

Abstract
Mutations in the presenilin 1 (PS1) gene are the most commonly recognized cause of familial Alzheimer's disease (FAD). Besides senile plaques, neurofibrillary tangles, and neuronal loss, Alzheimer's disease (AD) is also accompanied by vascular pathology. Here we describe an age-related vascular pathology in two lines of PS1 FAD-mutant transgenic mice that mimics many features of the vascular pathology seen in AD. The pathology was especially prominent in the microvasculature whose vessels became thinned and irregular with the appearance of many abnormally looped vessels as well as string vessels. Stereologic assessments revealed a reduction of the microvasculature in the hippocampus that was accompanied by hippocampal atrophy. The vascular changes were not congophilic. Yet, despite the lack of congophilia, penetrating vessels at the cortical surface were often abnormal morphologically and microhemorrhages sometimes occurred. Altered immunostaining of blood vessels with basement membrane-associated antigens was an early feature of the microangiopathy and was associated with thickening of the vascular basal laminae and endothelial cell alterations that were visible ultrastructurally. Interestingly, although the FAD-mutant transgene was expressed in neurons in both lines of mice, there was no detectable expression in vascular endothelial cells or glial cells. These studies thus have implications for the role of neuronal to vascular signaling in the pathogenesis of the vascular pathology associated with AD.
Whereas most cases of Alzheimer's disease (AD) occur sporadically, some are inherited in an autosomal dominant pattern and known as familial AD (FAD). These cases mimic the sporadic disease clinically and pathologically except for a typically earlier age of onset. Mutations in three genes, the amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2) are known to cause FAD,1 with mutations in PS1 being the most commonly recognized cause of early onset cases.2
In addition to senile plaques, neurofibrillary tangles, and neuron and synapse loss, AD is also accompanied by vascular pathology. In the most recognized form of this pathology, congophilic amyloid angiopathy (CAA), amyloid is deposited in the walls of blood vessels with leptomeningeal and neocortical arteries and arterioles being most affected.3,4 Vascular pathology, however, also occurs in the microvasculature leading to a decreased density and fragmentation of microvasculature.5 Microvessels appear less branched and thin atrophic vessels, known as string vessels, appear while other vessels become kinked and looped. The cause and relationship of microvascular pathology to cognitive decline in AD remains unclear although patients with Down syndrome display a similar vascular pathology that is present in young cases devoid of senile plaques and neurofibrillary tangles6 arguing that vascular changes may precede the development of neurofibrillary tangles and senile plaques. Vascular pathology also occurs in human PS1- and PS2-associated FAD cases seeming to in general mimic the pathology found in sporadic cases.2
Despite the relative rarity of human FAD cases, their known genetic etiology makes them ideal for modeling in transgenic animals, and many FAD-mutant lines exist. Congophilic vascular deposits have been described in APP or APP/PS FAD-mutant mice.7–12 There has been no description of vascular pathology in models expressing PS1 FAD mutants alone. Here we describe a microvascular pathology in two lines of PS1 FAD-mutant transgenic mice. Although the pathology lacks CAA, it mimics many of the other features of AD-related vascular pathology. In both lines, even though the mutant transgene was expressed in neurons, no detectable expression was found in vascular endothelial cells or glial cells suggesting a role for neuronal to vascular signaling in the pathology development.
Materials and Methods
Genetically Modified Mice
Transgenic mice expressing a wild-type human PS1 cDNA or a cDNA containing the P117L FAD mutation under the control of the neuron-specific enolase (NSE) promoter were generated by pronuclear injection and have been previously described (Table 1).13,14 These mice were generated on the C57BL/6 × DBA F1 hybrid background and have been maintained by breeding to C57BL/6 mice. Genotyping was performed as described previously.14
Table 1.
Groups of Mice Studied
| Strain | Transgene | Ages studied | Pathology observed |
|---|---|---|---|
| PS1 PAC wild-type Tg | P1 bacteriophage artificial chromosome containing human PS1 | 4 to 28 months | None |
| PS1 PAC M146V Tg | P1 bacteriophage artificial chromosome containing human PS1 retrofitted with M146V FAD mutation | 4 to 33 months | -Age-related microvascular pathology (thinning, irregularity, string vessels) |
| -Reduced microvascular length | |||
| -Noncongophilic pathology in cortical penetrating vessels | |||
| -Abnormal immunostaining of basement membrane-associated antigens | |||
| -Thickening of vascular basal laminae | |||
| -Hippocampal atrophy | |||
| -Altered dendritic structure | |||
| Non-Tg littermates of PAC mice | None | 4 to 37 months | None |
| NSE-PS1 wild-type Tg | Human wild-type PS1 cDNA under control of NSE promoter | 4 to 22 months | None |
| NSE-P117L Tg | Human PS1 cDNA containing the P117L FAD mutation under control of the NSE promoter | 4 to 24 months | Similar to PS1 PAC M146V |
| Non-Tg littermates of NSE mice | None | 4 to 24 months | None |
Tg, transgenic.
Transgenic mice expressing wild-type human PS1 from a P1 bacteriophage artificial chromosome (PAC) were produced using the clone RP1-54D12 (204 kb; accession number AC 006342) containing the entire human PS1 transcription unit (>75 kb;15). Founders were generated by injecting C57BL/6 × C3H F1 oocytes as described previously.16
To generate PAC transgenic mice expressing the M146V FAD mutation, the 54D12 clone was retrofitted using the Rec A-mediated homologous recombination system described by Ali Imam et al.17 A 1.6-kb fragment (nucleotides 135,630–137,240) containing PS1 exon 6 was amplified from PAC 54D12 DNA by PCR using the primer pair 5′-GGAGACCAAGGTGGGCAGAT-3′ and 5′-TGGAGCCCTAGCCTTCATTCT-3′ and subcloned into the pCR2.1 vector (Invitrogen, Carlsbad, CA). The M146V FAD mutation (ATG to GTG codon change) was introduced by mutating the A residue at position 136,459 in the 54D12 clone (corresponding to nucleotide 653 of the PS1 mRNA) to G using the Quikchange kit (Stratagene, La Jolla, CA) and the complementary mutagenic primer set 5′-CCAGGAGGATAGTCACGACAACAATGACACT-3′ and 5′-AGTGTCATTGTTGTCGTGAGCGGATAACAATTTCAC-3′. The presence of the M146V mutation was identified by nucleotide sequence analyses.
The M146V mutation was introduced into the 54D12 PAC clone by homologous recombination using the pDF26 vector (gift from Drs. A. M. Ali Imam and F. Grosveld, Erasmus University, Rotterdam, The Netherlands). This vector harbors the chloramphenicol resistance gene (CmR), an rpsL+ allele for counter selection (StpS), a temperature-sensitive replication initiation protein (RepAts), an origin of replication and the recA gene of Escherichia coli. The mutant 1.6-kb fragment was excised from the pCR2.1 vector by XhoI-HindIII digestion and subcloned into the XhoI-HindIII sites of pDF26 (resulting in the plasmid pDFPS1M146V), which was transformed into E. coli XL blue cells (Stratagene) and chloramphenicol-resistant clones were selected at 30°C. Plasmid DNA from a pDFPS1M146V-positive clone was transformed into E. coli DH10 cells harboring the KanR PAC 54D12. Double transformants (pDFPS1M146V/PAC54D12) were selected on plates containing kanamycin and chloramphenicol at 30°C and then transferred to fresh kanamycin/chloramphenicol plates and grown overnight at 43°C (the nonpermissive temperature). To select for recombinant clones harboring pDFPS1M146V integrated into the homologous region of PAC54D12 via recA activity, correct type I and II integrations were identified by Southern blot analyses (using the above 1.6-kb fragment harboring PS1 exon 6 sequences as probe). Positive clones were grown overnight at 43°C on kanamycin-containing plates to allow excision of the integrated vector sequences. In these recombination events, either the original copy or the targeted sequence containing the modified M146V mutation is left behind. Excised vector PAC clones were further selected at 43°C on kanamycin/streptomycin plates, permitting the growth of only bacterial clones that excised the pDF rpsL+ vector sequences. Identification of PAC clones harboring the M146V mutation (PACPS1M146V) was performed as follows: a 0.25-kb fragment harboring the PS1 exon 6 sequences of interest (nucleotides 136,351-136,600 of the reference PAC 54D12) was amplified from individual PAC clones with the primer pair 5′-TGACAAGAATACCCAACCAT-3′ and 5′-TCCATTAACACTGACCTAGG-3′, digested with BspHI and analyzed by agarose gel electrophoresis. The M146V mutation was detected by the loss of one of the two BspHI restriction sites in exon 6. About 50% of the clones tested harbored the M146V mutation. DNA from two individual mutant PS1PAC M146V clones was isolated and analyzed as previously described.16 The purified DNA was adjusted to a concentration of 1 μg/ml in 10 mmol/L Tris-HCl, pH 7.5, 0.5 mmol/L EDTA, 30 nmol/L spermine, 70 nmol/L spermidine, 0.1 M NaCl and transgenic mice were generated by pronuclear injection into C57BL/6 × C3H F1 oocytes. Genotyping of tail DNA by PCR/BspHI restriction analyses identified 5 founders out of 18 births. Routine genotyping was performed by PCR of genomic DNA from tail biopsies with the primer pair described above followed if necessary by BspHI digestion to confirm the presence of the wild-type or M146V-mutant allele. Lines were maintained by breeding to C57BL/6 mice.
The PS1−/− mice used were those generated by Shen et al.18 These animals were provided on a mixed genetic background and have been maintained by breeding to C57BL/6 wild-type mice. Genotypes were determined by PCR as described in Shen et al.18 Animals were housed on 12 hour light/dark cycles and received food and water ad libitum. Because of the mixed genetic background of all of the lines, nontransgenic littermates were used as controls. All protocols were approved by the Institutional Animal Care and Use Committees of the James J. Peters Department of Veterans Affairs Medical Center and the Mount Sinai School of Medicine and were conducted in conformance with Public Health Service policy on the humane care and use of laboratory animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Tissue Processing
Adult mice were anesthetized with 150 mg/kg ketamine and 30 mg/kg xylazine and sacrificed by transcardial perfusion with cold 4% paraformaldehyde in PBS. After perfusion, brains were removed and postfixed in 4% paraformaldehyde for 48 hours, transferred to PBS, and stored at 4°C until sectioning. Fifty-micrometer-thick coronal sections were cut using a Leica VT1000 S Vibratome (Leica, Wetzlar, Germany).
Histology and Immunohistochemistry
H&E staining was performed using standard methods. Thioflavin S staining was performed as described in Schmidt et al.19 Immunohistochemical staining was performed on free-floating sections. For immunofluorescence staining, the primary antibodies used were a rabbit polyclonal anti-collagen IV antiserum (1/500; Chemicon International, Temecula, CA), a rabbit polyclonal anti-laminin antiserum (1/100; Sigma-Aldrich, St. Louis, MO), a rabbit polyclonal anti-fibronectin antiserum (1:400; Sigma-Aldrich), a rat monoclonal anti-perlecan antiserum (1:500; Neomarkers, Fremont, CA), a rabbit polyclonal anti-Aβ antiserum (1/200 dilution; Chemicon International), and a mouse monoclonal anti-β-III tubulin (TuJ1, 1/500; Covance Research Products, Denver, PA). Sections were blocked with Tris-buffered saline (TBS; 50 mmol/L Tris-HCl, 0.15 M NaCl (pH 7.6), and 0.15 M NaCl)/0.1% Triton X-100/5% goat serum (TBS-TGS) for 1.5 hours, and the primary antibody was applied overnight in TBS-TGS at room temperature. Following washing in PBS for 1 hour, immunofluorescence staining was detected by incubation with species-specific AlexaFluor secondary antibody conjugates (1/400; Molecular Probes, Burlingame CA) for 2 hours in TBS-TGS. Nuclei were counterstained with 1 μg/ml 4′,6-diamidino-2-phenylindole. After washing in PBS, sections were mounted on slides using Gel/Mount (Biomeda, Foster City, CA). Sections incubated with secondary antibody only were used as controls.
