

Abstract
Hyperhomocysteinemia (HHcy) is a risk factor for neuroinflammatory and neurodegenerative diseases. Homocysteine (Hcy) induces redox stress, in part, by activating matrix metalloproteinase-9 (MMP-9), which degrades the matrix and leads to blood–brain barrier dysfunction. Hcy competitively binds to γ-aminbutyric acid (GABA) receptors, which are excitatory neurotransmitter receptors. However, the role of GABA-A receptor in Hcy-induced cerebrovascular remodeling is not clear. We hypothesized that Hcy causes cerebrovascular remodeling by increasing redox stress and MMP-9 activity via the extracellular signal-regulated kinase (ERK) signaling pathway and by inhibition of GABA-A receptors, thus behaving as an inhibitory neurotransmitter. Hcy-induced reactive oxygen species production was detected using the fluorescent probe, 2′–7′-dichlorodihydrofluorescein diacetate. Hcy increased nicotinamide adenine dinucleotide phosphate-oxidase-4 concomitantly suppressing thioredoxin. Hcy caused activation of MMP-9, measured by gelatin zymography. The GABA-A receptor agonist, muscimol ameliorated the Hcy-mediated MMP-9 activation. In parallel, Hcy caused phosphorylation of ERK and selectively decreased levels of tissue inhibitors of metalloproteinase-4 (TIMP-4). Treatment of the endothelial cell with muscimol restored the levels of TIMP-4 to the levels in control group. Hcy induced expression of iNOS and decreased eNOS expression, which lead to a decreased NO bioavailability. Furthermore muscimol attenuated Hcy-induced MMP-9 via ERK signaling pathway. These results suggest that Hcy competes with GABA-A receptors, inducing the oxidative stress transduction pathway and leading to ERK activation.
Homocysteine (Hcy) is a non-protein sulfur-containing amino acid formed during intracellular conversion of methionine to cysteine. Increased blood levels of Hcy, known as hyperhomocysteinemia (HHcy) displays various neurological abnormalities, such as mental retardation, seizures, and Alzheimer’s disease (Sachdev et al., 2002; Robert et al., 2005; Santiard-Baron et al., 2005). The thiol sulfhydryl in Hcy like most thiols, undergo oxidation to the disulfide at physiological pH (Jacobsen, 2000), thus causing the disruption of redox homeostasis and affect the redox signaling pathways in vascular and neuronal cells (Obeid and Herrmann, 2006). Reactive oxygen species (ROS) subsequently induce the synthesis of matrix metalloproteinases (MMPs) in endothelial cells (ECs) (Haorah et al., 2007), human monocytes (Lu and Wahl, 2005), or human coronary smooth muscle cells (Valentin et al., 2005). MMPs, membrane-bound, zinc-dependent endoproteinases are involved in extracellular matrix degradation and vascular remodeling, which disrupt the blood–brain barrier (BBB) (Rosell et al., 2005). Activation of MMP-9 by Hcy plays an important role in ischemic stroke and it was shown to degrade basement membrane proteins, resulting in loss of brain endothelial integrity, and causing a macromolecular leakage (Lominadze et al., 2006; Rosell et al., 2006).
The proteolytic activity of MMPs is tightly regulated by the tissue inhibitors of metalloproteinases (TIMPs), four of which have been identified (Brew et al., 2000; Mannello and Gazzanelli, 2001; Rosell et al., 2006). An undisrupted balance between MMPs and TIMPs is necessary for the maintenance of brain normal function. Changes in their expression causes various neurological disorders such as stroke and dementia (Sternlicht and Werb, 2001; Jourquin et al., 2003). Hcy alters the expression of TIMPs, which results in abnormal activities of MMPs (Mujumdar and Tyagi, 1999).
A number of studies suggest that MMP-9 contains activator protein-1 (AP-1) sites in its gene promoter region (Hashimoto et al., 2003). Furthermore, AP-1 transcriptional activity is specifically regulated by extracellular signal-regulated kinase-1/2 (ERK-1/2) (Moshal et al., 2006b). Taken together, these studies suggested that the ERK-AP-1 pathway may be the major activator of MMP-9 gene expression (Kang et al., 2008). Some studies have shown that ROS down-regulated ERK activity in vitro (Ku et al., 2007).
GABA-A receptors involve in ERK signaling pathway (Bulleit and Hsieh, 2000). GABA is a major inhibitory neurotransmitter and a natural ligand for the GABA-A receptor. Most of the GABA-A receptor ligands are structurally derived from its agonists muscimol, THIP (4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol, a selective GABAA receptor agonist), or isoguvacine (Frolund et al., 2002). We (Shastry et al., 2006) and others (Griffiths et al., 1983) have shown that Hcy competed with muscimol for binding to the GABA-A receptor. In addition, we have previously showed that Hcy acted as a GABA-A receptor antagonist (Shastry et al., 2006). Thus, in the context of the present study muscimol acted as a ligand for a GABA-A receptor. GABA receptors play a significant role in brain microvascular permeability, which causes alterations in neuronal environment and leads to EC matrix (ECM) degradation and subsequently, to edema formation (Lee et al., 1995; Limmroth et al., 1996; Fruscella et al., 2001; Lazzarini et al., 2001). HHcy is associated with lowering of bioavailability of endothelial nitric oxide (NO), a potent vasodilator. A recent study showed that NO played a significant role in mediating neuronal death (Wang et al., 1998). Another study suggested that GABA-A agonist, muscimol caused a decrease in blood pressure, which was associated with an increase in endothelial NO synthase (eNOS) (Kishi et al., 2001).
The present study was undertaken to determine the role of GABA-A receptor in Hcy-mediated cerebrovascular remodeling. Herein we demonstrate that at pathologically high levels, Hcy triggered oxidative stress causing activation of MMP-9 in cultured brain ECs, which led to ERK activation. All these effects were partially inhibited by GABA-A receptor. Furthermore, we present evidence that muscimol attenuated Hcy-induced MMP-9 via the ERK signaling pathway.
Materials and Methods
Materials
Polyclonal antibodies against phospho-ERK-1/2 and total ERK-1/2 were obtained from Cell Signaling Technology (Beverly, MA). Antibodies against GABA-A, MMP-9, TIMP-4, and β-actin were purchased from Sigma Chemical Co. (St. Louis, MO). Horseradish peroxidase (HRP)-conjugated antibodies, eNOS, iNOS, collogen-IV (Col-IV), and elastin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Nω-nitro-L-arginine methyl ester (L-NAME), NOS inhibitor (Laude et al., 2003), uric acid (UA; an antioxidant dissolved in 50 mmol/L NaHCO3), DL-homocysteine, muscimol, DCFH-DA (2′,7′-dichlorodihydrofluoresceindiacetate), Dulbecco’s modified Eagle’s medium (DMEM), dimethyl sulfoxide (DMSO), and gelatin were obtained from Sigma (St. Louis, MO). N-3-amino-ethyl benzy-acetamidine (1400W), selective inhibitor of iNOS activity (Garvey et al., 1997; Laude et al., 2003) was purchased from Calbiochem (San Diego, CA). Fetal bovine serum (FBS), phosphate-buffered saline (PBS), penicillin, and streptomycin were obtained from Gibco (Grand Island, NY).
