubMed] [Google Scholar]
- 38.Cowburn D. Structure. 1996;4:10051008–1008. doi: 10.1016/s0969-2126(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 39.Cowburn D. Current Opinion in Structural Biology. 1997;7:835–838. doi: 10.1016/s0959-440x(97)80155-8. [DOI] [PubMed] [Google Scholar]
- 40.Senften M, Schenker G, Sowadski JM, Ballmer-Hofer K. Oncogene. 1995;10:199–203. [PubMed] [Google Scholar]
- 41.Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- 42.Bowlby MR, Fadool DA, Holmes TC, Levitan IB. J Gen Physiol. 1997;110:601–610. doi: 10.1085/jgp.110.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmes TC, Fadool DA, Levitan IB. J Neurosci. 1996;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marom S, Goldstein SA, Kupper J, Levitan IB. Receptors and Channels. 1993;1:81–88. [PubMed] [Google Scholar]
- 45.Sambrook J, Russel DW. Molecular Cloning. Cold Spring Harbor Press; New York: 2001. [Google Scholar]
- 46.Maroney AC, Qureshi SA, Foster DA, Brugge JS. Oncogene. 1992;7:1207–1214. [PubMed] [Google Scholar]
- 47.Marshall J, Molloy R, Moss GW, Howe JR, Hughes TE. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 48.Shi G, Kleinklaus AK, Marrion NV, Trimmer JS. The Journal of Biological Chemistry. 1994;269:23204–23211. [PubMed] [Google Scholar]
- 49.White MF, Kahn CR. The Journal of Biological Chemistry. 1994;269:1–4. [PubMed] [Google Scholar]
- 50.McGlade J, Cheng A, Pelicci G, Pelicci PG, Pawson T. Proc Nat Acad Sci. 1992;89:8869–8873. doi: 10.1073/pnas.89.19.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato K, Gotoh N, Otsuki T, Kakumoto M, Aoto M, Tokmakov AA, Shibuya M, Fukami Y. Biochemical and Biophysical Research Communications. 1997;240:399–404. doi: 10.1006/bbrc.1997.7667. [DOI] [PubMed] [Google Scholar]
- 52.Marom S, Levitan IB. Biophysical Journal. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishihara H, Sasaoka T, Wada T, Ishiki M, Haruta T, Usui I, Iwata M, Takano A, Uno T, Ueno E, Kobayashi M. Biochemical and Biophysical Research Communications. 1998;252:139–144. doi: 10.1006/bbrc.1998.9621. [DOI] [PubMed] [Google Scholar]
- 54.Walk SF, March ME, Ravichandran KS. European Journal of Immunology. 1998;28:2265–2275. doi: 10.1002/(SICI)1521-4141(199808)28:08<2265::AID-IMMU2265>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 55.Gong J, Xu J, Bezanilla M, van Huizen R, Derin R, Li M. Science. 1999;285:1565–1569. doi: 10.1126/science.285.5433.1565. [DOI] [PubMed] [Google Scholar]
- 56.Liao GY, Kreitzer MA, Sweetman BJ, Leonard JP. Journal of Neurochemistry. 2000;75:282–287. doi: 10.1046/j.1471-4159.2000.0750282.x. [DOI] [PubMed] [Google Scholar]
- 57.Nehring RB, Wischmeyer E, Döring F, Veh RW, Sheng M, Karschin A. The Journal of Neuroscience. 2000;20:156–162. doi: 10.1523/JNEUROSCI.20-01-00156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shin H, Hsueh YP, Yang FC, Kim E, Sheng M. The Journal of Neuroscience. 2000;20:3580–3587. doi: 10.1523/JNEUROSCI.20-10-03580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanada T, Lin L, Chandy KG, Oh SS, Chishti AH. The Journal of Biological Chemistry. 1997;272:26899–26904. doi: 10.1074/jbc.272.43.26899. [DOI] [PubMed] [Google Scholar]
- 60.Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Eur J Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- 61.Nitabach MN, Llamas DA, Araneda RC, Intile JL, Thompson IJ, Zhou YI, Holmes TC. Proceedings of the National Academy of Sciences USA. 2001;98:705–710. doi: 10.1073/pnas.031446198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hille B. Ion channels of excitable membranes. Sinauer Associates; Sunderland: 2001. [Google Scholar]
- 63.Hubbard SR, Till JH. Annu Rev Biochem. 2000;69:398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 64.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, Kay BK. Proceedings of the National Academy of Sciences USA. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borg JP, Ooi J, Levy E, Margolis B. Molecular and Cellular Biology. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zambrano N, Buxbaum JD, Minopoli G, Fiore F, De Candia P, De Renzis S, Faraonio R, Sabo S, Cheetham J, Sudol M, Russo T. The Journal of Biological Chemistry. 1997;272:6399–6405. doi: 10.1074/jbc.272.10.6399. [DOI] [PubMed] [Google Scholar]
- 67.Margolis B. Trends in Medical Sciences. 1999;10:262–267. doi: 10.1016/s1043-2760(99)00168-x. [DOI] [PubMed] [Google Scholar]
- 68.Faux MC, Scott JD. Cell. 1996;85:9–12. doi: 10.1016/s0092-8674(00)81075-2. [DOI] [PubMed] [Google Scholar]
- 69.Bence-Hanulec KK, Marshall J, Blair LAC. Neuron. 2000;27:121–131. doi: 10.1016/s0896-6273(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 70.Balkowiec A, Katz DM. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tongiorgi E, Righi M, Cattaneo A. J Neurosci. 1997;17:9492–9505. doi: 10.1523/JNEUROSCI.17-24-09492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]