Immunoperoxidase staining for collagen IV was performed on pepsin-digested tissue using methods similar to those described previously.20 The sections were pretreated with 10% methanol/1% hydrogen peroxide in PBS for 10 minutes, washed in PBS and treated with 1 mg/ml pepsin (Dako, Carpinteria, CA) in 3% acetic acid for 50 minutes at 50°C. After washing in PBS, the primary antibody was applied as described above followed by a goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1/300, Santa Cruz Biotechnology, Santa Cruz, CA) for two hours. Staining was visualized using 3,3′-diaminobenzidine (DAB) in 50 mmol/L Tris-imidazole buffer, pH 7.6. Sections were mounted on slides, dried overnight and counterstained with 0.5% cresyl violet for 5 minutes followed by dehydration with a graded series of ethanol solutions (70, 85, 90, 100% for 1 minute each). Slides were then treated with Americlear (Fisher, Tustin, CA) for 1 minute, followed by xylene for 1 minute and coverslipped with Cytoseal 60 (Richard-Allan Scientific, Kalamazoo, MI).
Stained sections were photographed on a Zeiss Axioimager microscope using the AxioVision Release 4.3 program (Zeiss, Thornwood, NY), a Nikon Eclipse E400 connected to a DXC-390 CCD camera (Nikon, Melville, NY) or a Zeiss LSM 510 confocal microscope. Unstained sections of fixed brain were photographed on a Nikon SMZ1500 stereomicroscope equipped with an oblique coherent contrast illumination system and connected to a SPOT RT digital camera (Sterling Heights, MI). Digital images were color balanced using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
In Situ Hybridization
For in situ hybridization, brains were embedded in OCT compound (Tissue Tek, Elkhart, IN). Fifteen-micrometer-thick cryostat sections were air dried for 1 to 2 hours and fixed in 4% paraformaldehyde in PBS-diethylpyrocarbonate. Sections were treated with 1 mg/ml proteinase K for 5 minutes at room temperature and fixed in paraformaldehyde for an additional 20 minutes. After several washes with PBS-diethylpyrocarbonate, sections were acetylated with acetic anhydride in the presence of triethanolamine. The sections were then prehybridized for 2 hours in 50% formamide, 5× saline sodium citrate, 5× Denhardt's solution, 250 μg/ml yeast RNA, and 500 μg/ml herring sperm DNA and hybridized at 70°C for 16 hours with 400 ng/ml of a heat-denatured (80°C) digoxigenin-labeled anti-sense RNA probe. To prepare a human PS1-specific probe we amplified a fragment spanning nucleotides 1891-2315 of human PS1 3′ untranslated region (accession number NM_000021), an area of very low homology between human and mouse PS1 with the primers 5′-ACTACCAGATTTGAGGGACGAG-3′ and 5′-GGCAATCACAGACGGTAATGAG-3′. The resulting 424-bp human PS1 DNA product was cloned into the pDrive vector (Qiagen, Valencia, CA). An antisense human-specific cRNA probe was prepared with XhoI-linearized plasmid in the presence of digoxigenin-labeled uridine triphosphate in addition to the other nucleotides and T7 RNA polymerase. Slides were washed for 1 hour in 0.2× saline sodium citrate at 70°C and subsequently with 50 mmol/L Tris-HCl (pH 7.6) and 0.15 M NaCl (TBS) at room temperature. Slides were blocked with 10% heat-inactivated goat serum in TBS at room temperature and incubated overnight with a 1/250 dilution of anti-digoxigenin antibodies at 4°C (Roche, Basel, Switzerland). After several washes with TBS, slides were equilibrated in alkaline phosphatase buffer (0.1 M Tris-HCl (pH 9.5), 0.1 M NaCl, 50 mmol/L MgCl2, 0.01% Tween-20, and 0.25 mg/ml levamisole) for 30 minutes, followed by staining with 0.4 mg/ml nitroblue tetrazolium chloride, and 0.19 mg/ml 5-bromo-4-chloro-3-indolyl-phosphate in the same solution for 72 hours at 4°C.
Western Blotting
For determination of PS1 levels, brains were homogenized in 10 volumes of radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl (pH 7.6), 0.15 M NaCl, 1 mmol/L EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) containing HALT protease and phosphatase inhibitor mixtures (Pierce, Rockford, IL). The samples were centrifuged at 15,000 rpm for 20 minutes and the supernatant collected. Protein concentrations were determined using the BCA reagent assay (Pierce). Protein samples were mixed with Laemmli buffer and loaded without boiling onto SDS-PAGE gels. Gels were blotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). The blots were blocked with 50 mmol/L Tris-HCl (pH 7.6), 0.15 M NaCl, 0.1% Tween-20 (TBST), and 5% nonfat dry milk and probed with the mouse monoclonal antibody NT.1 (1/2000), which recognizes the human PS1 N-terminal fragment (NTF) or with the mouse monoclonal antibody 33B10 (1/3000), which recognizes both human and mouse PS1 C-terminal fragment (CTF) followed by secondary detection with horseradish peroxidase conjugated goat anti-mouse IgG. The bands were visualized with the ECL+ reagent (GE Healthcare Bio-Sciences, Piscataway, NJ) and exposure to CL-Xposure film (Pierce).
For blotting of basement membrane associated proteins, one cerebral hemisphere from each animal was homogenized in 50 mmol/L Na Phosphate pH 7.6, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton, 0.25% sodium deoxycholate, 0.5% SDS buffer supplemented with Halt protease and phosphatase inhibitor mixtures I and II (Sigma-Aldrich). The homogenates were centrifuged at 14,000 rpm for 20 minutes, and the supernantants were removed. Protein concentration was determined as above, and for blotting, 50 μg of protein per sample was run on 7.5% SDS-PAGE gels and blotted onto polyvinylidene difluoride membranes. For collagen IV analysis, the samples were mixed with Laemmli buffer without mercaptoethanol and not boiled before loading on the gel. The membranes were blocked for 1 hour as described above and incubated overnight at 4°C with primary antibodies diluted in blocking solution followed by secondary detection with horseradish peroxidase-conjugated anti-rabbit IgG (1/5000 in blocking solution with only 1% nonfat dry milk; GE Healthcare Bio-Sciences). The bands were visualized with ECL+ reagent (GE Healthcare Bio-Sciences) and exposure to HyBlot CL film (Denville Scientific, Metuchen, NJ) and/or imaging with a Kodak Image Station 4000R. As needed blots were stripped with Re-blot plus strong (Millipore) and reprobed. The primary antibodies used were a rabbit polyclonal anti-fibronectin (1/3000; Sigma-Aldrich), rabbit polyclonal anti-laminin (1/500; Sigma-Aldrich), and rabbit polyclonal anti-collagen IV (1/3000; Abcam, Cambridge, UK). A rabbit polyclonal anti-β-tubulin (1/3000; Abcam) was used as a loading control. Quantitation was performed with Image Quant TL software (GE Healthcare Bio-Sciences) with normalization to the level of β-tubulin.
Stereologic Analysis
Brains were cut in serial 50-μm-thick coronal sections using a Vibratome (Leica). Every sixth section was chosen, beginning with a random section between 1 and 6 and immunostained for collagen IV as described above. To reduce bias, all analyses were performed blind to genotype. For stereologic analyses, collagen IV-immunostained sections were first viewed at low magnification (×2.5) to outline the hippocampal borders using a Zeiss Axioplan 2 microscope and StereoInvestigator software (MBF Bioscience, Williston, VT). Vascular area and length was then examined using the software's Space Balls probe21–24 using a ×40 oil Zeiss PlanApochromat objective, 1.0 n.a. The dimensions of the random sampling grid used were set at x = 310 μm, y = 300 μm, and a hemisphere with a radius of 15 μm was used. A 0.5-μm guard zone was set at the top and bottom of the tissue to avoid counting in the compromised cut surface while the remainder of the tissue thickness represented the disector height; the mean section thickness was 18.3 μm. Sampling grids were systematically and randomly placed by the stereology software throughout the region of interest. Vascular length density was calculated from the total number of intersections between the vasculature and the hemispheres (mean number of counted intersections = 335), using the following formula derived from the description of the Space Balls method and using the area of the delineated region and section thickness after tissue processing:
where VLD represents the capillary length density, Σis the total number of intersections between the hemispheres and the microvessels, DX and DY the distance between the midpoints of the centers of the hemispheres in the x- and y-axes, t the actual average section thickness after histological processing, r the radius of the hemispheres and V the investigated tissue volume. When using stereologic techniques on processed tissue, tissue volume change is always of concern. In this study, tissue sections were mounted soon after cutting and before staining, which minimizes shrinkage in the x–y directions.22 Shrinkage in the z-direction can be substantial; the stereology software corrected for this by measuring the true section thickness at regular intervals and using this factor in calculating total vascular length and hippocampal volume.
Ultrastructural Analyses
The method used for electron microscopy has been described previously.25 Briefly, the embedding technique followed the procedure as described26 to study the ultrastructure of blood vessels. Mice from each genotype were anesthetized and perfused as described above with 4% paraformaldehyde containing 0.125% glutaraldehyde. Tissue was removed and postfixed in the same perfusate overnight. Brains were then removed and 250-μm-thick coronal sections were cut using a Vibratome and relevant cortical blocks were dissected out. Freeze substitution and low-temperature embedding of the specimens was performed as described elsewhere.26–28 The slices were cryoprotected by immersion in 4% d-glucose, followed with increasing concentrations of glycerol (from 10 to 30% in phosphate buffer; v/v). Sections were plunged rapidly into liquid propane cooled by liquid nitrogen (−190°C) in a Universal Cryofixation System KF80 (Reichert-Jung, Vienna, Austria). The samples were immersed in 0.5% uranyl acetate dissolved in anhydrous methanol (−90°C, 24 hours) in a cryosubstitution AFS unit (Leica, Vienna, Austria). The temperature was raised from −90°C to −45°C in steps of 4°C/h. After washing with anhydrous methanol, the samples were infiltrated with Lowicryl HM20 resin (Electron Microscopy Sciences, Fort Washington, PA) at −45°C. Polymerization with UV light (360 nm) was performed for 48 hours at −45°C, followed by 24 hours at 0°C. Ultrathin sections (70 nm) were cut with a diamond knife on a Reichert-Jung ultramicrotome and mounted on nickel grids using a Coat-Quick adhesive pen (Electron Microscopy Sciences). Ultrastructural analyses were performed on a JEOL 1200 EX electron microscope (Tokyo, Japan) and imaged with an advantage CCD camera (Advanced Microscopy Techniques, Danvers, MA). Electron microscopy images were produced using Adobe Photoshop 7.0 and adjusted for brightness and contrast.
Statistical Procedures
Results are presented as mean ± the SEM. For all comparisons the equality of variance was first assessed by Levene's test. As none of the variances were found to differ significantly (P > 0.05, Levene statistic) comparisons were made using a one-way analysis of variance followed by a Tukey honestly significant difference (HSD) test for multiple comparisons of all pairs of groups or using unpaired Student's t-tests when two groups were compared. Statistical analyses were performed using the program GraphPad Prism 4.0 (GraphPad Software, San Diego, CA) or SPSS 16.0 (SPSS, Chicago, IL).
Results
Generation of PAC Transgenic Mice Expressing Human Wild-Type or M146V FAD-Mutant PS1
Transgenic mice expressing human wild-type PS1 were generated using a P1 artificial chromosome (PAC) clone (54D12) containing the human PS1 transcription unit. To create transgenic mice expressing an FAD mutant, the PAC54D12 clone was retrofitted with the M146V PS1 FAD mutation using Rec-A mediated homologous recombination. We selected for study two lines of mice (one wild-type and one FAD mutant) that expressed similar levels of human PS1 in the brain as judged by Western blotting and that had similar patterns of transgene expression based on in situ hybridization. In Figure 1, Western blots of adult brain extracts are shown blotted with the human specific antibody NT.1, which recognizes the PS1 NTF or with the antibody 33B10, which recognizes the PS1 CTF. NT.1 recognized a unique band present in both transgenic lines that was not seen in non-transgenic brain (Figure 1A). Western blotting with 33B10 (Figure 1B), which was generated against a region of the PS1 protein that is identical in mouse and human,14 showed comparable levels of PS1 expression in the wild-type and FAD-mutant lines at levels about two- to threefold higher in transgenic compared with nontransgenic brain. Also of note is that the human CTF, which can be discriminated from the mouse CTF by its slower mobility, largely replaced the mouse PS1 (Figure 1C).
Figure 1.