Cell culture
bEnd3 is an immortalized mouse brain ECs line that was originally generated in 1990 (Montesano et al., 1990). The bEnd3 cells were purchased from American Type Culture Collection (Manassas, VA) and were grown according to the supplier’s instructions in DMEM, supplemented with 4.5 g/L glucose, 3.7 g/L sodium bicarbonate, 4 mM glutamine, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, pH 7.4. Cells were maintained at 37°C with 5% CO2 in a humidified environment and were used at the fifth or sixth passage for the experiments.
Protein extraction
The bEnd3 cells were grown to confluence in 6-well plates. The ECs were treated with Hcy (100 μM) in the presence or absence of muscimol (50 μM), L-NAME (100 μM), 1400W (100 μM) or UA (100 μM) for 24 h. After treatments cells were extracted by scraping off the monolayer from the plates, followed by centrifugation at 125g for 5 min at 4°C. Cell pellets were transferred to 1.5 ml tubes, washed twice with ice-cold PBS, and were lysed with 1.0 ml ice-cold lysis buffer (composition: 10 M Tris–HCl; pH 7.4, 20% glycerol, 0.5% Triton X-100, 100 mM NaCl2, and protease inhibitor cocktail). Lysates were centrifuged at 14,000g for 15 min at 4°C to remove cell debris. The supernatants (protein extracts) were used for Western blot analysis.
In situ labeling of ROS
The bEnd3 cells were grown on 8-well cover glass plates and serum-starved before the treatments, with following: 100 μM Hcy, 50 μM muscimol, or 50 μM muscimol + 100 μm Hcy for 24 h. The cells treated with medium alone were used as a control group. After treatment, the cells were washed twice with PBS (NaCl 137 mM, KH2PO4 1.47 mM, Na2HPO4·7H2O 10.8 mM, KCl 2.7 mM; pH 7.4). DCFH-DA (20 μM) was added and the cells were incubated in a CO2 incubator for 45 min at 37°C. Then cells were extensively washed with PBS and digital images of the cells were taken with the confocal microscope (Olympus FV1000, objective 100×). ROS formation was visualized using a multiline Argon (495 nm) to excite the dye, while emission was observed above 519 nm. Hcy-induced formation of ROS was assessed in each well by analyzing the total fluorescence intensity of four random fields of observation with image analysis software (Image-Pro Plus, Media Cybernetics, Bethesda, MD). All the experiments were done in duplicate (two wells for each treatment group).
Immunofluorescent staining for phospho ERK1/2
The bEnd3 cells were grown on 8-well coverglass plates coated with sure coat (Life Technologies, Highland Park, NJ). The cells were serum-starved before the treatments with following: 100 μM Hcy, 50 μM muscimol, or 50 μM muscimol + 100 μm Hcy for 24 h). The cells treated with medium alone were used as a control group. After treatment, the cells were washed twice with PBS and fixed in 3.7% paraformaldehyde/PBS at 4°C for 15 min. Cells were briefly washed with PBS, permeabilized with 0.02% Tween-20 in PBS for 10 min, followed by blocking with blocking buffer (PBS, 1% FBS, 0.1% Tween-20) for 30 min in a humidified chamber. The samples were then incubated for 1 h at room temperature with primary antibodies diluted (1:1,000) in blocking buffer. Primary antibodies were detected with respective secondary antibodies conjugated to Alexa-488 or Taxes Red for 1 h at room temperature in dark. Stained monolayers were mounted on Immuno-mounting media (Invitrogen, Carlsbad, CA) and examined using confocal microscope (Olympus FV1000). To enable the comparison of changes in fluorescence intensity and punctuate staining pattern, the images were taken under an identical microscope settings for all treatment groups. Hcy-induced ERK1/2 phosphorylation was assessed in each well by analyzing the total fluorescence intensity of four random fields with Image-Pro Plus. All the experiments were done in duplicate.
ERK activation
Cells were pretreated with or without muscimol (50 μM), followed by Hcy (100 μM) treatment for 1 h. The cells treated with medium alone were used as a control group. Cell extracts were prepared as mentioned above. Equal amounts (12 μg) of protein from each sample were separated by SDS–PAGE and transferred to a nitrocellulose membrane. The blots were first incubated with the primary antibody against phospho-ERK-1/2 (1:500) and then against total ERK-1/2 (1:500). Immunodetection was performed with ECL-plus kit using HRP-conjugated secondary antibody (1:3,000).
Westen blot analysis
Western blotting was performed as described previously (Tyagi et al., 2006). Briefly, 12 μg cell lysate protein was loaded onto 10–15% SDS–PAGE gels and electrophoresed under non-reducing conditions and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk in TBS-T (50 mM Tris–HCl, 150 mM NaCl, 0.1% Tween-20, pH 7.4), incubated with respective primary antibodies for 1 h at room temperature. After probing with appropriate secondary antibodies for 1 h at room temperature the blots were developed using X-ray film (RPI Corp, Inc., Mount Prospect, IL) with a Kodak 2000A developer (Eastman Kodak, Rochester, NY). Image analysis was performed using UMAX PowerLock II (Taiwan). Results were expressed as a ratio of the integrated optical density (IOD) of the target protein band to the IOD of β-actin band in the same lane.
MMP-9 activity by in-gel zymography
bEnd3 cells were cultured in 6-well plates. The cells were serum-starved before the treatments with following: medium alone, 100 μM Hcy, 50 μM muscimol, or 50 μM muscimol + 100 μm Hcy for 24 h. The cells treated with medium alone were used as control. MMP-9 activity in the different treatment groups was measured by in-gel gelatin zymography (Moshal et al., 2006b). Briefly, after the treatment, the supernatant from the cultured cells was collected and concentrated using minicon filters (Millipore, Billerica, MA) with 15 kDa cutoff. The conditioned media was assayed for protein concentration using Bradford’s protein measurement assay. The protein samples (20 μg) were electrophoresed under non-reducing conditions on 7.5% SDS–polyacrylamide gel containing 2 mg/ml gelatin. The gels were rinsed in renaturation buffer containing 2.5% Triton X-100, followed by overnight incubation in activation buffer (composition: 50 mM of Tris–HCl, pH 7.4; and 5 mM of CaCl2) at 37°C. The gels were stained in Coomassie blue R-250 and then distained appropriately with 10% acetic acid. The clear digested regions representing MMP-9 activity, as accessed by running pre-stained molecular weight markers, were quantified densitometrically using Un-Scan-It software (Silk Scientific Inc., Orem, UT).