Transgene expression in PS1 PAC wild-type and PS1 PAC M146V transgenic mice. Western blotting was performed on extracts (50 μg of protein) from 2-month-old nontransgenic (non-Tg) littermate controls, human PS1 PAC wild-type transgenic (PS1 Wt Tg), or PS1 PAC M146V transgenic mice (M146V). A: Blots were probed with NT.1, a human-specific antibody that recognizes the ∼30 kDa human but not the mouse NTF. Note the lack of immunoreaction in the nontransgenic brain. B: The blot was probed with antibody 33B10, which recognizes both the mouse and human PS1 CTFs of ∼20 kDa. Note the comparable expression levels in the PS1 PAC wild-type and M146V-mutant lines and the elevation of total PS1 CTF in the transgenic lines compared with the nontransgenic. C: Blotting with 33B10 was repeated with less protein loaded (25 μg). Note that the mouse (Mu) CTF is distinguishable from the human (Hu) CTF by its slightly faster migration and that in the transgenic lines the human protein has largely replaced the mouse.
We analyzed transgene expression pattern in adult PS1 PAC transgenic mice by in situ hybridization using a human specific cRNA probe and a digoxigenin detection system. In both PS1 wild-type and FAD-mutant transgenic lines, the human transgene was expressed in all brain regions and exhibited a neuronal expression pattern mimicking that of the endogenous PS1 in adult mouse brain.29 An example of labeling in the hippocampus is shown in Figure 2.
Figure 2.

Transgene expression in PS1 PAC transgenic mice. In situ hybridization was performed on 2-month-old adult animals using a human-specific PS1 antisense cRNA probe derived from the 3′ untranslated region of the gene. Shown are sagittal sections of hippocampus from a PS1 PAC wild-type transgenic (A), a PS1 PAC M146V transgenic (B), and a non-transgenic littermate control (C). Note the lack of hybridization signal in the nontransgenic control brain (C) and the prominent labeling of the granule cell and pyramidal cell layers in the transgenic animals (A and B). Scale bar: 100 μm.
To test whether the PS1 PAC transgenes could rescue the embryonic lethality of PS1 null-mutant mice (PS1−/−) we bred the transgenes from the PS1 PAC wild-type and PS1 PAC M146V lines onto the mouse PS1−/− background. Both wild-type and FAD-mutant transgenes rescued the embryonic lethality of the PS1 null mutant. Rescued adult animals appeared normal in size, were fertile, and grossly indistinguishable from their non-transgenic littermates. These results confirm the functionality of both the wild-type and FAD-mutant PAC transgenes in the mouse and are also consistent with other reports that PS1 FAD mutants can rescue the embryonic lethality of the PS1-null mouse.30,31
Age-Related Microvascular Pathology in PS1 FAD-Mutant Transgenic Mice
Vascular pathology in human AD includes CAA, which is observed mostly in small arteries and arterioles as well as a distinctive alteration of the cerebral microvasculature with vascular attrition and the appearance of a variety of abnormal vessels.5 To determine whether PS1 FAD-mutant mice exhibit vascular pathology, we examined PS1 PAC M146V transgenic mice as well as mice that express the PS1 P117L FAD mutation under the control of the NSE promoter. The NSE lines were developed previously and express a human cDNA containing the P117L FAD mutation.14 As controls, we generated lines that express a wild-type human PS1 cDNA driven by the same promoter. Control lines were selected that expressed similar levels and patterns of PS1 expression as the FAD mutants based on Western blotting and in situ hybridization.14 Like other PS1 FAD-mutant mice,32 NSE-P117L FAD-mutant mice have elevated levels of Aβ42 and an increased ratio of Aβ42/4013 compared with nontransgenic and NSE-PS1 wild-type transgenic mice, indicating the functionality of the P117L-mutant transgene.
Initially we examined vascular morphology using collagen IV immunostaining to visualize blood vessels. In young PS1 FAD-mutant M146V or P117L mice (6 months of age or less), we noted no abnormalities. However, we consistently observed vascular pathology with aging in both M146V and P117L FAD-mutant mice. Examples of the pathology in the hippocampus of PS1 PAC M146V transgenic mice are shown in Figure 3. Compared with controls, microvessels in the FAD-mutant mice were often thinner and irregular. In addition many string-like vessels (eg, Figure 3H) were observed while other vessels displayed a kinked or twisted morphology (eg, Figure 3G). The thinning and irregularity as well as the strings and twisted morphologies are highly reminiscent of the vascular pathology seen in AD.5 These changes were found widely in both cortical and subcortical regions. We consistently observed some degree of this pathology in mice 1 year of age or older with the pathology becoming more apparent in older mice. No morphological differences were seen with aging between the nontransgenic controls and the human PS1 PAC wild-type transgenic mice.
Figure 3.

Age-related vascular alterations in PS1 PAC M146V transgenic mice. Anti-collagen IV immunoperoxidase-stained sections of the hippocampus from 28-month-old nontransgenic control (A and D), 27-month-old PS1 PAC M146V transgenic (B, E, and G–J), and 28-month-old PS1 PAC wild-type transgenic (C and F) mice. Note the thinning of the microvessels in the FAD mutant (B and E). Higher magnification images of microvessels from the FAD mutant are shown in G–J. A twisted and kinked vessel is indicated by an arrowhead (G), and a string-like vessel is indicated by an arrow (H). Scale bar: 100 μm, A–C; 50 μm, D and E; 20 μm, G–J.
We also observed a similar age-related vascular pathology in NSE-P117L transgenic mice (Figure 4). This pathology has been consistently seen in NSE-P117L transgenic mice >1 year of age and is not found in age-matched nontransgenic littermates or in NSE-PS1 wild-type transgenic mice. Although some mild irregularities can be seen at times in normal vessels (eg, Figure 4C), these changes never approached the degree of irregularity seen in PS1 FAD-mutant vessels making both lines of PS1 FAD-mutant mice easily distinguishable from controls on a morphological basis.
Figure 4.

Microvascular pathology in NSE-P117L FAD mutant mice. Anti-collagen IV immunoperoxidase-stained microvessels in the entorhinal cortex of 2-year nontransgenic (A), NSE-P117L transgenic (B, D, and E), and NSE-PS1 wild-type transgenic (C) mice. Note the irregularity of the vascular surfaces in the FAD mutant (B). Arrows in D and E point to string-like vessels found in the FAD mutant. Scale bar: 50 μm, A–C; 25 μm, D and E.
Decreased Microvasculature and Brain Atrophy in Aging PS1 PAC M146V Transgenic Mice
Vascular loss is a hallmark of the microvascular pathology in AD.5 To determine whether the vascular pathology observed in PS1 PAC M146V transgenic mice was accompanied by a reduced microvasculature we performed stereologic assessments of collagen IV-immunostained microvessels in the hippocampus in groups of 15 month or older PS1 PAC M146V and PS1 PAC wild-type transgenic mice as well as nontransgenic littermate controls. As shown in Figure 5, compared with the control groups, total vascular length was lower by 40 to 45% in the FAD mutant: falling from 772 ± 130 cm (SEM) and 720 ± 43 cm in the nontransgenic and PS1 PAC wild-type transgenic mice, respectively, to 430 ± 28 cm in the FAD mutant (F2,12 = 7.646, P = 0.007, P < 0.05, PS1 PAC M146V versus both PS1 PAC wild-type transgenic mice and nontransgenic littermate controls, Tukey HSD). Mean microvessel area was 25 to 30% less in the FAD mutant, but this difference did not reach statistical significance (F2,12 = 2.863, P = 0.096). The microvascular density differed by <20% between the groups (F2,11 = 0.890, P = 0.43).
Figure 5.

Decrease in microvessel length in the hippocampus of PS1 PAC M146V transgenic mice. Vascular parameters were assessed stereologically in the hippocampus of PS1 M146V PAC mice (n = 7, age range 15 to 33 months, average 24.4 ± 2.6 months) and were compared with control groups of nontransgenic (Non-Tg) littermates (n = 4, age range 28 to 37 months, average 32.5 ± 1.8 months) and PS1 PAC wild-type transgenic (PS1Wt Tg) mice (n = 4, age range 18 to 28 months, average 22.5 ± 2.6). A one-way analysis of variance revealed that the groups did not significantly differ by age (F2,12 = 3.462, P = 0.06, P > 0.07 all comparisons, Tukey HSD). Data are presented as total vessel length (A and B) and area (C and D) per hippocampus (C and D) as well as microvessel density (E and F). *P = 0.01 versus non-Tg and P = 0.03 versus PS1 Wt Tg (Tukey HSD).
The reduced vascular length without any change in vascular density was suggestive of concomitant hippocampal atrophy, and indeed, there was frequently visible brain atrophy in M146V FAD-mutant mice, including gross atrophy of the hippocampus in the oldest mice (Figure 6). A quantitative analysis showed that hippocampal volume was reduced by ∼30% in the FAD-mutant group compared with a pooled control group of nontransgenic and PS1 PAC wild-type transgenic mice (Figure 6; P = 0.012, unpaired t-test). Whereas the basis for the brain atrophy remains to be explored in depth, one aspect of the pathology appears to be attrition of dendritic structure in the FAD-mutant animals. This attrition was most apparent in neocortical large pyramidal neurons. In Figure 7, β-III tubulin-immunostained sections of the neocortex are shown from a 7.5-month-old M146V FAD-mutant as well as control mice imaged by confocal microscopy. Marked alterations of the dendritic structure are apparent in the FAD mutant. Although it is not possible to infer a direct causal relationship, the microvascular pathology seen in PS1 PAC M146V transgenic mice appears to be associated with substantial brain atrophy.
Figure 6.

Age-related brain atrophy in PS1 PAC M146V transgenic mice. Coronal sections of unstained fixed brain from 3-year-old nontransgenic (A) and M146V PAC transgenic littermate mice (B) are shown. The lateral ventricle (LV), as well as the dorsal (D3V) and ventral (V3V), portions of the third ventricle are indicated. C and D: Hippocampal volumes of the same mice analyzed in Figure 5. Average hippocampal volume was 30% lower in the M146V FAD-mutant mice compared with either control group with a one-way analysis of variance revealing significant between group differences (F2,12 = 3.921, P = 0.049), although posttests did not reach statistical significance (P > 0.09, Tukey HSD). However, if the two control groups (non-Tg and PS1 Wt Tg), which did not differ (P = 1.00), were treated as a pooled control, hippocampal volumes (D) in the M146V FAD-mutant mice were significantly lower *P = 0.012, vs. control (unpaired t-test).
Figure 7.