RNA preparation and semi-quantitative RT-PCR
Expression of redox enzymes and MMPs were assessed with an expression of their respective mRNAs. The bEnd3 were cultured in 6-well plates and treated with medium alone, 100 μM Hcy, 50 μM muscimol, or 100 μm Hcy + 50 μM muscimol for 24 h. Total RNA was isolated from cells using Trizol (Invitrogen), as described earlier (Tyagi et al., 2005). Briefly, the concentration of total RNA was quantified by measuring the absorbance at 260 nm. Samples with a peak area ratio of 28S-to-18S rRNA >2.0 were used. Two micrograms of RNA was reverse-transcribed using oligo dT primers with a total reaction volume of 20 μl. The reverse transcription program was 25°C for 10 min, 42°C for 50 min, and then 70°C for 15 min. Polymerase chain reaction was performed using 2 μl of each RT product (cDNA) with a total reaction volume of 20 μl. The PCR thermal cycle was the following: 94°C for 2 min, 35 cycles at 94°C for 60 sec, at 57°C for 30 sec, at 72°C for 80 sec. This was followed by a final extension for 5 min at 72°C. Primers were: NOX-4 (forward) 5′-TGGAACTTGGGTTCTTCCAG-3′ and (reverse) 5′-CCAGAATGAGGATCCCAGAA-3′; TRX (forward) 5′-TGGATCCATTTCCATATGGT-3′ and (reverse) 5′-CCTTGTTAGCACCGGAGAAC-3′; MMP-9 (forward) 5′-TACAGGGCCCCTTCCTTACT-3′ and (reverse) 5′-CCACATTTGACGTCCAGAGA-3′; and GAPDH (forward) 5′-ATGGGAAGCTGGTCATCAAC-3′ and (reverse) 5′-TGTGAGGGAGATGCTCAGTG-3′. Each sample (20 μl) PCR product, together with negative controls, was subjected to electrophoresis on 1% agarose gel and stained with Ethidium bromide. All these primers were custom-designed from Invitrogen.
Statistical analysis
Results were expressed as means ± SEM; Paired or unpaired Student’s t-test were used, where appropriate, for comparing the mean values between the control and tested groups. The difference between mean values of multiple groups were analyzed by one-way analysis of variance (ANOVA) followed by a Scheffe’s post hoc analysis. Statistical significance was considered at P < 0.05. Number of experiments n = 6 in each experimental group unless mentioned otherwise.
Results
GABA-A receptor mitigated Hcy-induced oxidative stress
Confocal microscopy showed a significant increase in DCF fluorescence in Hcy-treated bEND3 cells in comparison to control cells (Fig. 1). Hcy-induced increase in DCF fluorescence was abated by the addition of muscimol (Fig. 1). Treatment of the cells with muscimol alone did not change ROS formation compared to that in control group (Fig. 1).
Fig. 1.

Hcy-induced formation of reactive oxygen species (ROS) in brain endothelial cells. Examples of images of Hcy-induced ROS formation in the cells treated with medium alone (CT), 100 μM of Hcy (Hcy), 50 μM of muscimol (MC), or 100 μM Hcy, and 50 μM of muscimol (Hcy + MC). The arrows indicate the fluorescence of ROS in the endothelial cell. n = 4 for all groups.
Expression of mRNAs for NOX-4 was significantly up-regulated in the presence of Hcy (Fig. 2A,B). Hcy-induced up-regulation of NOX-4 was prevented by muscimol (Fig. 2A,B). Muscimol alone significantly down-regulated the expression of NOX-4 in comparison to that in control group (Fig. 2A,B). The mRNA expression of thioredoxin (TRX) was significantly down-regulated in the presence of Hcy (Fig. 2A,C). This Hcy-induced down-regulation of TRX was not altered by muscimol leaving it at the level of lesser than in control (Fig. 2A,C). However, muscimol alone significantly up-regulated the expression of TRX compared to that in control (Fig. 2A,C).
Fig. 2.

Expression of mRNA of NOX-4 and TRX in brain endothelial cells(ECs). ECs were treated with: medium alone (CT),100μM of Hcy(Hcy), 50 of μM muscimol (MC), or 100 μM of Hcy, and 50 μM muscimo (Hcy + MC) for 24 h. A: Change in mRNA expression of NOX-4 and TRX shown by RT-PCR. B: Analysis of expression of mRNA of NOX-4 in each treatment group. C: Analysis of expression of mRNA of TRX in each treatment group. Data are presented as ratios of integrated optical density (IOD) of each band of the treatment group for NOX-4 or TRX to IOD of the respective GAPDH band. *P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. n = 4 for all groups.
Role of muscimol in Hcy-induced ERK activation
Hcy induced a significant increase in ERK1/2 phosphorylation in comparison to the control group (Fig. 3). This Hcy-induced ERK1/2 phosphorylation was inhibited by a GABA-A receptor agonist muscimol (Fig. 3). Phosphorylation of ERK1/2 induced by muscimol alone was greater than in the control group (Fig. 3). These results were further confirmed with immunofluorescent labeling; showing that phosphorylation of ERK-1/2 was significantly increased in Hcy-treated bEnd3 cells compared to the control (Fig. 4). Presence of muscimol inhibited this Hcy-induced increase in ERK-1/2 phosphorylation (Fig. 4). Muscimol alone did not cause apparent change in ERK-1/2 phosphorylation (Fig. 4).
Fig. 3.

Hcy-induced ERK phosphorylation in mouse brain endothelial cells. A: Phosphorylation of ERK-1/2 shown by Western blot analysis for the following treatment groups: medium alone (CT), 100 μM of Hcy (Hcy), 50 of μM muscimol (MC), or 100 μM of Hcy, and 50 μM muscimo (Hcy + MC). Membranes were re-probed for total ERK1/2, bottom gel. B: Analysis Hcy-induced ERK1/2 phosphorylation. The data are presented as ratios of integrated optical density (IOD) of each phoshorylated ERK1/2 band to IOD of the respective total ERK1/2 band. *P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. n = 4 for all groups.
Fig. 4.

Hcy-induced ERK phosphorylation in mouse brain microvascular endothelial cells (ECs) shown by confocal microscopy. Examples of images of Hcy-induced ERK phosphorylation in the cells treated with: medium alone (CT), 100 μM of Hcy (Hcy), 50 μM of muscimol (MC), or 100 μM Hcy, and 50 μM of muscimol (Hcy + MC). The arrows indicate the red fluorescence of phosphorylated ERK, while the nuclei are identified with DAPI staining (blue). n = 4 for all groups.
Role of GABA-A receptor in Hcy-induced MMP-9 activation
Hcy increased activity of MMP-9 (Fig. 5A,B,E,F) and its mRNA expression (Fig. 5C,D,G,H). The presence of muscimol (agonist of GABA-A receptor) and UA (antioxidant) significantly decreased this Hcy-induced MMP-9 activity (Fig. 5A,B,E,F) and mRNA expression (Fig. 5C,D,G,H). While muscimol alone did not have an effect on activity of MMP-9, its presence decreased Hcy-induced activity of MMP-9 in ECs (Fig. 5A,B). Muscimol decreased Hcy-induced mRNA overexpression of MMP-9 (Fig. 5C,D). The mRNA expression of MMP-9 was decreased by muscimol alone (Fig. 5C,D). Uric acid significantly decreased Hcy-induced MMP-9 activity and its mRNA expression (Fig. 5E–H). Uric acid alone had no effect on activity or mRNA expression of MMP-9 (Fig. 5E–H).
Fig. 5.