Altered dendritic structure in PS1 FAD-mutant mice. Shown are confocal laser scanning microscopy images of the neocortex from a 10 month old nontransgenic mouse (A), as well as 7.5-month-old PS1 M146V PAC (B), and PS1 PAC wild-type transgenic (C) mice. Sections were immunostained for β-III tubulin with the antibody Tuj (red) and counterstained with a 4′,6-diamidino-2-phenylindole nuclear stain (blue). Note that the dendrites in the FAD mutant (one of which is indicated by an arrowhead) appear shorter and thinner. Scale bar: 20 μm.
Vascular Pathology in PS1 FAD-Mutant Mice Is Not Congophilic
CAA involves the deposition of amyloid within blood vessels. It is the most recognized form of vascular pathology in human AD. To determine whether vascular pathology in PS1 FAD-mutant mice was associated with congophilic changes, we performed thioflavin S staining (Figure 8). We found no congophilic changes in either M146V or P117L FAD-mutant mice stained at various ages, even though congophilic changes were evident in a CRND8 transgenic mouse33 included as a positive control. We have also not found any evidence of Aβ deposition by immunostaining in blood vessels of the PS1 FAD-mutant mice (data not shown).
Figure 8.

Vascular pathology is not congophilic in PS1 FAD mutant mice. Nomarski-imaged (A and C) or thioflavin S-stained (B and D) sections from a 15-month-old NSE-P117L FAD-mutant transgenic mouse (A and B) or a 1-year-old CRND8 transgenic mouse (C and D) are shown. An arrow in B indicates a nonstained penetrating vessel at the cortical surface in the NSE-P117L FAD-mutant mouse, likely an arteriole based on size. A thioflavin S-stained vessel from a CRND8 transgenic mouse is shown as a positive control (D). An amyloid plaque in the CRND8 mouse is indicated by an arrowhead in D. Scale bar: 100 μm.
Interestingly, despite the lack of congophilia, penetrating vessels at the cortical surface, which are most prone to CAA, were often abnormal morphologically in FAD-mutant mice. Examples of penetrating cortical vessels stained with H&E are shown in Figure 9. These vessels in the FAD-mutant lines, presumably arterioles based on their size, often showed a “cracked” appearance or an image of a “vessel within a vessel” associated with a widened perivascular space (Figure 9, C–E). Microhemorrhages, which are a feature commonly associated with CAA, were also sometimes observed (Figure 9F). Interestingly, the description of a “double barreling” or “vessel within a vessel,” appearance has also been used to describe the appearance of thioflavin S-stained congophilic vessels found in human AD.34,35 However, the vessels described here did not exhibit congophilia or immunohistochemical evidence of Aβ deposition.
Figure 9.
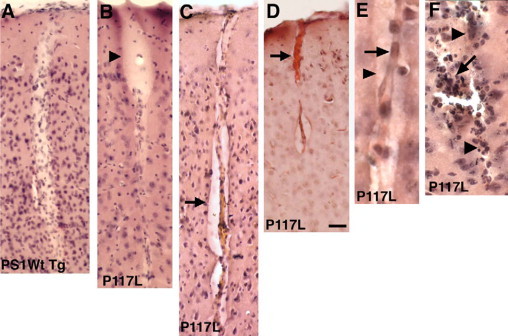
Histological abnormalities in penetrating cortical vessels in PS1 FAD-mutant mice. Shown are H&E-stained sections from NSE-PS1 wild-type transgenic (A) or NSE-P117L FAD mutant (B–F) mice. B: An abnormally expanded perivascular space at the cortical surface is indicated by the arrowhead. Microvessels in C and D have abnormally expanded perivascular spaces and a “vessel within a vessel” or “double-barreled” appearance with the vessel in D containing a thrombus (arrow). E: A higher power view of a double-barreled vessel with the internal cord indicated by an arrow and the outer vessel wall by an arrowhead. F: A vessel with evidence of prior hemorrhage. The outlines of the vessel are indicated by arrowheads. An arrow indicates an area of infiltration of hemosiderin-laden mononuclear cells and polymorphonuclear leukocytes. Scale bar: 50 μm, A–D; 25 μm, E and F.
Altered Immunostaining of Microvessels in PS1 FAD-Mutant Animals with Basement Membrane-Associated Proteins
Alterations of the vascular basement membranes have been suggested to be an early feature of the microvascular pathology in AD.36 The status of vascular basement membranes can be assessed by immunostaining for membrane-associated antigens. However, staining for many antigens including collagen IV occurs only inconsistently in perfusion-fixed materials from normal adult mice.20,37,38 We recently found that widespread staining of the brain vasculature in adult mice can be achieved with collagen IV as well as other antigens, if the immunostaining is preceded by a pepsin digestion step,20 a methodology that was used for the collagen IV staining described above.
Interestingly, however, vascular staining occurred in the brains of PS1 FAD-mutant mice with antigens such as collagen IV and perlecan even without pepsin digestion. In Figure 10, examples are shown of collagen IV vascular staining in PS1 PAC M146V-mutant and NSE-P117L transgenic lines. In contrast to non-transgenic or wild-type human transgenic lines, vascular staining was prominent in both FAD-mutant transgenic lines with collagen IV despite the lack of pepsin treatment. In Figure 11, examples of immunostaining of the hippocampus for the heparan sulfate proteogycan, perlecan, are shown in NSE-P117L FAD-mutant transgenic, NSE-PS1 wild-type transgenic and an age-matched nontransgenic mouse. Widespread vascular staining was seen with perlecan in the NSE-P117L brain. In contrast, little to no perlecan immunoreactivity was detected in the nontransgenic or NSE-PS1 wild-type transgenic animal.
Figure 10.

Abnormal vascular staining of collagen IV in PS1 FAD-mutant mice stained without pepsin pretreatment. Shown is collagen IV immunostaining (green) of the hippocampus (A and B) or entorhinal cortex (C and D) of 15-month-old nontransgenic (A) or NSE-P117L (B), as well as 10-month-old PS1 wild-type PAC (C) or M146V PAC (D) transgenic mice. Sections are counterstained with a nuclear stain (4′,6-diamidino-2-phenylindole, blue). Note the prominent labeling of the vasculature in the FAD-mutant transgenic mice (B and D). Scale bar: 100 μm.
Figure 11.

Abnormal vascular staining with perlecan in PS1 FAD-mutant mice stained without pepsin pretreatment. Anti-perlecan immunostaining is shown in the hippocampus from a 15-month-old nontransgenic (A), NSE-PS1 wild-type (B), and NSE-P117L transgenic (C). Note the prominent vascular staining in the NSE-P117L transgenic animal (C). Scale bar: 100 μm.
Unaltered Levels of Basement Membrane-Associated Proteins in PS1 FAD-Mutant Transgenic Mice
The increased immunostaining of the microvasculature for basement membrane-associated proteins in PS1 FAD-mutant mice could be attributed to either increased expression of basement membrane-associated proteins or an altered presentation of antigens due to a reorganization of the vascular basement membrane. To determine whether total levels of vascular membrane-associated proteins were altered in the brain of FAD-mutant mice, we compared levels of fibronectin, laminin, and collagen IV by Western blotting in NSE-P117L FAD-mutant to nontransgenic control mice. As shown in Figure 12, we did not find any difference in total levels of fibronectin, laminin, or collagen IV in FAD-mutant brain. In combination with the altered immunostaining properties for these same antigens noted above, these observations suggest that there is a reorganization of the vascular basement membrane in PS1 FAD-mutant mice.
Figure 12.