Hcy-induced MMP-9 formation in endothelial cells. The cells were treated with: medium alone (CT), 100 μM Hcy (Hcy), 50 μM muscimol (MC), or 100 μM Hcy, and 50 μM muscimol (Hcy + MC). Activity of MMP-9 in endothelial cells shown by zymography (A,E) and by RT-PCR (C,G). B,F: Analyses of Hcy-induced MMP-9 activity. Data are presented as ratios of integrated optical density (IOD) of each MMP-9 band to IOD of the respective loading control (same volume of each sample was run on SDS–PAGE and stained by Coomassie blue) in the treatment groups. D,H: Analyses of Hcy-induced MMP-9 mRNA expression. The data are presented as ratios of IOD of each MMP-9 band to IOD of the GAPDH band in the respective treatment groups. *P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. n = 4 for all groups.
Role of GABA-A receptor in expression of Col-IV and elastin
Hcy significantly increased protein expression of Col-IV (Fig. 6A,B) and decreased expression of elastin (Fig. 6A,C). This resulted in increase of Col-IV/elastin expression ratio (Fig. 6A,D). Presence of muscimol significantly lowered Hcy-induced overexpression of Col-IV (Fig. 6A,B) and restored Hcy-down-regulated elastin expression to its level in control group (Fig. 6A,C). This resulted in significantly lower Col-IV/elastin expression ratio compared to that in the group with Hcy alone (Fig. 6A,D). Muscimol alone did not change expression of Col-IV but it decreased elastin expression (Fig. 6A–C). Therefore, this led to a greater Col-IV/elastin expression ratio than in control, but it was significantly less than in Hcy treated group (Fig. 6A,D).
Fig. 6.

Expression of collogen IV(Col-IV)and elastin in endotheial cells. The cells were tretated with medium alone (CT),100μMHcy(Hcy),50μM muscimol (MC), or 100 μMHcy, and 50μM muscimol (Hcy + MC) for 24 h. A: Expression of Col-IV and elastin shown by Western blot analysis. B–D: Analyses of Col-IV and elastin expressions. For Col-IV (B) or elastin (C) expressions the data are presented as ratios of integrated optical density (IOD) of each band in the treatment group to IOD of the respective β-actin band. To determine changes in Col-IV/elastin ratio induced by Hcy (D) the data a represented asratios of IOD col-IV band to IOD of elastinb and of the respective treatment group.*P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. n = 4 for all groups.
Hcy decreased NO bioavailability by antagonizing the GABA-A receptor
At normal concentrations (5 μM) Hcy did not have an effect on expression of eNOS, iNOS, or TIMP4 (Fig. 7A,B). At higher concentration (100 μM), Hcy significantly decreased TIMP4 and eNOS, and increased iNOS expression (Fig. 7A,B). Hcy-induced decrease in TIMP4 or eNOS expressions were restored by addition of muscimol (Fig. 7A,B). Presence of muscimol significantly inhibited Hcy-induced overexpression of iNOS (Fig. 7A,B). Muscimol alone increased the protein expression of TIMP4 and eNOS and decreased expression of iNOS (Fig. 7A,B).
Fig. 7.
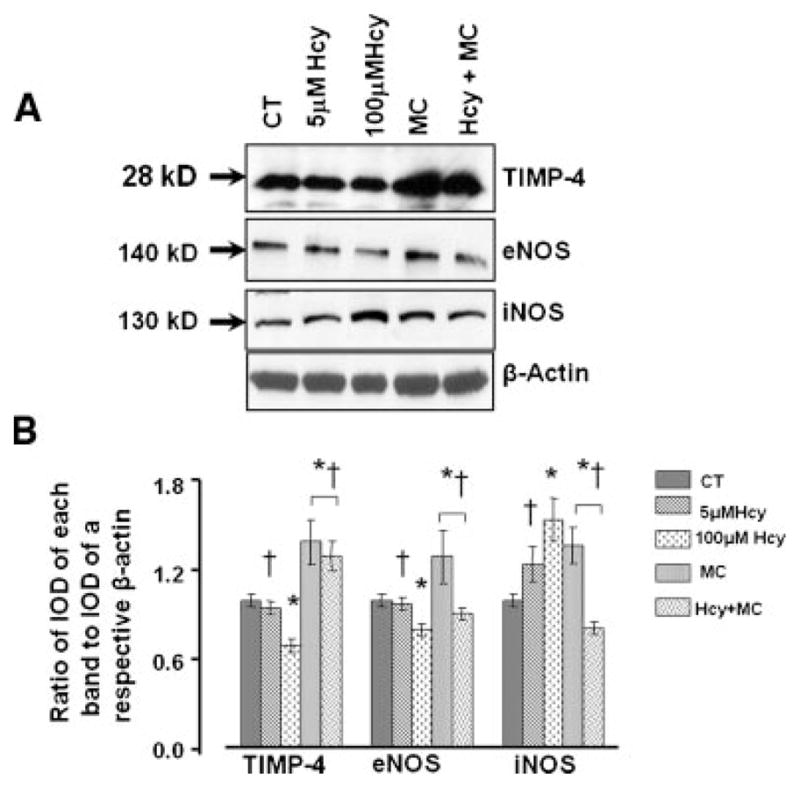
Hcy-induced protein expression of TIMP4, eNOS, and iNOS in endotheial cells. A: The cells were treated with medium alone (CT), 5 μM of Hcy, 100 μM of Hcy, 50 μM of muscimol (MC), or 100 μM of Hcy, and 50 μM of muscimol (Hcy + MC) for 24 h. B: Densitometric analyses of TIMP-4, eNOS, and iNOS expressions. The data are presented as ratios of IOD of each target protein band to IOD of β-actin band in the respective treatment group. *P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. n = 4 for all groups.
In parallel series of experiments designed to determine if Hcy affected iNOS more than eNOS, treatment of the ECs with 100 μM of Hcy significantly increased expression of iNOS in the cultured ECs (Fig. 8A,B). This increase was reversed by the presence of L-NAME and 1400W (a specific blocker of iNOS) had even greater effect in decreasing expression of iNOS in the presence of Hcy (Fig. 8A,B). Presence of muscimol along with L-NAME decreased further Hcy-induced iNOS expression (Fig. 8A,B). However, muscimol had no effect on Hcy-induced iNOS expression in the presence of 1400W (Fig. 8A,B). L-NAME or 1400W alone significantly decreased expression of iNOS in ECs compared to that in control or even in Hcy and L-NAME-treated groups (Fig. 8A,B).
Fig. 8.