Unaltered levels of extracellular matrix-related proteins in brain homogenates of PS1 FAD-mutant mice. A: Levels of fibronectin (FN), laminin (Lam), and collagen IV (Col IV) were determined by Western blotting in extracts from the cerebral hemispheres of 19- to 26-month-old NSE-P117L FAD-mutant mice (tg, n = 8) and their nontransgenic littermate controls (wt, n = 6). Average ages of the transgenic (24.8 ± 0.74 months) and nontransgenic controls (24.0 ± 0.73 months) did not differ (P = 0.74, unpaired t-test). The arrow in the laminin panel indicates the ∼200 kDa form. Blotting for fibronectin and laminin was performed sequentially on the same blot, followed by probing for β-tubulin as a loading control. As the samples for collagen IV quantification could not be boiled, these samples were electrophoresed separately and reprobed with β-tubulin. The β-tubulin panel shown is that which was used for fibronectin and laminin normalization. B: Quantification of the blots in A is shown with results normalized to the levels of β-tubulin (tub). There were no statistically significant differences between the groups (unpaired t-tests).
Ultrastructural Analysis of Microvascular Pathology in PS1 FAD-Mutant Transgenic Mice
To determine whether the immunostaining findings described above reflect alterations of the vascular basal laminae, we examined the microvasculature in the cerebral cortex of PS1 FAD-mutant transgenic mice by electron microscopy (EM). Figure 13 shows microvessels from the brains of 10- to 11-month-old PS1 PAC M146V or NSE-P117L FAD-mutant transgenic mice as well as from a non-transgenic littermate control or a mouse with the PS1 PAC wild-type transgene bred onto the PS1−/− background. Microvessels in the non-transgenic and human PS1 wild-type transgenic exhibited classic neurovascular ultrastructure with circular lumens, intact endothelial cells and smooth capillary walls (Figure 13, A and B). Despite the fact that vascular alterations at the light level are typically not apparent in 10–11 month old mice, many abnormal vascular profiles were apparent in both FAD-mutant lines (Figure 13, C–F). Luminal circularity was frequently lost and there were varying degrees of degeneration of the capillary wall. In addition, endothelial cell nuclei were distorted and swollen with in some cases white halos apparent around the nuclei (Figure 13, E and F).
Figure 13.

Ultrastructural analysis of cerebrovascular pathology in 10- to 11-month-old PS1 FAD-mutant transgenic mice. Transversely sectioned capillaries are shown from the prefrontal cortex of a nontransgenic littermate control (A), a mouse with the PS1 PAC wild-type transgene bred onto the PS1−/− background (B), a PS1 PAC M146V FAD mutant (C and D), or a NSE-P117L FAD mutant (E and F). The microvessels in A and B exhibit circular lumens, intact endothelial cells, and smooth capillary walls. An endothelial cell nucleus (N) is indicated in B. C and D: Note the distortions of luminal circularity and the altered endothelial cell nuclei that appear both distorted and swollen. Note the halo formation surrounding the endothelial nuclei in E and F. There were also varying degrees of degeneration of the capillary walls with amorphous material present in the capillary lumens (arrowheads in E and F). The surrounding neurophil appears intact. Scale bar: 2 μm.
By light microscopy, string vessels typically appear as abnormal connecting vessels between larger vessels that may extent for 30 μm or more between vessels (Figure 3). To our knowledge there have been no descriptions of the ultrastructural correlates of string vessels. We searched for vascular profiles that might represent string vessels. Despite the relative rarity of string vessels, we were able to observe several profiles that may represent early stages of string vessel formation (Figure 14). In two of the three examples in Figure 14, abnormal bridging structures can be seen between microvessels. In one, two transversely sectioned microvessels are shown connected by a dysmorphic process containing two endothelial cell nuclei (Figure 14A), whereas in another (Figure 14B) a relatively normal appearing microvessel is connected to a degenerated microvessel by a short bridge. Other longitudinally cut microvessels could be seen that contained degenerative strictures (Figure 14C) that may also represent early stages of string vessels.
Figure 14.

Abnormal vascular profiles in PS1 FAD-mutant transgenic mice. Shown are microvessels from the prefrontal cortex of the M146V FAD-mutant mouse shown in Figure 13. A: Two transversely sectioned microvessels are seen connected by a dysmorphic process containing two endothelial cell nuclei (indicated by asterisks). B: A relatively normal appearing microvessel at the bottom of the image is connected to a very dysmorphic microvessel by a short bridge (indicated by an arrowhead) that may represent a primitive string vessel. C: A longitudinally cut microvessel is shown that contains a degenerated segment with a stricture (indicated by an arrowhead). A dysmorphic endothelial cell nucleus (N) is indicated. The surrounding neuropil in each panel appears intact. Scale bar: 4 μm in A and C; 3 μm in B.
The altered immunostaining of microvessels in PS1 FAD-mutant animals with basement membrane-associated proteins shown above is also suggestive of an altered vascular basement membrane and indeed thickening and disruption of the vascular basal lamina was apparent in both lines. An example of basal lamina thickening from the NSE-P117L line is illustrated in Figure 15. Interestingly, despite the obvious microvascular pathology, the surrounding neuropil appeared intact (Figures 13–15).
Figure 15.

Thickening of the vascular basal lamina in NSE-P117L FAD-mutant transgenic mice. Transversely sectioned microvessels are shown from the prefrontal cortex of a nontransgenic littermate control (A) and an NSE-P117L FAD mutant (B). The basal laminae are indicated by asterisks in both panels. Note the thickened appearance of the basal lamina in the P117L FAD mutant as well as the altered chromatin structure in the nucleus (N) of the endothelial cell. Similar observations were made in the PS1 PAC M146V FAD mutant. Scale bar: 500 nm.
Neuronal Expression of the FAD-Mutant Transgenes
The pathology in the NSE-P117L is intriguing since the NSE transgene does not appear to be expressed in non-neuronal cells. Evidence for this is derived from our previous in situ hybridization studies where we observed a neuronal specific expression pattern of the NSE-driven transgenes.14 We have also examined PS1 expression in adult brain of the human PS1 PAC transgenic mice. Although hybridization with a human specific cRNA probe revealed widespread neuronal expression, we observed no expression in or around brain blood vessels (Figure 16). In addition, even if we relaxed the hybridization conditions so that the human probe cross-reacts with the endogenous mouse PS1, we still found no vascular hybridization (data not shown). These in situ findings on the expression of mouse PS1 are consistent with most other studies on the localization of PS1 in normal adult brain, which have reported an essentially neuronal specific expression pattern with no detectable expression in nonneuronal cells.29,39–41
Figure 16.

Lack of transgene expression in blood vessels of PS1 PAC transgenic mice. In situ hybridization was performed with a human-specific PS1 antisense cRNA probe. An example is shown of hybridization along the cortical surface in a PS1 PAC wild-type transgenic mouse. Note the strong hybridization in cells in the deeper cortical layers and the lack of hybridization in the microvessels in layer I, two of which are indicated by an arrow. Scale bar: 50 μm, A; 25 μm, B.
These studies thus suggest that limited neuronal expression of a PS1 FAD mutant may be sufficient to induce vascular pathology. One prediction of this model is that pathology should not occur in microvessels located in regions such as white matter where transgene expression does not occur. As expected, the white matter vessels did not show pathology in either PS1 PAC M146V or NSE-P117L FAD-mutant transgenic mice. In Figure 17, examples of white matter vessels in the corpus callosum of a two-year-old PS1 PAC M146V transgenic mouse are shown. In the white matter, the microvessels appeared normal in contrast to the dysmorphic vessels seen in the adjacent hippocampus. Microvessels in the white matter of NSE-P117L FAD-mutant mice also appeared normal (data not shown). Collectively, these observations suggest that PS1 FAD mutants disrupt neuronal to vascular signaling in the adult brain. They also suggest that vascular remodeling in white matter may be controlled by distinct regulatory mechanisms that do not involve PS1.
Figure 17.