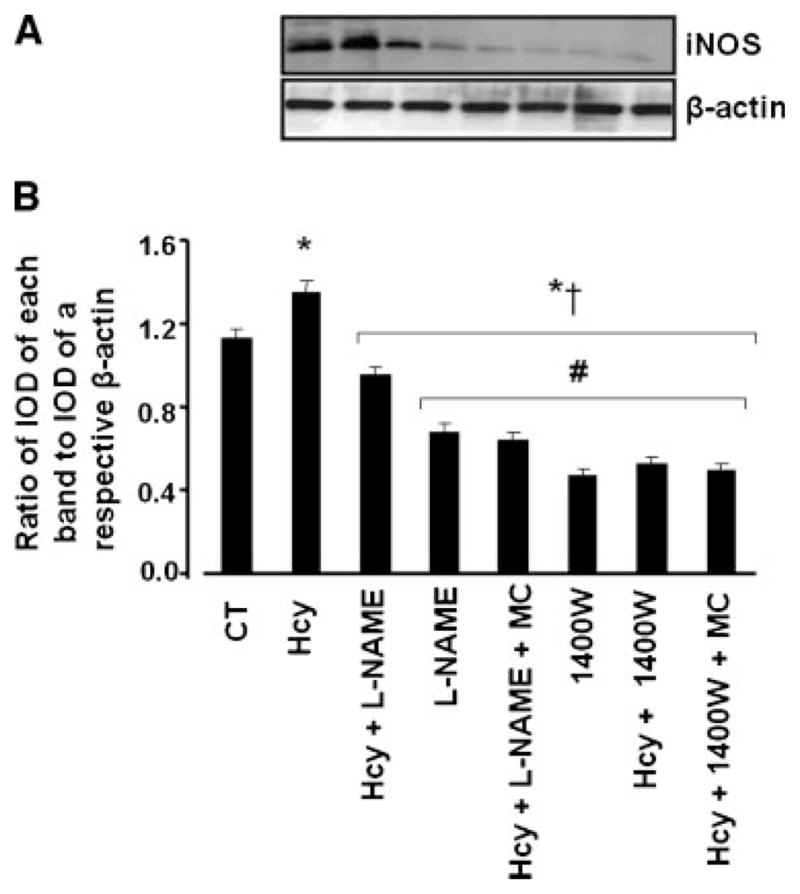
Hcy-induced changes in iNOS expression in endothelial cells. A: The cells were treated with medium alone (CT), 100 μM Hcy, 100 μM Hcy with 100 μM L-Name (Hcy + L-NAME), 100 μM L-NAME (L-NAME), 100 μM Hcy with 100 μM L-NAME and 50 μM muscimol (Hcy + L-NAME + MC), 100 μM 1400W (1400W), 100 μM Hcy with 100 μM 1400W (Hcy + 1400W), and 100 μM Hcy with 100 μM 1400W and 50 μM muscimol (Hcy + 1400W + MC) for 24 h. B: Densitometric analysis of iNOS expression. The data are presented as ratios of IOD of each iNOS band to IOD of β-actin band in the respective treatment group. *P < 0.05 versus control, †P < 0.05 versus 100 μM of Hcy. #P < 0.05 versus Hcy + L-NAME. n = 4 for all groups.
Hcy-induced NO production
Real-time measurements of NO content in brain ECs showed that Hcy induced significant decrease in NO production compared to that in control group (Fig. 9). Pre-treatment with muscimol reversed this Hcy-induced decrease in NO production (Fig. 9). Content of NO at its maximum level was higher in the cells treated with muscimol alone (30 ± 4.5 pA) compared to that in the control group (22.5 ± 3.0 pA, Fig. 9).
Fig. 9.

Effect of Hcy on production of nitric oxide (NO) in mouse brain endothelial cells. Confluent endothelial cells were treated with medium alone (CT), 100 μM of Hcy, or 100 μM of Hcy, and 50 μM of muscimol (Hcy + MC). Genreation of NO was recorded by Apollo 4000 free radical analyzer after administration of 10−5 M of acetylcholine (ACH) to each group of cells mentioned above. Before the measurements, the NO probe was standardized using nitroprusside as an NO donor. The moment of ACH treatment is indicated by the arrow. n = 4 for all groups.
Discussion
There are three ranges of HHcy: moderate (16–30 μM), intermediate (31–100 μM), and severe (>100 μM) (Ji and Kaplowitz, 2004). In homocyteineuric patients, one of the examples of severe HHcy, plasma level of Hcy up to 0.4 mM have been found (Perry et al., 1967). The concentrations used in the present study represent a severe HHcy.
Previously we showed that GABA-A receptor is involved in Hcy-induced EC layer integrity and permeability changes (Tyagi et al., 2007). The results of the present study demonstrate the role of GABA-A receptor as an inhibitory neurotransmitter in brain EC remodeling, which may be one of the mechanisms leading to an increase in EC layer permeability seen earlier (Tyagi et al., 2007). Pathologically high levels (100 μM) of Hcy triggered oxidative stress, leading to MMP-9 activation via ERK-1/2 signaling (Moshal et al., 2006a). Hcy caused a dramatic increase in ROS, in part by decreasing TRX and increasing NOX-4, while muscimol diminished these effects of Hcy. These data indicate that muscimol specifically competes with Hcy for binding to the GABA-A receptor affecting the redox enzymes.
In the present study, we found that UA (an antioxidant) decreased Hcy-induced both protein and mRNA overexpression of MMP-9. These results confirm the role of oxidative stress in activation of MMPs. High levels of Hcy activated the MMPs and therefore, altered collagen/elastin ratio leading to sub-endothelial matrix remodeling of the endothelial layer. Since UA decreased Hcy-induced activation of MMPs, this suggested a role of ROS in Hcy-induced sub-endothelial matrix remodeling.
In tissue and cells, NADPH oxidase is a primary source of ROS generation (Dworakowski et al., 2006). Activity of most of the oxidants (NOX) depends on levels of antioxidant enzyme, Trx (Dworakowski et al., 2006). The Trx system protects cells from oxidative injury under a variety of pathologic conditions including atherosclerosis, hypertension, vascular injury, and cardiovascular disease (Yamawaki et al., 2003). Trx is expressed ubiquitously in ECs (Okuda et al., 2001) and protects ECs from ROS-induced cytotoxicity (Nakamura et al., 1994). Trx appears to exert its ROS-scavenging properties through Trx peroxidase (Chae et al., 1994). We showed earlier that the levels of Trx in tissue and cardiac microvascular ECs decreased in result of increased levels of NOX-4 (Tyagi et al., 2005). Hcy itself undergo auto-oxidation causing the disruption of the redox enzymes (Trx/NOX) in vascular and neural cells (Obeid and Herrmann, 2006). It is known that disbalance of Trx/NOX enzymes may result in neurological dysfunction (Ho et al., 2001). The results of the present study show that Hcy increased NOX-4 and decreased expression of Trx, while muscimol reversed these effects of Hcy. These data suggest a role of GABA-A receptor activation in modulating the expression of the redox enzymes (Trx/NOX) disrupted by increased levels of Hcy. These results also suggest that Hcy serves as an antagonist to GABA-A receptor and increases oxidative stress by inhibiting GABA-A receptor indicating that oxidative stress was a GABA-A receptor activity dependent. These data coincided with results of another study, where it was suggested that Hcy-stimulated superoxide anion production in the vascular wall and was mediated through the activation of NADPH oxidase leading to the endothelial dysfunction during HHcy (Edirimanne et al., 2007). It has been shown that Hcy-induced ERK (MAPK) stimulation in cardiac ECs can be blocked by pretreatment with an antioxidant, N-acetylcysteine (NAC), suggesting a role of oxidative stress in signaling events (Bogoyevitch et al., 2000). In the present study, we found that Hcy increased ERK phosphorylation in brain ECs, which was suppressed by muscimol. These results indicate that Hcy antagonized the GABA-A receptor to evoke the activation of MAPK signaling. Thus, our study suggests that GABA-A receptor may be a key player in ERK modulation in ECs, and its agonists may play a protective role against Hcy-induced alterations in cerebrovascular remodeling by deactivating the MAPK signal transduction.
Previously, we showed that Hcy increased MMP-9 activity in aortic microvascular ECs (Moshal et al., 2006b). However, the role of GABA-A receptor in activation of MMP-9 was not defined. In the present study, for the first time, we demonstrated that mouse brain ECs contained the GABA-A receptor and its activity was suppressed by increased content of Hcy. Others have suggested that GABA-A receptor antagonist may increase levels of MMP-9 activity in rats (Zhang et al., 1998). In the present study, muscimol activated GABA-A receptor in the mouse brain ECs and ameliorated the Hcy-induced MMP-9 activation. These results suggest that latent MMPs were activated by Hcy-induced oxidative stress. Treatment with muscimol reduced the Hcy-induced MMP-9 activation and its mRNS expression suggesting a role of GABA-A receptor in activation of MMP-9. These data may also indicate that Hcy behaved as an antagonist to GABA-A receptor.
An imbalance between MMPs and TIMPS may lead to vascular pathological remodeling (Wald et al., 2002; Rosenberg, 2009) by altering the collagen/elastin content ratio (Castro et al., 2008). In accordance to our previous in vivo study (Lominadze et al., 2006), the present study showed an increase in MMP-9 activity due to increased Hcy concentration. In addition, we found an increased expression of Coll-IV and a decreased elastin content, which led to an increase in Coll-IV/elastin ratio in Hcy treated ECs. These data suggest that an increase in Hcy concentration may lead to a sub-endothelial matrix remodeling by altering Coll-IV/elastin ratio. Muscimol reversed Hcy-induced activation of MMP-9 and caused an increase in Coll-IV/elastin ratio indicating that muscimol may compete with Hcy for binding with GABA-A receptor leading to a lesser accumulation of Hcy. In addition, muscimol-induced activation of GABA-A receptor may inhibit the Hcy-induced sub-endothelial matrix remodeling suggesting a role of GABA-A receptor in vascular remodeling. HHcy inactivated TIMP-4 and increased activities of MMP-9 and MMP-2 in hearts of hyperhomocysteinmic rats (Sood et al., 2002). The results of the present study show that Hcy decreased protein levels of TIMP-4, while muscimol prevented this effect of Hcy in mouse brain ECs. This may be a strong indication that Hcy affected balance of MMP-9 and TIMP-4 activity levels, secondary to induction of GABA-A receptor.
Pathologically high levels of Hcy attenuated and impaired the inhibitory neurotransmitter (GABA-A receptor) mechanism, preventing decrease in blood pressure (Kishi et al., 2001). It is known that muscimol decreases arterial blood pressure and has been used as an antihypertensive agent (Unger et al., 1984). Muscimol-induced decrease in blood pressure was associated with an increased production of NO from ECs (Baum and Becker, 1982). We showed earlier that decrease in eNOS expression ameliorated Hcy-induced vascular remodeling (Shastry et al., 2005). The results of the present study indicate that activation of GABA-A receptor induced generation of NO in bEND3 cells. Activation of GABA-A receptor ameliorated Hcy-induced a decrease in eNOS and an increase in iNOS expressions. A non-selective NOS inhibitor, L-NAME (Laude et al., 2003) reduced Hcy-induced expression of iNOS to the control level while 1400W, a selective iNOS inhibitor (Garvey et al., 1997; Laude et al., 2003), completely abrogated the Hcy-induced iNOS expression. Muscimol did not have an additional effect on Hcy-induced iNOS expression that was already abrogated by 1400W. However, activation of GABA-A receptor with muscimol further decreased Hcy-induced iNOS expression already reduced by L-NAME. Since muscimol ameliorated Hcy-induced decrease in eNOS expression and enhanced NO production even in the presence of Hcy, suggested a role of GABA-A receptor in production of NO by ECs through eNOS activation. In addition, our data indicate a role of muscimol in reversing the Hcy-induced an increase in NOX-4 and a decrease in TRX expressions suggesting a role of GABA-A receptor in modulating oxidative stress induced by an increased content of Hcy. Taken together these results suggest that activity of GABA-A receptor may have a significant functional role in modulating NO bioavailability by mainly affecting eNOS expression. In addition, our findings coincide with the notion that NO keeps MMP-9 in a latent form (Mujumdar et al., 2001). HHcy caused an increase in oxidative stress and activation of the latent MMP leading to generation of peroxynitrite and inactivation of TIMPs (Hunt et al., 2002). Therefore, our study may indicate that GABA-A receptor can be involved in prevention of Hcy-induced vascular remodeling.
Our observations define a novel role of GABA-A receptor in Hcy-induced MMP-9 activation and support the hypothesis presented in Figure 10. At a pathologically high-level, Hcy competes with GABA-A receptor and affects its activity resulting in up-regulation of NOX-4 and down-regulation of TRX mRNA expressions. This disbalance of NOX-4/TRX enzymes leads to increased production of ROS and activation of MMP-9 via ERK1/2 signaling pathway, in parallel to the decrease of TIMP-4 expression. Enhanced production of ROS may cause decrease in eNOS and increase in iNOS protein levels, which may lead a decreased bioavailability of NO. Thus, our study unequivocally underscores the essential role of GABA-A receptor in Hcy-induced alterations leading to the cerebrovascular remodeling.
Fig. 10.

Schematic representation of the proposed hypothesis for Hcy-induced cerebrovascular remodeling in mouse brain endothelial cells. At pathologically high levels (100 μM) of Hcy induces oxidative stress, in part, by decreasing TRX and increasing NOX-4 expression levels, and decreasing NO bioavailability. This leads to activation of MMP-9 via ERK-1/2 signaling pathway and by partial inhibition of GABA-A receptor. This is confirmed by an effect of GABA-A receptor agonist, muscimol, which ameliorates Hcy-induced oxidative stress, NO production, and MMP-9 activity/expression, in part by dephosphorylation of ERK.
HHcy is a major risk factor for neuroinflammatory and neurodegenerative diseases. This study specifically highlights the findings on contribution of GABA-A receptor in Hcy-induced MMP activation. GABA-A receptor could be a beneficial therapeutic candidate for the treatment of HHcy-associated pathologies, such as stroke, dementia, and neurological disorders.
Limitations of the study
GABA-A receptor is a Cl-ion channel (Liu et al., 2002; Anuradha et al., 2008). Although we did not investigate in the present study, defining the Hcy-induced possible changes in GABA-A receptor function as a Cl-ion channel would have been very interesting. Our data did not allow any conclusion regarding the GABA-A receptor function as a Cl-ion channel during increased content of Hcy. However, it has been shown that muscimol can affect function of GABA-A receptor as a Cl-ion channel (Reis et al., 2007). Therefore, it is plausible that if an increased content of Hcy inhibits function of GABA-A receptor as a Cl-ion channel, muscimol may abrogate this detrimental effect of Hcy. The separate studies that may address this hypothesis are needed.
Acknowledgments
Contract grant sponsor: NIH;
Contract grant numbers: HL-71010, NS-51568, HL-80394.
Supported in part by the NIH grants HL-71010 and NS-51568 to S.C.T. and HL-80394 to D.L.