Lack of microvascular pathology in white matter of PS1 FAD-mutant mice. Shown are anti-collagen IV immunoperoxidase-stained microvessels combined with a Nissl counterstain in a 2-year-old PS1 PAC M146V transgenic mouse. Note the normal vascular profiles in the corpus callosum (CC). In contrast, abnormal vascular profiles are apparent in the stratum oriens (SO) of the hippocampus. Within the stratum oriens, kinked (arrow) and thinned (arrowhead) microvessels are indicated. Scale bar: 50 μm.
Discussion
Vascular pathology is a consistent feature of AD. Here we show that two lines of PS1 FAD-mutant transgenic mice develop a vascular pathology that shares features of the vascular pathology in AD. Although vascular pathology has been described in other AD transgenic models that overexpress FAD mutations associated with APP or combinations of APP with PS1 or PS2 FAD mutations,7–12 this report is to our knowledge the first description of vascular pathology in mice expressing PS FAD mutations alone.
Several features of the pathology merit comment. First, the pathology is not congophilic. CAA is the most recognized vascular pathology in AD, and although its severity can be highly variable, CAA is present to some degree in nearly all autopsied cases3 and is likewise present in human PS1 FAD cases to varying degrees.2 The lack of congophilia could be interpreted to suggest that vascular pathology in PS1 FAD-mutant mice may not be related to altered amyloid production. However, it could also suggest that soluble Aβ oligomers42 may be toxic to blood vessels in addition to Aβ deposits. Whereas PS FAD-mutant mice do not develop amyloid plaques, they do consistently exhibit altered Aβ production. Indeed, the NSE-P117L line studied here has been previously shown to have high Aβ42 levels13 and the M146V FAD mutation consistently exhibits elevated Aβ42 in both fibroblasts from human FAD patients,43 as well as in transgenic mice.32 Soluble monomers and oligomers are also more difficult to detect by immunohistochemical methods. Thus, it remains possible that soluble or oligomeric forms that cannot be detected by immunohistochemical methods and do not lead to frank amyloid deposits are nevertheless driving the pathology. Interestingly, Kumar-Singh et al7 have reported that pathological changes can be found in both amyloid-laden and nonamyloidotic microvessels in Tg2576 and PS/APP mice, suggesting that even in the setting of frank CAA, vascular pathology still occurs in non-affected vessels. The present study can thus be seen as either further support for the Aβ oligomer hypothesis or as a challenge to the amyloid hypothesis itself. Additional studies are needed to determine whether oligomeric forms of Aβ deposits within these vessels.
A second notable feature of the pathology is that alterations in the vascular basement membranes occur early in PS1 FAD-mutant vessels. Vascular membrane alterations are also characteristic of the microangiopathy in AD.36 Ultrastructurally, the capillary basement membranes appeared thickened and dystrophic in PS1 FAD-mutant mice before other morphological manifestations of vascular pathology were evident. The basement membrane is a complex mixture of extracellular matrix proteins, proteoglycans, and structural proteins. Despite their presence in normal adult mouse brain, staining for many basement membrane antigens, including collagen IV, occurs only inconsistently in standard perfusion-fixed tissues unless an antigen retrieval method is used.20 Interestingly however, vascular staining occurred in the brains of PS1 FAD-mutant mice when using collagen IV as well as perlecan antisera even without pepsin digestion.
Enhanced vascular staining could reflect increased production of extracellular matrix-related proteins. Thickening of the vascular basal lamina was observed by electron microscopy. However, we did not find any increase in levels of collagen IV, laminin, or fibronectin in the brains of NSE-P117L FAD mutants by Western blotting, arguing that there is no increase in expression of these extracellular matrix-related proteins and that, instead, the increased vascular staining likely represents a reorganization of the vascular extracellular matrix with increased accessibility of these antigens to immunostaining. Interestingly, staining of vessels with antigens such as collagen IV without pepsin treatment is also typical of immature vessels,20 and is also seen in vessels in areas of injured adult brain following mechanical trauma.44 Whereas the physiological basis for these differing patterns of staining are incompletely understood, they seem to correlate best with the tightness of the gliovascular junctions (reviewed in Ref. 20). Indeed, one major structural difference between immature (which stain well for these antigens without pepsin treatment) and mature microvessels in brain is the large perivascular spaces present in immature microvessels that separate the outer glial basal lamina and the inner vascular one.44,45 In fact, the tightness of the gliovascular junctions seems to correlate with the intensity of laminin immunostaining following mechanical injury.44 Thus, by analogy, an early feature of the pathology in FAD-mutant mice may be a loss of the normal tightness of the gliovascular junctions. Importantly, widened perivascular spaces and glial processes filled with amorphous material were observed in FAD-mutant mice at an age before prominent vascular pathology was apparent at the light microscopy level.
Altered vascular basement membranes and gliovascular junctions could be interpreted as increased vascular remodeling in PS1 FAD-mutant mice, perhaps stimulated as a response to oligomeric Aβ. Alternatively, altered trophic signaling could be stimulating vascular remodeling independent of Aβ production. In this context, two important vasculotrophic factors, vascular endothelial growth factor and transforming growth factor β1 have been reported to be increased in AD brain.46–49 It has also been recently reported that expression of αvβ3 integrin, which is regarded as a marker of angiogenesis, is increased in microvessels of AD brain.50 This observation further supports the notion that active vascular remodeling is occurring in AD. Interestingly, transgenic mice expressing a constitutively active form of transforming growth factor β1 have also been reported to develop CAA with an early feature of the pathology being thickening of the basement membranes.51 Aβ itself is cleared from brain both through receptor-mediated transport across endothelial cells as well as by interstitial transport through perivascular spaces.52,53 Whether induced by Aβ or other factors, thickened vascular basement membranes with altered gliovascular junctions could in turn impair Aβ transport and perhaps create a cycle in which further membrane remodeling leads to progressive pathology.
A third notable feature of the vascular pathology in PS1 FAD-mutant mice is that the pathophysiological basis for the vascular pathology appears to originate in neurons as in both lines, the FAD-mutant transgenes are expressed in neurons but not in glia or blood vessels. Our previous in situ hybridization studies have documented a neuron-specific expression pattern of the NSE-driven wild-type and P117L transgenes.14 In fact, whereas PS1 is expressed widely during development and in nonneural tissues in the adult, immunohistochemical studies of PS1 expression have generally found a neuronal expression pattern in adult brain without detectable expression in glial cells or vascular endothelial cells.29 PS1 is expressed in reactive astrocytes but is otherwise not readily detected in astrocytes in uninjured brain.54,55 Our in situ hybridization studies of PS1 PAC mice with a human-specific cRNA probe also revealed widespread neuronal expression, but no expression in or around brain blood vessels.
Altogether these lines of evidence suggest that the vascular pathology has a neuronal origin. This could argue for a neuronal origin of toxic oligomeric Aβ, and these studies in fact parallel other studies showing that CAA can be induced by mutant APP transgenes driven by the neuron-specific Thy1 promoter.8–10 Alternatively, these studies may suggest that PS1 influences neuronal signals that regulate vascular homeostasis in adult brain with FAD-mutant PS1 disturbing these signals. This also provides an interesting parallel to our studies showing that during development, selective expression of PS1 on the neuroepithelial side is sufficient to rescue vascular development in the PS1 null mutant,56 an effect that cannot be explained on the basis of Aβ production. Indeed, based on the restricted expression of the PS1 FAD mutants to neurons one may postulate that the FAD mutant initially disrupts a neuronal-to-vascular signal that is required for normal vascular homeostasis. With time this could lead to a gradual age-related vascular compromise and with further aging, the vascular defect could compromise the integrity of the neurovascular unit until metabolic or trophic support of the neuron becomes affected leading to neuronal dysfunction.
Independent of what the mechanisms for the early alterations in the vascular basement membranes in PS1 FAD mutant might be, we show here that PS1 FAD mutant mice develop a microangiopathy that mimics many of the core features of the vascular pathology seen in AD but without CAA. These studies have implications both for the form of Aβ that may be crucial in producing vascular pathology and the role of neuronal to vascular signaling in the production of pathology. Future studies of these mice offer the opportunity to uncover the role of PS1 FAD mutants in neurovascular signaling and provide insights into how neurovascular signaling may be disrupted in sporadic AD as well.
Acknowledgements
Transgenic mice were generated through the Mount Sinai School of Medicine Mouse Genetics Shared Resource Facility. TgCRND8 mice were a gift from Drs. David Westaway and Paul Fraser, and we thank Drs. Michelle Ehrlich and Sam Gandy as well as Ms. Loren Khan for maintaining the Mount Sinai colony. Drs. Paul Matthews and Nikolaos Robakis are thanked for gifts of antibodies and Drs Ali Imam and Frank Grosveld for providing plasmids. We thank Bridget Wicinski for expert technical assistance.
Footnotes
Supported by the National Institute on Aging (grants AG20139, AG02219, AG05138, AG010491, and AG029361), the Alzheimer's Association (IIRG-07-57318), and a merit award from the Department of Veterans Affairs (5I01BX000342-02).
References
- 1.Ertekin-Taner N. Genetics of Alzheimer's disease: a centennial review. Neurol Clin. 2007;25:611–667. doi: 10.1016/j.ncl.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am J Geriatr Psychiatry. 2004;12:146–156. doi: 10.1097/00019442-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Kumar-Singh S. Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008;7(Suppl 1):67–82. doi: 10.1111/j.1601-183X.2007.00380.x. [DOI] [PubMed] [Google Scholar]
- 4.Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62:885–898. doi: 10.1093/jnen/62.9.885. [DOI] [PubMed] [Google Scholar]
- 5.Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer's disease. Neurol Res. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- 6.Buée L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM. Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol (Berl) 1994;87:469–480. doi: 10.1007/BF00294173. [DOI] [PubMed] [Google Scholar]
- 7.Kumar-Singh S, Pirici D, McGowan E, Serneels S, Ceuterick C, Hardy J, Duff K, Dickson D, Van Broeckhoven C. Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am J Pathol. 2005;167:527–543. doi: 10.1016/S0002-9440(10)62995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 11.Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross-talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer's disease. J Neurosci. 2007;27:824–831. doi: 10.1523/JNEUROSCI.4345-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Wen PH, Shao X, Shao Z, Hof PR, Wisniewski T, Kelley K, Friedrich VL, Jr, Ho L, Pasinetti GM, Shioi J, Robakis NK, Elder GA. Overexpression of wild type but not an FAD mutant Presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol Dis. 2002;10:8–19. doi: 10.1006/nbdi.2002.0490. [DOI] [PubMed] [Google Scholar]
- 15.Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, Rogaeva EA, Ikeda M, Holman K, Lin C, Lukiw WJ, de Jong PJ, Fraser PE, Rommens JM, St George-Hyslop P. Analysis of the 5′ sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics. 1997;40:415–424. doi: 10.1006/geno.1996.4523. [DOI] [PubMed] [Google Scholar]
- 16.Gama Sosa MA, DeGasperi R, Gonzalez EA, Kelley K, Lazzarini R, Elder GA. BAC and PAC DNA for the generation of transgenic animals. Biotechniques. 2002;33:51–53. doi: 10.2144/02331bm07. [DOI] [PubMed] [Google Scholar]
- 17.Ali Imam AM, Patrinos GP, de Krom M, Bottardi S, Janssens RJ, Katsantoni E, Wai AWK, Sherrat DJ, Grosveld FG. Modification of human β-globin locus PAC clones by homologous recombination in Escherichia coli. Nucleic Acids Res. 2000;28:e65. doi: 10.1093/nar/28.12.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt ML, Robinson KA, Lee VM, Trojanowski JQ. Chemical and immunological heterogeneity of fibrillar amyloid in plaques of Alzheimer's disease and Down's syndrome brains revealed by confocal microscopy. Am J Pathol. 1995;147:503–515. [PMC free article] [PubMed] [Google Scholar]
- 20.Franciosi S, De Gasperi R, Dickstein DL, English DF, Rocher AB, Janssen WG, Christoffel D, Sosa MA, Hof PR, Buxbaum JD, Elder GA. Pepsin pretreatment allows collagen IV immunostaining of blood vessels in adult mouse brain. J Neurosci Methods. 2007;163:76–82. doi: 10.1016/j.jneumeth.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calhoun ME, Mouton PR. Length measurement: new developments in neurostereology and 3D imagery. J Chem Neuroanat. 2001;21:257–265. doi: 10.1016/s0891-0618(01)00093-x. [DOI] [PubMed] [Google Scholar]
- 22.Kreczmanski P, Schmidt-Kastner R, Heinsen H, Steinbusch HW, Hof PR, Schmitz C. Stereological studies of capillary length density in the frontal cortex of schizophrenics. Acta Neuropathol (Berl) 2005;109:510–518. doi: 10.1007/s00401-005-1003-y. [DOI] [PubMed] [Google Scholar]
- 23.Mouton PR, Gokhale AM, Ward NL, West MJ. Stereological length estimation using spherical probes. J Microsc. 2002;206:54–64. doi: 10.1046/j.1365-2818.2002.01006.x. [DOI] [PubMed] [Google Scholar]
- 24.Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–831. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 25.Janssen WG, Vissavajjhala P, Andrews G, Moran T, Hof PR, Morrison JH. Cellular and synaptic distribution of NR2A and NR2B in macaque monkey and rat hippocampus as visualized with subunit-specific monoclonal antibodies. Exp Neurol. 2005;191(Suppl 1):S28–S44. doi: 10.1016/j.expneurol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 27.Hjelle OP, Chaudhry FA, Ottersen OP. Antisera to glutathione: characterization and immunocytochemical application to the rat cerebellum. Eur J Neurosci. 1994;6:793–804. doi: 10.1111/j.1460-9568.1994.tb00990.x. [DOI] [PubMed] [Google Scholar]
- 28.van Lookeren Campagne M, Oestreicher AB, van der Krift TP, Gispen WH, Verkleij AJ. Freeze-substitution and Lowicryl HM20 embedding of fixed rat brain: suitability for immunogold ultrastructural localization of neural antigens. J Histochem Cytochem. 1991;39:1267–1279. doi: 10.1177/39.9.1833448. [DOI] [PubMed] [Google Scholar]
- 29.Elder GA, Tezapsidis N, Carter J, Shioi J, Bouras C, Li H-C, Johnston JM, Efthimiopoulos S, Friedrich VL, Robakis NK. Identification and neuron specific expression of the S182/presenilin I protein in human and rodent brains. J Neurosci Res. 1996;45:308–320. doi: 10.1002/(SICI)1097-4547(19960801)45:3<308::AID-JNR13>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 30.Qian S, Jiang P, Guan XM, Singh G, Trumbauer ME, Yu H, Chen HY, Van de Ploeg LH, Zheng H. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Aβ1-42/43 expression. Neuron. 1998;20:611–617. doi: 10.1016/s0896-6273(00)80999-x. [DOI] [PubMed] [Google Scholar]
- 31.Davis JA, Naruse S, Chen H, Eckman C, Younkin S, Price DL, Borchelt DR, Sisodia SS, Wong PC. An Alzheimer's disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998;20:603–609. doi: 10.1016/s0896-6273(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 32.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 33.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 34.Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol. 1986;45:79–90. [PubMed] [Google Scholar]
- 35.Vinters HV. Cerebral amyloid angiopathy: a critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 36.Zarow C, Barron E, Chui HC, Perlmutter LS. Vascular basement membrane pathology and Alzheimer's disease. Ann NY Acad Sci. 1997;826:147–160. doi: 10.1111/j.1749-6632.1997.tb48467.x. [DOI] [PubMed] [Google Scholar]
- 37.Jucker M, Bialobok P, Hagg T, Ingram DK. Laminin immunohistochemistry in brain is dependent on method of tissue fixation. Brain Res. 1992;586:166–170. doi: 10.1016/0006-8993(92)91390-z. [DOI] [PubMed] [Google Scholar]
- 38.Mori S, Sternberger NH, Herman MM, Sternberger LA. Variability of laminin immunoreactivity in human autopsy brain. Histochemistry. 1992;97:237–241. doi: 10.1007/BF00267633. [DOI] [PubMed] [Google Scholar]
- 39.Busciglio J, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA. Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J Neurosci. 1997;17:5101–5107. doi: 10.1523/JNEUROSCI.17-13-05101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cribbs DH, Chen LS, Bende SM, LaFerla FM. Widespread neuronal expression of the presenilin-1 early-onset Alzheimer's disease gene in the murine brain. Am J Pathol. 1996;148:1797–1806. [PMC free article] [PubMed] [Google Scholar]
- 41.Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh DM, Selkoe DJ. Aβ oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 43.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 44.Szabo A, Kalman M. Disappearance of the post-lesional laminin immunopositivity of brain vessels is parallel with the formation of gliovascular junctions and common basal lamina: a double-labelling immunohistochemical study. Neuropathol Appl Neurobiol. 2004;30:169–177. doi: 10.1046/j.0305-1846.2003.00524.x. [DOI] [PubMed] [Google Scholar]
- 45.Caley DW, Maxwell DS. Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J Comp Neurol. 1970;138:31–47. doi: 10.1002/cne.901380104. [DOI] [PubMed] [Google Scholar]
- 46.Chiappelli M, Borroni B, Archetti S, Calabrese E, Corsi MM, Franceschi M, Padovani A, Licastro F. VEGF gene and phenotype relation with Alzheimer's disease and mild cognitive impairment. Rejuvenation Res. 2006;9:485–493. doi: 10.1089/rej.2006.9.485. [DOI] [PubMed] [Google Scholar]
- 47.Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer's disease. J Alzheimers Dis. 2006;10:111–118. doi: 10.3233/jad-2006-10114. [DOI] [PubMed] [Google Scholar]
- 48.Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002;23:237–243. doi: 10.1016/s0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- 49.Grammas P, Ovase R. Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer's disease brain. Am J Pathol. 2002;160:1583–1587. doi: 10.1016/s0002-9440(10)61105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B. Evidence of angiogenic vessels in Alzheimer's disease. J Neural Transm. 2009;116:587–597. doi: 10.1007/s00702-009-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Cribbs DH, Chen LS, Cotman CW, LaFerla FM. Injury induces presenilin-1 gene expression in mouse brain. Neuroreport. 1996;7:1773–1776. doi: 10.1097/00001756-199607290-00016. [DOI] [PubMed] [Google Scholar]
- 55.Huynh DP, Vinters HV, Ho DH, Ho VV, Pulst SM. Neuronal expression and intracellular localization of presenilins in normal and Alzheimer disease brains. J Neuropathol Exp Neurol. 1997;56:1009–1017. doi: 10.1097/00005072-199709000-00006. [DOI] [PubMed] [Google Scholar]
- 56.Wen PH, De Gasperi R, Sosa MA, Rocher AB, Friedrich VL, Jr, Hof PR, Elder GA. Selective expression of presenilin 1 in neural progenitor cells rescues the cerebral hemorrhages and cortical lamination defects in presenilin 1-null mutant mice. Development. 2005;132:3873–3883. doi: 10.1242/dev.01946. [DOI] [PMC free article] [PubMed] [Google Scholar]