Literature Cited
- Anuradha H, Srikumar BN, Shankaranarayana Rao BS, Lakshmana M. Euphorbia hirta reverses chronic stress-induced anxiety and mediates its action through the GABA(A) receptor benzodiazepine receptor-Cl(−) channel complex. J Neural Transm. 2008;115:35–42. doi: 10.1007/s00702-007-0821-6. [DOI] [PubMed] [Google Scholar]
- Baum T, Becker F. Hypotensive and postural effects of the gamma-aminobutyric acid agonist muscimol and of clonidine. J Cardiovasc Pharmacol. 1982;4:165–169. doi: 10.1097/00005344-198203000-00001. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Ng DCH, Court N, Draper KA, Dhillon A, Abas L. Intact mitochondrial electron transport function is essential for signalling by hydrogen peroxide in cardiac myocytes. J Mol Cell Cardiol. 2000;32:1469–1480. doi: 10.1006/jmcc.2000.1187. [DOI] [PubMed] [Google Scholar]
- Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- Bulleit RF, Hsieh T. MEK inhibitors block BDNF-dependent and -independent expression of GABA(A) receptor subunit mRNAs in cultured mouse cerebellar granule neurons. Brain Res Dev Brain Res. 2000;119:1–10. doi: 10.1016/s0165-3806(99)00119-4. [DOI] [PubMed] [Google Scholar]
- Castro MM, Rizzi E, Figueiredo-Lopes L, Fernandes K, Bendhack LM, Pitol DL, Gerlach RF, Tanus-Santos JE. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis. 2008;198:320–331. doi: 10.1016/j.atherosclerosis.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem. 1994;269:27670–27678. [PubMed] [Google Scholar]
- Dworakowski R, Anilkumar N, Zhang M, Shah AM. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem Soc Trans. 2006;34:960–964. doi: 10.1042/BST0340960. [DOI] [PubMed] [Google Scholar]
- Edirimanne VER, Woo CWH, Siow YL, Pierce GN, Xie JYOK. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can J Physiol Pharmacol. 2007;85:1236–1247. doi: 10.1139/Y07-112. [DOI] [PubMed] [Google Scholar]
- Frolund B, Ebert B, Kristiansen U, Liljefors T, Krogsgaard-Larsen P. GABA-A receptor ligands and their therapeutic potentials. Curr Top Med Chem. 2002;2:817. doi: 10.2174/1568026023393525. [DOI] [PubMed] [Google Scholar]
- Fruscella P, Sottocorno M, Di BM, Diomede L, Piccardi N, Cagnotto A, Grossi G, Romano M, Mennini T, Roma G. 1, 5-Benzodiazepine tricyclic derivatives exerting anti-inflammatory effects in mice by inhibiting interleukin-6 and prostaglandinE(2)production. Pharmacol Res. 2001;43:445–452. doi: 10.1006/phrs.2001.0800. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJR, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- Griffiths R, Williams DC, O’Neill C, Dewhurst IC, Ekuwem CE, Sinclair CD. Synergistic inhibition of [3H]muscimol binding to calf-brain synaptic membranes in the presence of L-homocysteine and pyridoxal 5′-phosphate. A possible mechanism for homocysteine-induced seizures. Eur J Biochem. 1983;137:467–478. doi: 10.1111/j.1432-1033.1983.tb07850.x. [DOI] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Wen G, Lawton MT, Boudreau NJ, Bollen AW, Yang GY, Barbaro NM, Higashida RT, Dowd CF, Halbach VV, Young WL. Abnormal expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in brain arteriovenous malformations. Stroke. 2003;34:925–931. doi: 10.1161/01.STR.0000061888.71524.DF. [DOI] [PubMed] [Google Scholar]
- Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB. Homocysteine potentiates β-amyloid neurotoxicity: Role of oxidative stress. J Neurochem. 2001;78:249–253. doi: 10.1046/j.1471-4159.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- Hunt MJ, Aru GM, Hayden MR, Moore CK, Hoit BD, Tyagi SC. Induction of oxidative stress and disintegrin metalloproteinase in human heart end-stage failure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L239–L245. doi: 10.1152/ajplung.00001.2002. [DOI] [PubMed] [Google Scholar]
- Jacobsen DW. Hyperhomocysteinemia and oxidative stress: Time for a reality check? Arterioscler Thromb Vasc Biol. 2000;20:1182–1184. doi: 10.1161/01.atv.20.5.1182. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourquin J, Tremblay E, Decanis N, Charton G, Hanessian S, Chollet AM, Le Diguardher T, Khrestchatisky M, Rivera S. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur J Neurosci. 2003;18:1507. doi: 10.1046/j.1460-9568.2003.02876.x. [DOI] [PubMed] [Google Scholar]
- Kang KA, Zhang R, Piao MJ, Ko DO, Wang ZH, Lee K, Kim BJ, Shin T, Park JW, Lee NH, Yoo BS, Hyun JW. Inhibitory effects of triphlorethol-A on MMP-1 induced by oxidative stress in human keratinocytes via ERK and AP-1 inhibition. J Toxicol Environ Health Part A. 2008;71:992–9999. doi: 10.1080/01932690801934653. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Sakai K, Shigematsu H, Shimokawa H, Takeshita A. Overexpression of eNOS in the RVLM causes hypotension and bradycardia via GABA release. Hypertension. 2001;38:896–901. [PubMed] [Google Scholar]
- Ku BM, Lee YK, Jeong JY, Mun J, Han JY, Roh GS, Kim HJ, Cho GJ, Choi WS, Yi GS, Kang SS. Ethanol-induced oxidative stress is mediated by p38 MAPK pathway in mouse hippocampal cells. Neurosci Lett. 2007;419:64–67. doi: 10.1016/j.neulet.2007.03.049. [DOI] [PubMed] [Google Scholar]
- Laude K, Favre J, Thuillez C, Richard V. NO produced by endothelial NO synthase is a mediator of delayed preconditioning-induced endothelial protection. Am J Physiol Heart Circ Physiol. 2003;284:H2053–H2060. doi: 10.1152/ajpheart.00627.2002. [DOI] [PubMed] [Google Scholar]
- Lazzarini R, Malucelli BE, Palermo-Neto J. Reduction of acute inflammation in rats by diazepam: Role of peripheral benzodiazepine receptors and corticosterone. Immunopharmacol Immunotoxicol. 2001;23:253–265. doi: 10.1081/iph-100103864. [DOI] [PubMed] [Google Scholar]
- Lee WS, Limmroth V, Ayata C, Cutrer FM, Waeber C, Yu X, Moskowitz MA. Peripheral GABAA receptor-mediated effects of sodium valproate on dural plasma protein extravasation to substance P and trigeminal stimulation. Br J Pharmacol. 1995;116:1661–1667. doi: 10.1111/j.1476-5381.1995.tb16388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limmroth V, Lee WS, Moskowitz MA. GABAA-receptor-mediated effects of progesterone, its ring-A-reduced metabolites and synthetic neuroactive steroids on neurogenic oedema in the rat meninges. Br J Pharmacol. 1996;117:99–104. doi: 10.1111/j.1476-5381.1996.tb15160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QY, Chang YH, Schaffner AE, Smith SV, Barker JL. Allopregnanolone activates GABAA receptor/Cl− channels in a multiphasic manner in embryonic rat hippocampal neurons. J Neurophysiol. 2002;88:1147–1158. doi: 10.1152/jn.00942.2001. [DOI] [PubMed] [Google Scholar]
- Lominadze D, Roberts A, Tyagi N, Tyagi S. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Circ Physiol. 2006;290:H1206–H1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wahl LM. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-{kappa}B activity in lipopolysaccharide-activated human primary monocytes. J Immunol. 2005;175:5423–5429. doi: 10.4049/jimmunol.175.8.5423. [DOI] [PubMed] [Google Scholar]
- Mannello F, Gazzanelli G. Tissue inhibitors of metalloproteinases and programmed cell death: Conundrums, controversies, and potential implications. Apoptosis. 2001;6:479–482. doi: 10.1023/a:1012493808790. [DOI] [PubMed] [Google Scholar]
- Montesano R, Pepper M, Mohle-Steinlein U, Risau W, Wagner E, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435–445. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- Moshal KS, Sen U, Tyagi N, Henderson B, Steed M, Ovechkin AV, Tyagi SC. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. Am J Physiol Cell Physiol. 2006a;290:C883–C891. doi: 10.1152/ajpcell.00359.2005. [DOI] [PubMed] [Google Scholar]
- Moshal K, Singh M, Sen U, Rosenberger D, Henderson B, Tyagi N, Zhang H, Tyagi SC. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol. 2006b;291:H2825–H2835. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- Mujumdar V, Tyagi S. Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertens. 1999;17:261–270. doi: 10.1097/00004872-199917020-00011. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int J Cardiol. 2001;79:277–286. doi: 10.1016/s0167-5273(01)00449-1. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Matsuda M, Furuke K, Kitaoka Y, Iwata S, Toda K, Inamoto T, Yamaoka Y, Ozawa K, Yodoi J. Adult T cell leukemia-derived factor/human thioredoxin protects endothelial F-2 cell injury caused by activated neutrophils or hydrogen peroxide. Immunol Lett. 1994;42:75–80. doi: 10.1016/0165-2478(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- Okuda M, Inoue N, Azumi H, Seno T, Sumi Y, Hirata KI, Kawashima S, Hayashi Y, Itoh H, Yodoi J, Yokoyama M. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1483–1487. doi: 10.1161/hq0901.095550. [DOI] [PubMed] [Google Scholar]
- Perry TL, Hansen S, MacDougall L, Warrington PD. Sulfur-containing amino acids in the plasma and urine of homocystinurics. Clinica Chimica Acta. 1967;15:409–420. doi: 10.1016/0009-8981(67)90005-8. [DOI] [PubMed] [Google Scholar]
- Reis G, Pacheco D, Francischi J, Castro M, Perez A, Duarte I. Involvement of GABAA receptor-associated chloride channels in the peripheral antinociceptive effect induced by GABAA receptor agonist muscimol. Eur J Pharmacol. 2007;564:112–115. doi: 10.1016/j.ejphar.2007.02.043. [DOI] [PubMed] [Google Scholar]
- Robert K, Pages C, Ledru A, Delabar J, Caboche J, Janel N. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience. 2005;133:925–935. doi: 10.1016/j.neuroscience.2005.03.034. [DOI] [PubMed] [Google Scholar]
- Rosell A, Alvarez-Sabin J, Arenillas JF, Rovira A, Delgado P, Fernandez-Cadenas I, Penalba A, Molina CA, Montaner J. A matrix metalloproteinase protein array reveals a strong relation between MMP-9 and MMP-13 with diffusion-weighted image lesion increase in human stroke. Stroke. 2005;36:1415–1420. doi: 10.1161/01.STR.0000170641.01047.cc. [DOI] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2009;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Sachdev PS, Valenzuela M, Wang XL, Looi JCL, Brodaty H. Relationship between plasma homocysteine levels and brain atrophy in healthy elderly individuals. Neurology. 2002;58:1539–1541. doi: 10.1212/wnl.58.10.1539. [DOI] [PubMed] [Google Scholar]
- Santiard-Baron D, Aupetit J, Janel N. Plasma homocysteine levels are not increased in murine models of Alzheimer’s disease. Neurosci Res. 2005;53:447–449. doi: 10.1016/j.neures.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Shastry S, Moning L, Tyagi N, Steed M, Tyagi S. GABA receptors and Nitric oxide ameliorate constrictive collagen remodeling on hyperhomocysteinemia. J Cell Physiol. 2005;205:422–427. doi: 10.1002/jcp.20416. [DOI] [PubMed] [Google Scholar]
- Shastry S, Tyagi N, Moshal K, Lominadze D, Hayden M, Tyagi S. GABA receptors ameliorate Hcy-mediated integrin shedding and constrictive collagen remodeling in microvascular endothelial cells. Cell Biochem Biophys. 2006;45:157–165. doi: 10.1385/CBB:45:2:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood HS, Cox MJ, Tyagi SC. Generation of nitrotyrosine precedes activation of metalloproteinase in myocardium of hyperhomocysteinemic rats. Antioxidants Redox Signal. 2002;4:799–804. doi: 10.1089/152308602760598954. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:H2649–H2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- Tyagi N, Ovechkin A, Lominadze D, Moshal K, Tyagi S. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J Cell Biochem. 2006;98:1150–1162. doi: 10.1002/jcb.20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Moshal KS, Tyagi SC, Lominadze D. γ-aminbutyric acid A receptor mitigates homocysteine-induced endothelial cell permeability. Endothelium. 2007;14:315–323. doi: 10.1080/10623320701746164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T, Becker H, Dietz R, Ganten D, Lang RE, Rettig R, Schomig A, Schwab NA. Antihypertensive effect of the GABA receptor agonist muscimol in spontaneously hypertensive rats. Role of the sympathoadrenal axis. Circ Res. 1984;54:30–37. doi: 10.1161/01.res.54.1.30. [DOI] [PubMed] [Google Scholar]
- Valentin F, Bueb JL, Kieffer P, Tschirhart E, Atkinson J. Oxidative stress activates MMP-2 in cultured human coronary smooth muscle cells. Fundam Clin Pharmacol. 2005;19:661–667. doi: 10.1111/j.1472-8206.2005.00371.x. [DOI] [PubMed] [Google Scholar]
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. BMJ. 2002;325:1202–1206. doi: 10.1136/bmj.325.7374.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Ho YS, Pan MH, Lin JK. Mechanisms of cell death induced by nitric oxide and peroxynitrite in Calu-1 cells. Environ Toxicol Pharmacol. 1998;6:35–44. doi: 10.1016/s1382-6689(98)00016-7. [DOI] [PubMed] [Google Scholar]
- Yamawaki H, Haendeler J, Berk BC. Thioredoxin: A key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- Zhang JW, Deb S, Gottschall PE. Regional and differential expression of gelatinases in rat brain after systemic kainic acid or bicuculline administration. Eur J Neurosci. 1998;10:3358–3368. doi: 10.1046/j.1460-9568.1998.00347.x. [DOI] [PubMed] [Google Scholar]